
Abstract
A new multistage synthesis of the N-borylated dipeptide B12-PheGlyOH has been proposed. The approach is based on the reaction of nucleophilic addition of amino acid derivatives to the [B12H11NCCH3]– anion. The products of each stage have been characterized by multinuclear NMR spectroscopy, IR absorption spectroscopy, and ESI mass spectrometry.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
INTRODUCTION
Neutron capture therapy (NCT) is a binary method of non-invasive treatment of malignant tumors [1]; however, the development of this method is directly related with the search for new effective therapeutic agents [2]. An important requirement for compounds used for 10B-NCT is a high selectivity of accumulation in tumor tissues and a high content of boron in molecules to create the required therapeutic concentration [3]. closo-Dodecaborate anion [B12H12]2– attracts close attention of researchers when creating new agents for 10B-NCT due to its low toxicity and high boron content [4–6].
The effectiveness of drugs for 10B-NCT depends on the selectivity of the accumulation of boron compounds in tumor cells, which requires the introduction of vector groups into the cluster cage. Amino acids and peptides are important transport groups due to their ability to bind selectively to various receptors, mainly expressed by tumor cells [7–11].
It was previously shown that the addition of nucleophilic reagents of various natures to nitrile functional groups in anionic boron clusters can be considered as a convenient way of directed synthesis of their substituted derivatives [12–24], including those based on biologically active substances [6, 25] and biomimetic systems [26, 27]. It was found that the addition of tert-butyl esters of proteinogenic amino acids to nitrile derivatives of the closo-decaborate anion and their subsequent selective hydrolysis is a convenient method for the preparation of N-borylated peptides [28]. tert-Butyl esters of amino acids are used due to the stability of amidine-closo-borates under the conditions of their hydrolysis [29].
The development of methods for the preparation of nitrile derivatives of the closo-dodecaborate anion [30, 31] will expand the possibilities for the creation of new promising preparations for 10B-NCT on their basis.
The aim of this work is to obtain conjugates of the nitrile derivative of the closo-dodecaborate anion as a starting compound for the synthesis of borylated peptides with phenylalanine for use in 10B-NCT.
EXPERIMENTAL
Elemental analysis for carbon, hydrogen, and nitrogen was performed on a CHNS-3 FA 1108 Elemental Analyzer (Carlo Erba). Boron was determined by ICP MS on an iCAP 6300 Duo inductively coupled plasma atomic emission spectrometer at the Center for Collective Use of the Scientific Analytical Center of the Federal State Unitary Enterprise IREA of the National Research Center “Kurchatov Institute.”
IR spectra of compounds were recorded on an Infralum FT-08 IR Fourier spectrophotometer (NPF AP Lumex) in the range 4000–400 cm–1 with a resolution of 1 cm–1. Samples were prepared as chloroform solutions.
1H, 11B, and 13C NMR spectra of solutions of the investigated compounds in CD3CN were recorded on a Bruker MSL-300-pulsed Fourier spectrometer (Germany) at frequencies of 300.3, 96.32, and 75.49 MHz, respectively, with internal deuterium stabilization. Tetramethylsilane or boron trifluoride etherate were used as external standards.Footnote 1
Preparative high performance liquid chromatography (HPLC) was performed on a STAYER isocratic HPLC system (NPKF Akvilon) with a spectrophotometric detector (UVV 104 with a variable wavelength of 200–360 nm) on a Knauer Eurospher 110 Si column.
Solvents of special quality grade (Russian State Standard) were used without further purification. Esters of amino acids in the form of hydrochlorides GlyOtBu and PheOMe were also used without further purification.
(NBu4)[B12H11(NCCH3)] (1) was obtained according to the method described [31]. Trifluoroacetic acid (1 mL) was added to a solution of (NBu4)2[B12H12] (0.94 g, 1.5 mmol) in acetonitrile (20 mL). The reaction mixture was heated in an autoclave at a temperature of 100°C for 30 min. The solvent was distilled off on a rotary evaporator; the solid residue was dissolved in CH3CN (30 mL) and CF3COOH (1 mL) was added. The solution was heated to 80°C in a dry argon atmosphere in an autoclave of the Minotavr-2 microwave synthesis system (NPF AP Lumex) at a radiation power of 100 W for 1 h. The solution cooled to room temperature was concentrated on a rotary evaporator and acetic acid (20 mL) was added. The resulting product was separated by decantation and washed with diethyl ether. Yield, 80% (0.5 g, 1.2 mmol).
(NBu4)[B12H11NHC(CH3)HNCH(CH2C6H5)COOСH3] (2). A suspension of HCl∙PheOMe (0.322 g, 1.5 mmol) in THF (15 mL) was prepared, and 4-dimethylaminopyridine (0.177 g, 1.45 mmol) was added. The suspension was stirred for 15 min and filtered through a Schott filter. Compound 1 (0.424 g, 1 mmol) was added to the resulting solution. The reaction mixture was stirred at room temperature for 1 h, then concentrated on a rotary evaporator, and diluted with dichloromethane. The resulting reaction mixture was washed with 0.1 N hydrochloric acid solution (15 mL) and twice with distilled water. A solution of the target compound was evaporated on a rotary evaporator; the solid residue was dried in a desiccator over P2O5. Yield, 95% (0.573 g).
IR spectrum (CHCl3, ν, cm–1): 3404, 3306, 3251 (ν(N–H)), 2489 (ν(B–H)), 1743 (ν(C=O)), 1645 (ν(С=N)), 1049 (δ(B–B–H)); 11B NMR (CD3CN, δ, ppm): –7.8 (s, 1B, B(1)), –16.5 (d, 12B, B(2–12), JB–H = 128 Hz); 1Н NMR (CD3CN, δ, ppm): –1.01–1.55 (m, 12Н, В12Н11), 8.25 (d, 1H NH–C=NH, J = 9.7 Hz), 7.38–7.25 (m, 5H, CHCH2C6H5), 6.56 (s, 1H, NH–C=NH), 4.36 (td, 1H, CHCH2C6H5, J = 9.7, 4.1 Hz), 3.78 (s, 3H, OCH3), 3.27 (dd, 1H, CHCH2C6H5, J = 13.7, 4.2 Hz), 3.10 (m, 8H, NBu4), 2.93 (dd, 1H, CHCH2C6H5, J = 13.7, 9.7 Hz), 1.59 (m, 8H, NBu4), 1.57 (s, 3H, NH=C–CH3), 1.35 (m, 8H, NBu4), 0.96 (m, 12H, NBu4); 13С NMR (CD3CN, δ, ppm): 171.9 (СOOCH3), 165.2 (С=NH), 137.3, 130.9, 129.6, 128.0 (CH2C6H5), 59.4 (NBu4), 55.1 (CHCH2C6H5), 52.4 (СOOCH3), 40.3 (CHCH2C6H5), 24.30 (NBu4), 20.30 (NBu4), 18.9 (CH3–C=NH), 13.8 (NBu4).
For 2 anal. calcd. (%): C, 55.72; H, 10.52; B, 21.5; N, 6.96. Found (%): C, 55.21; H, 11.56; B, 21.2; N, 6.88.
(NBu4)[B12H11NHC(CH3)HNCH(CH2C6H5)COOH] (3). A weighed portion of compound 2 (0.573 g) was dissolved in methanol (30 mL). 0.1 N NaOH solution (0.9 mL) was added dropwise under vigorous stirring. The reaction mixture was stirred at room temperature for 5 h. Then the resulting reaction solution was diluted with distilled water and acidified to pH 2. The target product was extracted with dichloromethane and recrystallized from ethyl alcohol. Yield, 75% (0.398 g).
IR spectrum (CHCl3, ν, cm–1): 3506 (ν(O–H)), 3405, 3310, 3254 (ν(N–H)), 2490 (ν(B–H)), 1739 (ν(C=O)), 1644 (ν(S=N)), 1049 (δ(B–B–H)); 11B NMR (CD3CN, δ, ppm): –7.8 (s, 1B, B(1)), –16.5 (d, 12B, B(2–12), JB–H = 128 Hz); 1Н NMR (CD3CN), δ, ppm: –1.01–1.55 (m, 12Н, В12Н11), 8.25 (d, 1H, NH–C=NH, J = 9.7 Hz), 7.38–7.25 (m, 5H, CHCH2C6H5), 6.56 (s, 1H, NH–C=NH), 4.36 (td, 1H, CHCH2C6H5, J = 9.7. 4.1 Hz), 3.27 (dd, 1H, CHCH2C6H5, J = 13.7. 4.2 Hz), 3.10 (m, 8H, NBu4), 2.93 (dd, 1H, CHCH2C6H5, J = 13.7, 9.7 Hz), 1.59 (m, 8H, NBu4), 1.57 (s, 3H, NH=C–CH3), 1.35 (m, 8H, NBu4), 0.96 (m, 12H, NBu4); 13S NMR (CD3CN), δ, ppm: 171.9 (SOOCH3), 165.2 (S=NH), 137.3, 130.9, 129.6, 128.0 (CH2C6H5), 59.4 (NBu4), 55.1 (CHCH2C6H5), 40.3 (CHCH2C6H5), 24.30 (NBu4), 20.30 (NBu4), 18.91 (CH3–C=NH), 13.8 (NBu4).
For 3 anal. calcd. (%): C, 55.00; H, 10.42; B, 22.0; N, 7.12. Found (%): C, 54.69; H, 10.23; B, 22.7; N, 7.17.
(NBu4)[B12H11NHC(CH3)HNCH(CH2C6H5)COHNCH2COOC(CH3)3] (4). N,N'-Dicyclohexylcarbodiimide (DCC) (0.110 g, 0.53 mmol) and 4-dimethylaminopyridine (0.122 g, 1 mmol) were added with vigorous stirring to derivative 3 (0.295 g, 0.5 mmol) in CH2Cl2 (10 mL). The reaction mixture was stirred for 1 h, and GlyOtBu (1.5 mmol) in THF (10 mL) was added. The solution was stirred overnight, and evaporated using a rotor. The product was isolated by preparative HPLC in the chloroform–THF system at a 4 : 1 ratio. Yield, 42%.
IR spectrum (CHCl3, ν, cm–1): 3406, 3360, 3306, 3256 (ν(N–H)), 2490 (ν(B–H)), 1740 (ν(C=O)), 1688 (ν(C=O)), 1641 (ν(С=N)), 1055 (δ(B–B–H)). 11B NMR (CD3CN, δ, ppm): –7.7 (s, 1B, B(1)), ‒16.4 (d, 12B, B(2–12), JB–H = 128 Hz); 1Н NMR (CD3CN, δ, ppm): –1.01–1.55 (m, 12Н, В12Н11), 8.32 (d, 1H NH–C=NH, J = 9.4 Hz), 7.39–7.22 (m, 5H, CHCH2C6H5), 6.90 (s, 1H, CONH), 6.60 (s, 1H, NH–C=NH), 4.27 (td, 1H, CHCH2C6H5, J = 9.6, 4.4 Hz), 3.83 (dd, 2H, NHCH2, J = 5.8, 2.2 Hz), 3.17 (dd, 1H, CHCH2C6H5, J = 13.6, 4.4 Hz), 3.10 (m, 8H, NBu4), 2.93 (dd, 1H, CHCH2C6H5, J = 13.7, 9.7 Hz), 1.59 (m, 8H, NBu4), 1.57 (s, 3H, NH=C–CH3), 1.45 (s, 9H, COOC(CH3)3), 1.35 (m, 8H, NBu4), 0.96 (m, 12H, NBu4); 13S NMR (CD3CN, δ, ppm): 170.5 (COOC(CH3)3), 169.4 (SONH), 165.1 (S=NH), 137.3, 130.9, 129.6, 128.0, 126.4 (CH2C6H5), 82.4, (COOC(CH3)3), 60.0 (CHCH2C6H5), 59.3 (NBu4), 42.7 (NH–CH2), 40.8. (CHCH2C6H5), 28.2 (COOC(CH3)3), 24.30 (NBu4), 20.30 (NBu4), 19.9 (CH3–C=NH), 13.8 (NBu4).
For 4 anal. calcd. (%): C, 56.40; H, 10.32; B, 18.5; N, 7.97. Found (%): C, 57.01; H, 10.22; B, 18.0; N, 7.99.
(NBu4)[B12H11NHC(CH3)HNCH(CH2C6H5)COHNCH2COOH] (5). Derivative 4 (0.100 g, 0.2 mmol) was dissolved in acetonitrile (5 mL); concentrated hydrochloric acid (2 mL) was added dropwise, and the mixture was refluxed for 2 h. After cooling, the reaction mixture was extracted three times with 10 mL portions of dichloromethane. The obtained extracts were combined and evaporated on a rotary evaporator. The target product was recrystallized from ethyl alcohol and dried in a desiccator over P2O5. Yield, 85% (0.110 g).
IR spectrum (CHCl3, ν, cm–1): 3503 (ν(O–H)), 3404, 3356, 3308, 3243 (ν(N–H)), 2493 (ν(B–H)), 1717 (ν(C=O)), 1682 (ν(C=O)), 1643 (ν(S=N)), 1050 (δ(B–B–H)); 11B NMR (CD3CN, δ, ppm): –7.7 (s, 1B, B(1)), –16.4 (d, 12B, B(2–12), JB–H = 129 Hz); 1Н NMR (CD3CN, δ, ppm): –1.01–1.55 (m, 12Н, В12Н11), 8.29 (d, 1H, NH–C=NH, J = 9.4 Hz), 7.40–7.19 (m, 6H, CHCH2C6H5, CONH), 6.62 (s, 1H, NH–C=NH), 4.33 (td, 1H, CHCH2C6H5, J = 9.6. 4.5 Hz), 3.83 (d, 2H, NHCH2, J = 5.7), 3.17 (dd, 1H, CHCH2C6H5, J = 13.6, 4.4 Hz), 3.10 (m, 8H, NBu4), 2.93 (dd, 1H, CHCH2C6H5, J = 13.7, 9.7 Hz), 1.59 (m, 8H, NBu4), 1.57 (s, 3H, NH=C–CH3), 1.35 (m, 8H, NBu4), 0.96 (m, 12H, NBu4); 13S NMR (CD3CN, δ, ppm): 171.6 (COOH), 170.8 (SONH), 165.2 (S=NH), 137.2, 130.9, 129.5, 128.0 (CH2C6H5), 59.8 (CHCH2C6H5), 59.2 (NBu4), 41.7 (NH–CH2), 40.8 (CHCH2C6H5), 24.30 (NBu4), 20.30 (NBu4), 19.9 (CH3–C=NH), 13.8 (NBu4).
For 5 anal. calcd. (%): C, 53.87; H, 9.97; B, 20.1; N, 8.67. Found (%): C, 53.99; H, 10.04; B, 19.5; N, 8.66.
RESULTS AND DISCUSSION
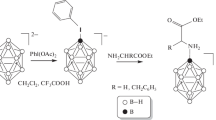
At the first stage of the work, the reaction of nucleophilic addition of tert-butyl esters of amino acids to nitrile derivatives of the closo-dodecaborate anion was studied. In contrast to the nitrile derivatives of the closo-decaborate anion, this reaction does not proceed. As a result, a number of products with unknown structures are formed.
As an alternative approach, the reaction between nitrile derivatives of the closo-dodecaborate anion with methyl esters of amino acids was investigated. This process is shown in Scheme 1a.
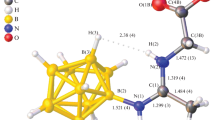
The progress of the reaction was monitored and the structure of the target compound was determined using multinuclear 1H, 11B, and 13C NMR spectroscopy. In the 11B NMR spectra of the obtained derivative, a signal from the boron atom bound to the nitrogen atom of the nitrile substituent is observed in the region of –7.8 ppm [B(1), I = 1] and a complex signal from unsubstituted boron atoms are observed at –16.5 ppm [B(2–12), I = 11, JB–H = 128 Hz]. In the 1H NMR spectrum of compound 2, the fragment of the nitrile substituent is represented by signals at 6.56 ppm assigned to the hydrogen atom bonded to the nitrogen atom, and at 1.57 ppm assigned to the methyl group located in a stronger field compared to similar conjugates of the closo-decaborate anion [31]. The amino acid fragment is represented by the signals of protons bound to nitrogen atoms at 8.25 ppm, with hydrogen atoms of the benzene ring in the range 7.38–7.25 ppm, signals of protons bound to the α-carbon atom at 4.36 ppm, protons of the methyl group at 3.78 ppm, and two complex signals of nonequivalent protons of the methylene group at 3.27 and 2.93 ppm. The tetrabutylammonium cation is represented by four signals at 3.10, 1.59, 1.35, and 0.96 ppm. In the 13C NMR spectrum of the obtained product, the nitrile fragment is represented by the signals of the carbon atom bonded to the nitrogen of the imino group at 165.2 ppm, and the signal of the carbon atom of the methyl group at 18.9 ppm. The amino acid residue is represented by signals from the ester group at 171.9, 52.4 ppm, signals from the α-carbon atom at 55.1 ppm, β-carbon atom at 40.3 ppm, and four signals of nonequivalent carbon atoms of the benzene ring at 137.3, 130.9, 129.6, and 128.0 ppm.

Scheme 1 . General scheme for the synthesis of N-borylated dipeptides.
Esters do not react to form peptide bonds. Thus, the next stage of our work was the hydrolysis of the methyl ester of the resulting conjugate. For this, the approach described earlier [32] (Scheme 1b) was used. The progress of the reaction was monitored and the structure of the resulting product was determined using multinuclear 1H, 11B, and 13C NMR spectroscopy. In the 1H and 13C NMR spectra, there are no signals from the protons and carbon of the methyl group of the ester, which confirms the completeness of the removal of the protective group.
The resulting conjugate containing a free carboxyl group can be used as a C-component to obtain a borylated dipeptide (Scheme 1c). Both components are readily soluble in organic solvents, which makes it possible to use standard peptide synthesis techniques using dicyclohexylcarbodiimide as a cross-linking reagent. Acid-labile protecting groups are preferred for the preparation of peptide conjugates with nitrile derivatives of the closo-dodecaborate anion to prevent destruction of the amidine moiety upon unblocking of the peptide’s carboxyl group.
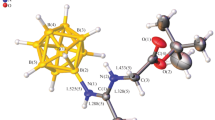
The structure of the resulting dipeptide was established using multinuclear NMR spectroscopy. In the 1H NMR spectrum, the glycine fragment is represented by signals of amide protons at 6.90 ppm, α‑atom at 3.83 ppm, and signals of methyl protons of tert-butyl ether at 1.45 ppm. In the 13C NMR spectrum, the glycine fragment is represented by signals from the carboxyl carbon atom at 170.5 ppm, and the α-carbon atom at 42.7 ppm. The tert-butoxycarbonyl fragment is represented by signals at 82.4 and 28.2 ppm.
The next step was the removal of the tert-butyl group (Scheme 1d) as described [28]. The process does not take place in a two-phase system dichloromethane–hydrochloric acid, regardless of the reaction temperature. Carrying out this reaction in a homogeneous acetonitrile–hydrochloric acid system allows the complete unblocking of the carboxyl group without the formation of by-products. The progress of the reaction was monitored and the structure of the resulting product was determined using multinuclear 1H, 11B, and 13C NMR spectroscopy. In the 1H and 13C NMR spectra, there are no signals of protons and carbon of the tert-butyl group of the ester, which confirms the completeness of the removal of the protective group.
CONCLUSIONS
An approach to the preparation of dipeptides conjugated with nitrile derivatives of the closo-dodecaborate anion is proposed. It has been shown that to obtain conjugates of nitrile derivatives of the closo-dodecaborate and free amino acids, methyl esters of amino acids must be used as starting compounds, since the addition of tert-butyl amino acid esters to the acetonitrile derivative [B12H11NCCH3]– does not lead to the formation of the amidine-closo-dodecaborates.
In the future, these conjugates can be used to obtain borylated peptides by peptide synthesis methods according to the Fmoc protocol.
Notes
The studies were carried out using the equipment of the Center for Collective Use of the Kurnakov Institute RAS, functioning within the framework of the State Assignment of the Kurnakov Institute RAS in the field of fundamental scientific research.
REFERENCES
Boron Science: New Technologies and Applications, Ed. by N. S. Hosmane (CRC Press, 2011).
C. V. T. Hey-Hawkins, Boron-Based Compounds: Potential and Emerging Applications in Medicine (John Wiley & Sons Ltd, 2018).
T. Tsurubuchi, M. Shirakawa, W. Kurosawa, et al., Cells 9, 5 (2020). https://doi.org/10.3390/cells9051277
Y. Hattori, S. Kusaka, M. Mukumoto, et al., J. Med. Chem. 55, 6980 (2012). https://doi.org/10.1021/jm300749q
Y. Hattori, S. Kusaka, M. Mukumoto, et al., Amino Acids 46, 2715 (2014). https://doi.org/10.1007/s00726-014-1829-5
K. Hu, Z. Yang, L. Zhang, et al., Coord. Chem. Rev. 405, 213139 (2020). https://doi.org/10.1016/j.ccr.2019.213139
J. Capala, R. F. Barth, M. Bendayan, et al., Bioconjug. Chem. 7, 7 (1996). https://doi.org/10.1021/bc950077q
F. Issa, M. Kassiou, and L. M. Rendina, Chem. Rev. 111, 5701 (2011). https://doi.org/10.1021/cr2000866
S. Hofmann, J. Lindner, A. G. Beck-Sickinger, et al., Chem. Bio. Chem. 19, 2300 (2018). https://doi.org/10.1002/cbic.201800343
T. He, J. C. Misuraca, and R. A. Musah, Sci. Rep. 7, 16995 (2017). https://doi.org/10.1038/s41598-017-16926-w
A. R. Genady, J. A. Ioppolo, M. M. Azaam, et al., Eur. J. Med. Chem. 93, 574 (2015). https://doi.org/10.1016/j.ejmech.2015.02.033
A. P. Zhdanov, M. V. Lisovsky, L. V. Goeva, et al., Russ. Chem. Bull. 58, 1694 (2009). https://doi.org/10.1007/s11172-009-0234-9
V. Sícha, J. Plesek, M. Kvícalová, et al., Dalton Trans. 5, 851 (2009). https://doi.org/10.1039/b814941k
M. Y. Stogniy, S. A. Erokhina, I. B. Sivaev, et al., Phosphorus Sulfur Silicon Relat. Elem. 194, 983 (2019). https://doi.org/10.1080/10426507.2019.1631312
S. A. Anufriev, A. V. Shmal’ko, M. Y. Stogniy, et al., Phosphorus Sulfur Silicon Relat. Elem. 195, 901 (2020). https://doi.org/10.1080/10426507.2020.1804148
E. V. Bogdanova, M. Y. Stogniy, L. A. Chekulaeva, et al., New J. Chem. 44, 15836 (2020). https://doi.org/10.1039/d0nj03017a
A. P. Zhdanov, I. N. Polyakova, G. A. Razgonyaeva, et al., Russ. J. Inorg. Chem. 56, 847 (2011). https://doi.org/10.1134/S003602361106026X
A. L. Mindich, N. A. Bokach, F. M. Dolgushin, et al., Organometallics 31, 1716 (2012). https://doi.org/10.1021/om200993f
A. L. Mindich, N. A. Bokach, M. L. Kuznetsov, et al., Organometallics 32, 6576 (2013). https://doi.org/10.1021/om400892x
A. P. Zhdanov, I. N. Klyukin, A. Y. Bykov, et al., Polyhedron 123, 176 (2017). https://doi.org/10.1016/j.poly.2016.11.035
V. K. Burianova, D. S. Bolotin, A. S. Novikov, et al., Inorg. Chim. Acta 482, 838 (2018). https://doi.org/10.1016/j.ica.2018.07.038
V. K. Burianova, D. S. Bolotin, A. S. Mikherdov, et al., New J. Chem. 42, 8693 (2018). https://doi.org/10.1039/c8nj01018h
M. Y. Stogniy, S. A. Erokhina, K. Y. Suponitsky, et al., New J. Chem. 42, 17958 (2018). https://doi.org/10.1039/c8nj04192j
A. P. Zhdanov, A. V. Nelyubin, I. N. Klyukin, et al., Russ. J. Inorg. Chem. 64, 841 (2019). https://doi.org/10.1134/S0036023619070180
A. V. Nelyubin, I. N. Klyukin, A. S. Novikov, et al., Mendeleev Commun. 31, 201 (2021). https://doi.org/10.1016/j.mencom.2021.03.018
K. A. Zhdanova, A. P. Zhdanov, A. V. Ezhov, et al., Macroheterocycles 7, 394 (2014). https://doi.org/10.6060/mhc140494z
A. V. Ezhov, F. Y. Vyal’ba, I. N. Kluykin, et al., Macroheterocycles 10, 505 (2017). https://doi.org/10.6060/mhc171254z
A. V. Nelyubin, I. N. Klyukin, A. P. Zhdanov, et al., Russ. J. Inorg. Chem. 64, 1499 (2019). https://doi.org/10.1134/S003602361912012X
Protective Groups in Organic Chemistry, Ed. by J. F. W. McOmie (Springer US, Boston, MA, 1995). https://doi.org/10.1007/978-1-4684-7218-9
A. V. Nelyubin, N. A. Selivanov, A. Y. Bykov, et al., Russ. J. Inorg. Chem. 65, 795 (2020). https://doi.org/10.1134/S0036023620060133
A. V. Nelyubin, I. N. Klyukin, A. P. Zhdanov, et al., Russ. J. Inorg. Chem. 66, 139 (2021). https://doi.org/10.1134/S0036023621020133
A. K. Chakraborti, Basak, and V. Grover, J. Org. Chem. 64, 8014 (1999). https://doi.org/10.1021/jo9900351
Funding
The study was supported by the Russian Foundation for Basic Research, projects. nos. 20-33-90119 and 20-03-00763.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflicts of interest.
Additional information
Translated by V. Avdeeva
Rights and permissions
Open Access. This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nelyubin, A.V., Selivanov, N.A., Klyukin, I.N. et al. New Method for Synthesis of Substituted N-Borylated Dipeptides Based on Acetonitrile Derivative of the closo-Dodecaborate Anion. Russ. J. Inorg. Chem. 66, 1390–1395 (2021). https://doi.org/10.1134/S0036023621090096
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0036023621090096