
Abstract
Despite the importance of phages in driving horizontal gene transfer (HGT) among pathogenic bacteria, the underlying molecular mechanisms mediating phage adsorption to S. aureus are still unclear. Phage ϕ11 is a siphovirus with a high transducing efficiency. Here, we show that the tail protein Gp45 localized within the ϕ11 baseplate. Phage ϕ11 was efficiently neutralized by anti-Gp45 serum and its adsorption to host cells was inhibited by recombinant Gp45 in a dose-dependent manner. Flow cytometry analysis demonstrated that biotin-labelled Gp45 efficiently stained the wild-type S. aureus cell but not the double knockout mutant ΔtarM/S, which lacks both α- and β-O-GlcNAc residues on its wall teichoic acids (WTAs). Additionally, adsorption assays indicate that GlcNAc residues on WTAs and O-acetyl groups at the 6-position of muramic acid residues in peptidoglycan are essential components of the ϕ11 receptor. The elucidation of Gp45-involved molecular interactions not only broadens our understanding of siphovirus-mediated HGT, but also lays the groundwork for the development of sensitive affinity-based diagnostics and therapeutics for S. aureus infection.
Similar content being viewed by others
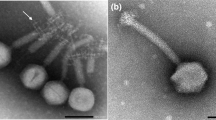
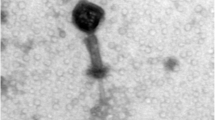
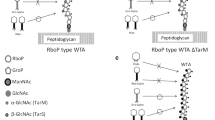
Introduction
Recently, there has been a renewed interest in phage-bacteria interactions because phages have not only profound influence on the biology of bacterial pathogens1,2. Despite comprehensive studies on phage genomes15 and the role of S. aureus phages in horizontal transfer of resistance and virulence genes among clones and species12, the molecular interactions mediating phage adsorption to the staphylococcal cell surface remain poorly understood.
The mechanism underlying S. aureus phage adsorption has often been assumed to be similar to that of phages infecting gram-negative bacteria. However, as gram-positive bacteria have a very different cell wall structure compared to that of gram-negative bacteria, phages infecting gram-positive bacteria may employ adsorption mechanisms different from those infecting gram-negative bacteria. Accounting for over 50% of the cell wall mass, WTAs are the most abundant surface molecules in the cell wall of bacteria belonging to the order Bacillales, which includes genera such as Bacillus, Listeria and Staphylococcus7,35. Hence, it is most likely that phages infecting bacteria of these genera need to interact with WTAs for successful adsorption.
In this study, we demonstrated that GlcNAc residues on WTAs are essential for ϕ11 adsorption regardless of their anomeric configurations. We also found that 6-O-acetylation of muramic acid residues in peptidoglycan is involved in ϕ11 adsorption. We showed that Gp45 and Gp54 are two baseplate proteins critical for ϕ11 infection, as both antisera can neutralize ϕ11 infection dose-dependently. Recombinant Gp45 inhibits ϕ11 adsorption in a dose-dependent manner and binds to glycosylated WTAs, demonstrating that Gp45 is the RBP of ϕ11. Unfortunately, recombinant Gp54 purified from E. coli was not stable and hence unsuitable for cell wall binding studies and its functions could not be tested.
Staphylococcal pathogenicity islands (SaPIs) have an intimate relationship with temperate staphylococcal phages. Phages can induce the SaPI cycle, which allows the SaPIs to be efficiently encapsidated into special small phage heads commensurate with their size10. Previous mutational analyses of the genes present in the morphogenesis cluster of ϕ11 demonstrated that the Gp45 was essential for both the phage infectivity and transduction of its cognate SaPI13. Of note, Δgp54 seemed to lose its baseplates and failed to plate on S. aureus. Surprisingly, Δgp54 was still able to transduce SaPIs, although with a 100-fold reduction in transduction efficiency when compared with wild-type ϕ11. These results highlight that Gp45 is essential for the recognition process, while the presence of the Gp54 significantly increases the binding affinity between the phage and its receptor. As the N-terminus of Gp54 was predicted to be similar to that of BppU, which maintains the attachment of RBP to the baseplate core in TP901-1, it is tempting to speculate that Gp54 plays an important role in anchoring RBP in the baseplate.
Previously, a tail protein ORF636 from a serogroup A phage phiSLT was characterized as an adhesion protein that binds to poly-glycerolphosphate (GroP) chain of lipoteichoic acids (LTAs)25. Notably, ORF636 shares high homology with Gp45 (62% similarity) and the ORF636 sequence exists in all known serogroup A phages infecting S. aureus. However, it was shown that all tested serogroup A phages can still form plaques on a S. aureus mutant deficient in LTAs, but not on a mutant deficient in WTAs11, suggesting that WTAs but not LTAs are required for S. aureus phage infection. The tight binding of glycerol and glycerolphosphate for the RBPs suggested that LTAs could act as receptors for lactococcal phages36, however the structure of LTAs is well conserved and thought to be too simple to explain the different host specificities of various lactococcal phages. Recently, by mutational analysis, it was demonstrated that cell wall polysaccharide (CWPS) is the host cell surface receptor of tested lactococcus phages of different groups and that differences between the CWPS structures play a crucial role in determining phage host range37.
It is noteworthy that many phages need a protein receptor for adsorption, for example, Fuh A, OmpA, OmpC, LamB for E. coli phages, GamR, YueB for Bacillus phages and Pip for Lactococcus phage38. Interestingly, all these protein receptors are non-essential and many of them were identified by transposon mutagenesis. However, by screening a mutant library of S. aureus we were unable to isolate ϕ11-resistant mutants, which carry transposon insertions in genes encoding membrane proteins39. It is now generally acknowledged that carbohydrate recognizing phages possess a broad baseplate structure with multiple receptor binding sites. Conversely, phages with stubby ends or tail fibres, including the lactococcal c2 phages and the Bacillus phage SPP1, may recognize protein receptors on the cell surface40. The crystal structure of Gp45 was solved and it was found that Gp45 forms six trimers in the baseplates of ϕ11 and that each monomer of Gp45 contains a five-bladed propeller domain with a cavity that could accommodate a GlcNAc moiety (Koc et al., unpublished data). Hence, the presence of 18 receptor binding sites in the baseplate of ϕ11 suggests that its receptors are saccharides but not proteins.
Accounting for over 50% of the cell wall mass, WTAs are considered to be the most abundant surface molecules in S. aureus and have been implicated in various critical processes and interactions such as staphylococcal cell division, biofilm formation, β-lactam resistance and staphylococcal pathogenesis41,42. Due to the in-homogeneity of WTA, its analysis has proven to be very challenging. Unlike research carried out on DNA, RNA or protein, methods available for studying WTA function are very limited. Despite technical limitations, a few WTA-interacting proteins such as FmtA43, WTA antibody, MBL44 and SREC-145 have recently been identified. Here, we report Gp45 as a new WTA-interacting protein. Our results may eventually provide new tools for labelling and detecting the subdomain structures in the cell wall of S. aureus. Additionally, this study establishes a solid basis for the development of sensitive affinity-based infection diagnostics46 and therapeutics for MRSA infection.
Materials and Methods
Bacterial strains and growth conditions
S. aureus strains used in these studies are listed in Table 1. Bacteria were grown at 37 °C in BM broth (1% tryptone, 0.5% yeast extract, 0.5% NaCl, 0.1% K2HPO4, 0.1% glucose) under agitation.
Construction of S. aureus mutants
The deletion mutants ∆pip1, ∆pip2, and ∆pip1/2 were constructed by allelic exchange. For knockout plasmid construction, the primers listed in Table S1 in the Supplementary Information were used. For deletion of pip1, flanking regions were amplified with primer pairs pip1-F1-up/pip1-F1-dn and pip1-F2-up/pip1-F2-dn. Purified PCR products were digested with SalI/NheI and NheI/EcoRI respectively and subsequently ligated into the SalI/EcoRI digested knockout vector pBASE647. The resulting plasmid was used for allelic exchange48. For the construction of the pip2 deletion mutant, a similar approach was pursued. The flanking regions of pip2 were amplified with primer pairs pip2-F1-up/pip2-F1-dn and pip2-F2-up/pip2-F2-dn, digested with XbaI and ligated. Afterwards this marker-less knockout cassette was subcloned into pKOR-1 and the resulting plasmid was used for mutant construction via allelic exchange48.
Overexpression and purification of the recombinant Gp45 and Gp54
Both gp45 and gp54 were amplified by PCR from S. aureus strain SA113, which is a ϕ11 lysogen. The primers used for the PCR reaction are listed in Table S1 in the Supplementary Information. The amplified gp45 or gp54 genes were subcloned into the expression vector pET28a between the NheI and XhoI sites. The resulting plasmids were transformed into E. coli BL21 for overexpression of Gp45 or Gp54. Both proteins were fused to a hexa-histidine-tag at the N-terminus to facilitate purification. After IPTG induction of the host cells, recombinant Gp45 was extracted and purified according to the procedure described previously49. Briefly, cells were lysed via ultrasonication (Digital Sonifier, Branson). After centrifugation at 38.000 × g for 55 min, cell debris was removed and the supernatant containing recombinant Gp45 protein was loaded on a 5 mL Ni-NTA-column (GE Healthcare). Fractions containing Gp45 were pooled and concentrated to 1 mg/mL using Vivaspin 20 centrifugal concentrators with a molecular size cut-off of 50,000 (Sartorius, Göttingen, Germany). The concentrated sample was then loaded on a size-exclusion chromatography column SD200 pre-equilibrated with SEC-buffer containing 25 mM HEPES, 150 mM NaCl, 1 mM DTT. Fractions containing Gp45 were pooled and concentrated as pure Gp45 preparations. The purity and folding of the recombinant Gp45 were assessed with SDS-PAGE, Circular dichroism (CD) spectroscopy and dynamic light scattering (DLS). Gp54 was purified by the same procedure as for Gp45.
Preparation of cell wall from S. aureus strains
The cell wall was extracted according to the procedure described previously50. Briefly, S. aureus overnight cultures were harvested by centrifugation at 5000 × g for 10 minutes. The cells were washed with 20 mM NH4Ac buffer (pH 4.8) and re-suspended in the same buffer. After disruption in a cell disrupter (Euler, Frankfurt am Main, Germany), the cell lysates were centrifuged at 5000 × g to remove the intact cells. The supernatant was collected as a crude extract of cell wall and mixed well with 5 mM MgSO4, 40 U/mL DNase and 80 U/mL RNase at final concentrations before overnight incubation at 37 °C. Next, to remove any cell membrane contamination, SDS was added to a final concentration of 2%, followed by ultra-sonication for 15 min. After heating at 65 °C for one hour, the cell wall preparations were washed six times with 20 mM NH4Ac buffer by centrifugation at 12,000 × g. Finally, the cell wall preparations were re-suspended in distilled water and quantified by measuring the amount of inorganic phosphate using the QuantiChromTM Phosphate Assay Kit (BioAssay Systems, USA) as described previously39.
Bacteriophage experiments
Using the double layer soft agar method, ϕ11 was propagated with the indicator strain, S. aureus strain RN4220, as a host.
Phage plating efficiencies were determined to investigate the effects of Gp45, Gp54 anti-sera and cell wall preparations on the inactivation of ϕ11. In brief, 100 μL of ϕ11 (3 × 106 PFU/mL) was mixed with 100 μL of cell wall preparations or antisera of certain concentrations and incubated at 37 °C for 10 min. Samples pre-incubated without any cell wall preparations or sera served as controls. Next, the mixtures were diluted before plating on the indicator strain (S. aureus strain RN4220) using double agar overlay methods. After overnight incubation at 37 °C, the plaques were enumerated. The efficiency of plating was calculated relative to that of plating of ϕ11 pre-incubated without any sera or cell wall preparations.
Adsorption assays were performed according to the procedure described previously11. Briefly, 200 μL of S. aureus wild-type or mutant cells containing 8 × 107 CFU were mixed with 100 μL of ϕ11 containing 3 × 105 PFU and incubated at 37 °C for 15 min. The bound phages were separated from the free phages by centrifugation at 13,000 × g for 5 min. Adsorption was calculated by determining the number of PFU of the unbound phage in the supernatant and subtracting it from the total number of input PFU. Adsorption efficiency was expressed relative to the adsorption of wild-type strain RN4220. Each adsorption assay was repeated at least three times. To study the inhibition of adsorption by Gp45, cells were pre-incubated with the purified recombinant Gp45 of indicated concentrations for 15 min before adding phages to the host cells.
Purification of ϕ11 and electron microscopy methods
Phage ϕ11 lysate was centrifuged at 73000 × g, 4 °C for two hours (Beckman Optima XL-80K). The resulting pellet was re-suspended in 500 μL of TMN buffer containing 10 mM Tris-HCl, pH 7.5, 10 mM MgSO4, 500 mM NaCl. The sample was then mixed well with 55% CsCl in TMN-buffer to give a final concentration of 42% CsCl and subjected to ultracentrifugation at 245,000 × g, 15 °C for 20 hours (Beckman). The visible phage band on the CsCl gradient was collected and sequentially dialyzed for two hours each in a D-Tube Dialyzer Mini (Novagen®, Merck Millipore, Darmstadt, Germany) against decreasing concentrations of NaCl in TMN buffer (10 mM Tris-HCl, pH 7.5, 10 mM MgCl2, 4 M NaCl) until the NaCl concentration after each round of dialysis was at 4 M, 2 M, 1 M and 10 mM NaCl, respectively.
For immunogold labelling, purified phage samples were adsorbed to glow discharged, pioloform and carbon-coated grids. The grids were then blocked with 0.2% gelatin in phosphate-buffered saline for 10 min followed by incubation with rabbit anti-Gp45 or rabbit anti-Gp54 serum, which were diluted in blocking buffer at 1:20 and 1:100, respectively. Polyclonal rabbit antisera were raised against purified recombinant Gp45 or Gp54 using a custom antibody service, Speedy 28-Day polyclonal program from Eurogentec (Brussels, Belgium). After blocking at room temperature for 60 min, the grids were washed six times with blocking buffer for a total time of 15 min before incubation with goat anti-rabbit IgG coupled with 12 nm gold colloids (Dianova, Hamburg), which was diluted with blocking buffer at 1:30. After incubation at room temperature for 60 min, the grids were washed three times with blocking buffer for 10 min and three times with phosphate-buffered saline for 10 min, followed by washing four times with double-distilled water for 2 min. Finally, the grids were negatively stained with 1% (w/v) aqueous uranyl acetate before examination with a JEM-1400Plus transmission electron microscope (JEOL, Japan)
Flow cytometry analysis
Flow cytometry was carried out to evaluate the binding of recombinant Gp45 to the S. aureus cell surface. Purified recombinant Gp45 was labelled with biotin using the EZ-Link™ NHS-Biotin kit (Thermo Fisher Scientific). Biotin-labelled Gp45 was then incubated with S. aureus wild-type or mutant cells for 30 min with shaking at room temperature. Cells were washed and stained with strep-Alu488 (Invitrogen) for one hour at 4 °C. Finally, cells were fixed for flow cytometry analysis.
Statistical analysis
Results are expressed as the means ± standard deviations from at least three independent experiments. Statistical analysis was performed using GraphPad Prism (GraphPad Software, Inc., La Jolla, USA, Version 5.04). Statistically significant differences were calculated with two-tailed Student’s t-test or one-way ANOVA with Bonferroni’s post-test as indicated.
Additional Information
How to cite this article: Li, X. et al. An essential role for the baseplate protein Gp45 in phage adsorption to Staphylococcus aureus. Sci. Rep. 6, 26455; doi: 10.1038/srep26455 (2016).
References
Lindsay, J. A. Staphylococcus aureus genomics and the impact of horizontal gene transfer. Int J Med Microbiol 304, 103–109, doi: 10.1016/j.ijmm.2013.11.010 (2014).
**a, G. & Wolz, C. Phages of Staphylococcus aureus and their impact on host evolution. Infection, genetics and evolution: journal of molecular epidemiology and evolutionary genetics in infectious diseases 21, 593–601, doi: 10.1016/j.meegid.2013.04.022 (2014).
Hagens, S. & Loessner, M. J. Bacteriophage for biocontrol of foodborne pathogens: calculations and considerations. Current pharmaceutical biotechnology 11, 58–68 (2010).
Fischetti, V. A., Nelson, D. & Schuch, R. Reinventing phage therapy: are the parts greater than the sum? Nature biotechnology 24, 1508–1511, doi: 10.1038/nbt1206-1508 (2006).
Duplessis, M. & Moineau, S. Identification of a genetic determinant responsible for host specificity in Streptococcus thermophilus bacteriophages. Molecular microbiology 41, 325–336 (2001).
Spinelli, S., Veesler, D., Bebeacua, C. & Cambillau, C. Structures and host-adhesion mechanisms of lactococcal siphophages. Frontiers in microbiology 5, 3, doi: 10.3389/fmicb.2014.00003 (2014).
**a, G., Kohler, T. & Peschel, A. The wall teichoic acid and lipoteichoic acid polymers of Staphylococcus aureus. International journal of medical microbiology: IJMM 300, 148–154, doi: 10.1016/j.ijmm.2009.10.001 (2010).
Baptista, C., Santos, M. A. & Sao-Jose, C. Phage SPP1 reversible adsorption to Bacillus subtilis cell wall teichoic acids accelerates virus recognition of membrane receptor YueB. Journal of bacteriology 190, 4989–4996, doi: 10.1128/jb.00349-08 (2008).
Sao-Jose, C. et al. The ectodomain of the viral receptor YueB forms a fiber that triggers ejection of bacteriophage SPP1 DNA. J Biol Chem 281, 11464–11470, doi: 10.1074/jbc.M513625200 (2006).
Penades, J. R. & Christie, G. E. The phage-inducible chromosomal islands: a family of highly evolved molecular parasites. Annual Review of Virology 2, 181–201, doi: 10.1146/annurev-virology-031413-085446 (2015).
**a, G. et al. Wall teichoic Acid-dependent adsorption of staphylococcal siphovirus and myovirus. Journal of bacteriology 193, 4006–4009, doi: 10.1128/jb.01412-10 (2011).
Winstel, V. et al. Wall teichoic acid structure governs horizontal gene transfer between major bacterial pathogens. Nature communications 4, 2345, doi: 10.1038/ncomms3345 (2013).
Tormo, M. A. et al. Staphylococcus aureus pathogenicity island DNA is packaged in particles composed of phage proteins. Journal of bacteriology 190, 2434–2440, doi: 10.1128/jb.01349-07 (2008).
Tallent, S. M., Langston, T. B., Moran, R. G. & Christie, G. E. Transducing particles of Staphylococcus aureus pathogenicity island SaPI1 are comprised of helper phage-encoded proteins. Journal of bacteriology 189, 7520–7524, doi: 10.1128/jb.00738-07 (2007).
Kwan, T., Liu, J., DuBow, M., Gros, P. & Pelletier, J. The complete genomes and proteomes of 27 Staphylococcus aureus bacteriophages. Proceedings of the National Academy of Sciences of the United States of America 102, 5174–5179, doi: 10.1073/pnas.0501140102 (2005).
Quiles-Puchalt, N., Martinez-Rubio, R., Ram, G., Lasa, I. & Penades, J. R. Unravelling bacteriophage varphi11 requirements for packaging and transfer of mobile genetic elements in Staphylococcus aureus. Molecular microbiology 91, 423–437, doi: 10.1111/mmi.12445 (2014).
Iandolo, J. J. et al. Comparative analysis of the genomes of the temperate bacteriophages phi 11, phi 12 and phi 13 of Staphylococcus aureus 8325. Gene 289, 109–118 (2002).
Soding, J., Biegert, A. & Lupas, A. N. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res 33, W244–248, doi: 10.1093/nar/gki408 (2005).
Veesler, D. et al. Crystal structure of bacteriophage SPP1 distal tail protein (gp19.1): a baseplate hub paradigm in gram-positive infecting phages. J Biol Chem 285, 36666–36673, doi: 10.1074/jbc.M110.157529 (2010).
Veesler, D. & Cambillau, C. A common evolutionary origin for tailed-bacteriophage functional modules and bacterial machineries. Microbiology and molecular biology reviews: MMBR 75, 423–433, first page of table of contents, doi: 10.1128/mmbr.00014-11 (2011).
Flayhan, A. et al. Crystal Structure of pb9, the Distal Tail Protein of Bacteriophage T5: a Conserved Structural Motif among All Siphophages. J Virol 88, 820–828, doi: 10.1128/JVI.02135-13 (2014).
Kondou, Y. et al. Structure of the central hub of bacteriophage Mu baseplate determined by X-ray crystallography of gp44. Journal of molecular biology 352, 976–985, doi: 10.1016/j.jmb.2005.07.044 (2005).
Kanamaru, S. et al. Structure of the cell-puncturing device of bacteriophage T4. Nature 415, 553–557 (2002).
Rodriguez-Rubio, L. et al. The peptidoglycan hydrolase of Staphylococcus aureus bacteriophage 11 plays a structural role in the viral particle. Applied and environmental microbiology 79, 6187–6190, doi: 10.1128/aem.01388-13 (2013).
Kaneko, J., Narita-Yamada, S., Wakabayashi, Y. & Kamio, Y. Identification of ORF636 in phage phiSLT carrying Panton-Valentine leukocidin genes, acting as an adhesion protein for a poly(glycerophosphate) chain of lipoteichoic acid on the cell surface of Staphylococcus aureus. Journal of bacteriology 191, 4674–4680, doi: 10.1128/jb.01793-08 (2009).
Cole, C., Barber, J. D. & Barton, G. J. The Jpred 3 secondary structure prediction server. Nucleic Acids Res 36, W197–201, doi: 10.1093/nar/gkn238 (2008).
Veesler, D. et al. Structure of the phage TP901-1 1.8 MDa baseplate suggests an alternative host adhesion mechanism. Proceedings of the National Academy of Sciences of the United States of America 109, 8954–8958, doi: 10.1073/pnas.1200966109 (2012).
Coyette, J. & Ghuysen, J. M. Structure of the cell wall of Staphylococcus aureus, strain Copenhagen. IX. Teichoic acid and phage adsorption. Biochemistry 7, 2385–2389 (1968).
Lindberg, A. A. Bacteriophage receptors. Annual review of microbiology 27, 205–241, doi: 10.1146/annurev.mi.27.100173.001225 (1973).
Murayama, Y., Kotani, S. & Kato, K. Solubilization of phage receptor substances from cell walls of Staphylococcus aureus (strain Ceopenhagen) by cell wall lytic enzymes. Biken journal 11, 269–291 (1968).
Shaw, D. R. & Chatterjee, A. N. O-Acetyl groups as a component of the bacteriophage receptor on Staphylococcus aureus cell walls. Journal of bacteriology 108, 584–585 (1971).
Bera, A., Herbert, S., Jakob, A., Vollmer, W. & Gotz, F. Why are pathogenic staphylococci so lysozyme resistant? The peptidoglycan O-acetyltransferase OatA is the major determinant for lysozyme resistance of Staphylococcus aureus. Molecular microbiology 55, 778–787, doi: 10.1111/j.1365-2958.2004.04446.x (2005).
Geller, B. L., Ivey, R. G., Trempy, J. E. & Hettinger-Smith, B. Cloning of a chromosomal gene required for phage infection of Lactococcus lactis subsp. lactis C2. J Bacteriol 175, 5510–5519 (1993).
Sao-Jose, C., Baptista, C. & Santos, M. A. Bacillus subtilis operon encoding a membrane receptor for bacteriophage SPP1. Journal of bacteriology 186, 8337–8346, doi: 10.1128/jb.186.24.8337-8346.2004 (2004).
Brown, S., Santa Maria, J. P., Jr. & Walker, S. Wall teichoic acids of gram-positive bacteria. Annual review of microbiology 67, 313–336, doi: 10.1146/annurev-micro-092412-155620 (2013).
Spinelli, S. et al. Modular structure of the receptor binding proteins of Lactococcus lactis phages. The RBP structure of the temperate phage TP901-1. J Biol Chem 281, 14256–14262, doi: 10.1074/jbc.M600666200 (2006).
Ainsworth, S. et al. Differences in lactococcal cell wall polysaccharide structure are major determining factors in bacteriophage sensitivity. mBio 5, e00880–00814, doi: 10.1128/mBio.00880-14 (2014).
Bertozzi Silva, J., Storms, Z. & Sauvageau, D. Host receptors for bacteriophage adsorption. FEMS microbiology letters 363, doi: 10.1093/femsle/fnw002 (2016).
**a, G. et al. Glycosylation of wall teichoic acid in Staphylococcus aureus by TarM. J Biol Chem 285, 13405–13415, doi: 10.1074/jbc.M109.096172 (2010).
Mahony, J., McDonnell, B., Casey, E. & van Sinderen, D. Phage-Host Interactions of Cheese-Making Lactic Acid Bacteria. Annual review of food science and technology, doi: 10.1146/annurev-food-041715-033322 (2016).
Winstel, V., **a, G. & Peschel, A. Pathways and roles of wall teichoic acid glycosylation in Staphylococcus aureus. International journal of medical microbiology: IJMM 304, 215–221, doi: 10.1016/j.ijmm.2013.10.009 (2014).
Misawa, Y. et al. Staphylococcus aureus Colonization of the Mouse Gastrointestinal Tract Is Modulated by Wall Teichoic Acid, Capsule and Surface Proteins. Plos pathogens 11, e1005061, doi: 10.1371/journal.ppat.1005061 (2015).
Qamar, A. & Golemi-Kotra, D. Dual roles of FmtA in Staphylococcus aureus cell wall biosynthesis and autolysis. Antimicrobial agents and chemotherapy 56, 3797–3805, doi: 10.1128/aac.00187-12 (2012).
Kurokawa, K. et al. Glycoepitopes of staphylococcal wall teichoic acid govern complement-mediated opsonophagocytosis via human serum antibody and mannose-binding lectin. J Biol Chem 288, 30956–30968, doi: 10.1074/jbc.M113.509893 (2013).
Baur, S. et al. A nasal epithelial receptor for Staphylococcus aureus WTA governs adhesion to epithelial cells and modulates nasal colonization. Plos pathogens 10, e1004089, doi: 10.1371/journal.ppat.1004089 (2014).
Schmelcher, M. & Loessner, M. J. Application of bacteriophages for detection of foodborne pathogens. Bacteriophage 4, e28137, doi: 10.4161/bact.28137 (2014).
Geiger, T. et al. The stringent response of Staphylococcus aureus and its impact on survival after phagocytosis through the induction of intracellular PSMs expression. Plos pathogens 8, e1003016, doi: 10.1371/journal.ppat.1003016 (2012).
Bae, T. & Schneewind, O. Allelic replacement in Staphylococcus aureus with inducible counter-selection. Plasmid 55, 58–63, doi: 10.1016/j.plasmid.2005.05.005 (2006).
Koc, C. et al. Structural and enzymatic analysis of TarM glycosyltransferase from Staphylococcus aureus reveals an oligomeric protein specific for the glycosylation of wall teichoic acid. J Biol Chem 290, 9874–9885, doi: 10.1074/jbc.M114.619924 (2015).
Weidenmaier, C. et al. Role of teichoic acids in Staphylococcus aureus nasal colonization, a major risk factor in nosocomial infections. Nature medicine 10, 243–245, doi: 10.1038/nm991 (2004).
Kreiswirth, B. N. et al. The toxic shock syndrome exotoxin structural gene is not detectably transmitted by a prophage. Nature 305, 709–712 (1983).
Brown, S. et al. Methicillin resistance in Staphylococcus aureus requires glycosylated wall teichoic acids. Proceedings of the National Academy of Sciences of the United States of America 109, 18909–18914, doi: 10.1073/pnas.1209126109 (2012).
Iordanescu, S. & Surdeanu, M. Two restriction and modification systems in Staphylococcus aureus NCTC8325. Journal of general microbiology 96, 277–281, doi: 10.1099/00221287-96-2-277 (1976).
Acknowledgements
This work was supported by SFB766 to T.S., A.P. and G.X. from the German Research Foundation (DFG).
Author information
Authors and Affiliations
Contributions
X.L. and G.X. designed this study; X.L., C.K., P.K., Y.S. and B.K. performed the experiments; X.L., Y.S., M.E., J.P., C.W., T.S., C.C., A.P. and G.X. analysed the data; C.C., M.E. and G.X. wrote the manuscript. Every author reviewed the manuscript prior to submission.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Li, X., Koç, C., Kühner, P. et al. An essential role for the baseplate protein Gp45 in phage adsorption to Staphylococcus aureus. Sci Rep 6, 26455 (2016). https://doi.org/10.1038/srep26455
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep26455
- Springer Nature Limited
This article is cited by
-
Proteomic analysis revealed the biofilm-degradation abilities of the bacteriophage UPMK_1 and UPMK_2 against Methicillin-resistant Staphylococcus aureus
Biotechnology Letters (2022)
-
Analysis host-recognition mechanism of staphylococcal kayvirus ɸSA039 reveals a novel strategy that protects Staphylococcus aureus against infection by Staphylococcus pseudintermedius Siphoviridae phages
Applied Microbiology and Biotechnology (2019)
-
Peculiarities of Staphylococcus aureus phages and their possible application in phage therapy
Applied Microbiology and Biotechnology (2019)
-
Bacteriophage-host arm race: an update on the mechanism of phage resistance in bacteria and revenge of the phage with the perspective for phage therapy
Applied Microbiology and Biotechnology (2019)
-
Investigation of recombination-intense viral groups and their genes in the Earth’s virome
Scientific Reports (2018)