
Abstract
Surface defect of nanomaterials is an important physical parameter which significantly influences their physical and chemical performances. In this work, high concentration of surface oxygen vancancies (SOVs) are successfully introduced on {001} facets exposed BiOBr nanosheets via a simple surface modification using polybasic carboxylic acids. The chelation interaction between carboxylic acid anions and Bi3+ results in the weakness of Bi-O bond of BiOBr. Afterwards, under visible-light irradiation, the oxygen atoms would absorb the photo-energy and then be released from the surface of BiOBr, leaving SOVs. The electron spin resonance (ESR), high-resolution transmission electron microscopy (HRTEM) and UV–vis diffuse reflectance spectra (DRS) measurements confirm the existence of SOVs. The SOVs can enhance the absorption in visible light region and improve the separation efficiency of photo-generated charges. Hence, the transformation rate of adsorbed O2 on the as-prepared BiOBr with SOVs to superoxide anion radicals (•O2−) and the photocatalytic activity are greatly enhanced. Based on the modification by several carboxylic acids and the photocatalytic results, we propose that carboxylic acids with natural bond orbital (NBO) electrostatic charges absolute values greater than 0.830 are effective in modifying BiOBr.
Similar content being viewed by others
Introduction
Semiconductor photocatalysis has attracted a wide attention for its potential in solving energy and environmental issues1,2,3,4. In a typical photocatalytic process, charge kinetics mainly comprises three key steps: the generation of charges under photoexcitation, transfer to the surface and consumption for redox reactions on the surface, which drive the conversion from solar to chemical energy16,17,18, deposition of noble metals19, have been developed for improving the performance of semiconductor photocatalysts. At present, researchers have shown that the activity of photocatalysts is related not only to the crystal size and morphology, but also to the separation and transfer efficiency of the charges20.
In recent years, two-dimensional layer-structured inorganic photocatalysts such as carbon-containing compounds, transition metal oxides and transition metal dichalcogenides, have received massive research interest due to their unique structures and promising properties21,22,23,24,25,26. As a new class of layered materials, BiOX (X=F, Cl, Br and I) compounds exhibited their extensive photocatalytic applications in energy conversion and environmental remediation27,28,Fig. 1.

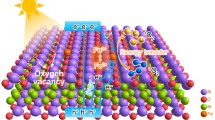
Schematic illustration of in-situ fabrication of surface oxygen vacancies.
XRD patterns of bare BiOBr and oxalic acid modified BiOBr (BiOBr-OA0.01) samples are shown in Fig. 2a. Firstly, it can be seen that all of the as-prepared BiOBr samples have clear and intense diffraction peaks implying the good crystallinity of the samples. All the diffraction peaks are in good agreement with the tetragonal BiOBr phase (JCPDS NO. 01-078-0348), no obvious impurity peak obtained when the oxalic acid is used as the additive. Secondly, compared with the standard spectrum of BiOBr shown in the bottom of Fig. 2a, the as-prepared pristine BiOBr and BiOBr-OA0.01 have stronger relative peak intensity at (001) series planes, such as (002), (003) and (004), exhibiting the highly exposed {001} facets of BiOBr.

(a) XRD patterns and (b) Raman spectrum of the as-synthesized BiOBr and BiOBr-OA0.01 samples.
Raman spectrum was carried out to indentify the interaction between oxalic acid anions and BiOBr. Typically, Raman signals of pristine BiOBr have three kinds with A1g, B1g and Eg. The positions of the Raman signals are relative to the short-range order of BiOBr crystal. The three characteristic bands at 112.3, 161.8 and 385.2 cm−1 are assigned to the A1g internal Bi−Br stretching mode, the Eg internal Bi−Br stretching mode and the B1g bands generated by the motion of oxygen atoms, respectively10. As shown in Fig. 2b, in comparison with BiOBr, a red shift of the bands at 112.3 cm−1 in BiOBr-OA0.01 crystals was obtained, which indicates the corresponding structural change. This implies a feeble variation of lattice symmetry induced by the do** of oxalic acid.
In order to further confirm the existence of oxalic acid anions and the interaction between oxalic acid anions and BiOBr, XPS analysis was performed. The high-resolution spectra of Bi 4f are shown in Fig. 3a. The two strong peaks centered at 158.8 and 164.1 eV for BiOBr are associated core lines of Bi 4f5/2 and 4f7/2. As for BiOBr-OA0.01, the Bi 4f5/2 and 4f7/2 peaks shift to 159.2 and 164.4 eV, respectively, indicating there are interactions between Bi and oxalic acid anions. The high-resolution spectra of Br 3d are shown in Fig. 3b. There are two peaks at 69.0 and 67.9 eV, which can be ascribed to Br 3d 5/2 and Br 3d, respectively14. However, the peaks in the BiOBr-OA0.01 sample are observed at 68.7 eV and 67.6 eV, which are lower than those for pure BiOBr. Such shifts can be attributed to the atomic interaction between Br− and oxalic acid anions. Because of bilayer structure of BiOBr, some adsorbed oxalic acid anions can be reserved in the surface bilayer structure of [Bi2O2]2+ slabs, resulting in the shifts of Bi 4f and Br 3d peaks. The C1s of the samples are shown in Fig. 3c. The peak at 284.6eV is attributed to the contaminated carbon. Besides, two new peak at 286.0 and 288.4 were obtained in BiOBr-OA0.01 sample, which are assigned to sp2 hybridized C–O and C=O bonds, respectively10. This indicates the existence of oxalic acid anions on BiOBr-OA0.01 surface.

X-Ray photoelectron spectra of (a) Bi 4f, (b) Br 3d, (c) C1s and (d) Time-dependent high resolution C 1s XPS spectra upon Ar+ sputtering for BiOBr-OA0.01.
To further clarify the spatial distributions of oxalic acid anions in the modified BiOBr, XPS depth profiling technique with Ar+ sputtering was employed. As shown in Fig. 3d, the peak intensity of carbon–oxygen bonds (at about 288.4 eV) in BiOBr-OA0.01 is decreased during the Ar+ sputtering from 0 to 40 s and vanished at 80 s. This observation suggests that the addition of oxalic acid anions during hydrothermal treatment leads to their existence on the surface of BiOBr, instead of uniform do** in the crystal. As a result, the chelation strategy of surface modification for BiOBr would supply a basis for the generation of SOVs and would not induce the occurance of bulk oxygen vacancies during the following light irradiation.
Based on the strong chelation ability of polybasic carboxylic acids, we design the introduction of oxalic acid for modifying BiOBr aiming at weakening the bond of Bi–O and facilitating the escape of the O atoms on the surface under visible-light irradiation, which acts as a kind of energy. To verify this hypothesis, the structure of irradiated BiOBr and BiOBr-OA0.01 samples were characterized by electron paramagnetic resonance at 77K in liquid N2, which is a direct and sensitive method to monitor the presence of oxygen defects. As shown in Fig. 4a, BiOBr-OA0.01 exhibits the apparent peak at g = 2.001, while the irradiated BiOBr does not. This signal has been reported previously, which can be attributed to surface oxygen vacancies11,38. Therefore, from the ESR result, the SOVs have been possibly formed.

(a) ESR spectra, (b) UV-vis absorption spectra, (c) X-ray photoelectron spectra of Bi 4f and (d) XRD patterns of the as-prepared irradiated BiOBr and BiOBr-OA0.01 photocatalysts.
The UV-vis diffuse reflectance spectra of irradiated BiOBr and BiOBr-OA0.01 are shown in Fig. 4b. Compared with BiOBr, the absorption sharp edge of BiOBr-OA0.01 has feeble shift to visible region and the absorbance is enhanced in the range of 450 to 800nm, which is induced by oxygen vacancies. From the inset of Fig. 4b, the color of the as-prepared photocatalysts changes from pale yellow of BiOBr to gray of BiOBr-OA0.01. The Eg of BiOBr is estimated by the following formula15:

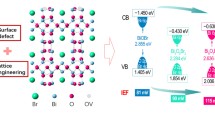
where α, h, ν, A and Eg are the absorption coefficient; Planck constant; the light frequency, a constant; and the band-gap energy, respectively. The value of n is 4 for BiOBr, determined by an indirect transition occurring in the semiconductor. The Eg of BiOBr can be estimated from a plot of (αhυ)1/2 versus energy (hυ). The Eg of BiOBr obtained from the tangent intercept is 2.71 eV (as shown in Fig. S1). To further elucidate the electronic structure of BiOBr, electrochemical flat potential measurements were carried out and the data were plotted in Mott–Schottky coordinates. As shown in Fig. S2, the flat band potentials of BiOBr is roughly −0.93V (vs. Ag/AgCl). Accordingly, as a n-type semiconductor, the flat band potentials of BiOBr is estimated to be −0.71 V (pH = 7) after correction of the Ag/AgCl potential of 0.22 V (vs. NHE)44. It is generally known that the conduction band (CB) positions of n-type semiconductors are about 0.1 V higher than their flat potentials. Thus it is deduced that the CB of BiOBr is about −0.81 V, which was more negative than the standard redox potential of O2/•O2− (−0.28 V vs NHE)45. Combining the Eg of 2.71eV, the relative valence band (VB) position of BiOBr can be inferred at 1.90V.
Figure 4c shows the high-resolution Bi 4f spectra of BiOBr and BiOBr-OA0.01. The binding energies of the Bi 4f7/2 and Bi 4f5/2 peaks of irradiated BiOBr are located at 158.8 and 164.1eV. In comparison to pure BiOBr, the Bi 4f5/2 and Bi 4f7/2 peaks of BiOBr-OA0.01 exhibit a shift of about 0.2 eV toward lower binding energy, which is indicative of the presence of lower charge Bi ions in BiOBr-OA0.0146,47. This result clearly verifies that the Bi3+ ions can be reduced to low-charge Bi ions, which is correlated with oxygen vacancies. The XRD pattern of BiOBr-OA0.01 is shown in Fig. 4d. Comparing to bare BiOBr, no other obvious impurity peaks can be detected when oxygen vacancies are introduced. A similar pattern of the adsorption-desorption isotherms for representative BiOBr and BiOBr-OA0.01 were also obtained (as shown in Fig. S3). There are no significant changes in the SBET value (BiOBr: 4.88 m2/g, BiOBr-OA0.01: 6.26 m2/g).
The microscopic information of the representative samples were examined by SEM and TEM. As shown from Fig. 5a–d, both the pristine BiOBr and BiOBr-OA0.01 display an aggregated morphology of nanosheets. The {001} facet exposure characteristic of BiOBr and BiOBr-OA0.01 are reflected by the set of diffraction spots in the corresponding fast Fourier transform pattern indexed as the [001] zone axis of tetragonal BiOBr and further confirmed by the 45o angle between (110) and (200) planes (inset of Fig. 5c,d)10,11. The high-resolution TEM image revealed the nanosheets are of high crystallinity (Fig. 5e,f). The lattice fringes with an inter-planar lattice spacing of 0.278 nm is indexed as the (110) atomic plane perpendicular to the (001) plane. Furthermore, pristine BiOBr displays perfect lattice features, however the edge of BiOBr-OA0.01 particles becomes dim and disordered, which indicates that the surface structure of BiOBr-OA0.01 is damaged and SOVs are formed38.

SEM and TEM images (a),(c),(e) of BiOBr and (b),(d),(f) of BiOBr-OA0.01.
The photocatalytic activities of pristine irradiated BiOBr and BiOBr-OA samples on enhancing the activation of O2 to •O2− radicals are fristly investigated by the photocatalytic process of nitro-tetrazolium blue chloride (NBT) under visible light. NBT exhibits an absorption maximum at 265 nm, which can be detected by UV-vis spectrophotometer; however, the product of •O2− with NBT does not. Hence, the removal efficiency of NBT can reflect the level of activation on O2 to •O2− radicals. Figure 6a,b show the transformation percentage of NBT in 20 min irradiation over pristine BiOBr and BiOBr-OA0.01, respectively. It is clear that the photocatalytic activation of O2 of BiOBr-OA samples gradually increases till the BiOBr-OA0.01 displays the highest activity. When the concentration of oxalic acid is further increased, the activity begins to decrease. Thus, the activity of modified BiOBr photocatalysts are greatly influenced by the concentration of oxygen vacancies, which can be controlled by the concentration of oxalic acid.

(a) Transformation percentage of NBT concentration after 20 min visible light irradiation; (b) Transformation process of NBT under visible light irradiation; (c,d) ESR spectra of DMPO–•O2− adducts after visible light irradiation for 6 min over BiOBr and BiOBr-OA0.01.
To further directly observe the generation of •O2− radicals, ESR technique was employed to monitor the •O2− radicals under visible light irradiation. Figure 6c,d show the results of pristine BiOBr and BiOBr-OA0.01 samples employing dimethyl sulphoxide (DMSO) as solvent48. It can be seen that the intensity of •O2− radicals in BiOBr-OA0.01 suspension is obviously intensive, while the signal in pristine BiOBr is very weak. The result is consistent with the NBT tests. In the BiOBr photocatalytic system, •O2− radicals are often considered as the products of photon-generated electron. Thus, this phenomenon indicates that the introduction of SOVs on BiOBr has enhanced the number of effective photon-generated electrons on the CB of BiOBr. Moreover, ** of carboxylic acids. The interaction between Bi3+ and carboxylic acid anions should play an important role and the atomic charges of the carboxylic oxygen should be the most critical factor responsible for the surface chelation. Natural bond orbital (NBO) analysis is a well-established tool to predict the atomic charges in molecules. Therefore, we carried out NBO analysis via the procedures contained with Gaussian 03 package. The NBO electrostatic charges of the carboxylic oxygen atom in these carboxylic acid ions are shown in Table 1. From the photocatalytic activity, the NBO values of the carboxylic oxygen listed in Table 1 and the molecular weight and dimension of the additives, it can be concluded that the enhancement of photocatalytic activity is closely related with the NBO values of the carboxylic oxygen and has no relationship with molecular weight and dimension of the modifiers. Carboxylic acids with higher NBO absolute values, especially greater than 0.830, are effective in modifying BiOBr. This result illustrates that our hypothesis about chelation function of carboxylic acids for metal ions are right, which would be broadened for other metal oxide nanomaterials.
Base on the successful introduction of SOVs on BiOBr and their highly oxidation properties, we attempt to clarify the underlying mechanism of the enhancement photocatalytic activity. It is well known that photocatalytic activity is mainly governed by the adsorption ability, crystal phase structure and separation efficiency of photo-generated charges. As mentioned above, there are no obvious changes of the BET surface area and crystal phase structures for BiOBr with and without SOVs. Thus, the separation efficiency of photoinduced charges may play an important role in the photocatalysis. Thus, in order to understand the effect of SOVs, surface photovoltage spectroscopy (SPV) technique is employed firstly to investigate the photo-generated charges transfer properties since a SPV response can be detected49 after the separation of photo-generated carriers The carboxylic acids modified BiOBr and pure BiOBr were synthesized by a hydrothermal route. In a typical procedure, 0.5 mmol Bi(NO3)3·5H2O was added to 40 mL oxalic acid solution with different concentrations. The mixture solution was magnetically stirred for 2h and then 0.5 mmol NaBr was added to this solution. After magnetically stirring for 3h, the solution was transferred into a 50 mL Teflon-lined stainless autoclave. The autoclave was heated at 160 °C for 8h. The resulting precipitates were collected, thoroughly washed with deionized water and dried at 80 °C in air. The corresponding samples are designated as BiOBr-OAx, (x is the concentration of oxalic acid solution). Pure BiOBr was prepared according to the same procedure with pure water as the reaction mediator in the absence of carboxylic acid. The SOVs on BiOBr samples were fabricated by irradiating BiOBr-OA in pure water under a 300 W xenon arc lamp with a 400 nm cutoff filter for 2 hours. The corresponding samples are designated as BiOBr-OAx. X-ray diffraction (XRD) patterns were measured on a Rigaku D/MAX 2500 X-ray diffractometer equipped with a Cu Kα radiation (λ = 0.154 nm) source. Raman measurements were carried out by Thermo DXR Microscope with a 633 nm laser. The Brunauer–Emmett–Teller (BET) specific surface areas (SBET) of the samples were analyzed by a Quanta chrome NOVA2000 nitrogen adsorption/desorption apparatus. Surface morphology was observed by scanning electron microscopy (FESEM, HITACHI S-4800). High-resolution transmission electron microscopy (HRTEM) images were collected with a field emission transmission electron microscope (JEOL JEM-2010). UV–Vis diffuse reflectance spectra (DRS) were recorded on a spectrophotometer (Thermofisher Evolution 220). X-ray photoelectron spectroscopy (XPS) measurements were performed on a PHI 1600 ESCA XPS system. All binding energies were calibrated using the contaminant carbon (C 1s = 284.6 eV) as the reference. Electron spin resonance (ESR) measurements were analyzed on a Bruker EMX-8/2.7 X-band ESR spectrometer operating in the X-band at 9.86 GHz and 2.005 mW. The surface photovoltage spectroscopy (SPV) measurements were carried out on a homemade apparatus, which was constituted by a source of monochromatic light with a light chopper and a lock-in amplifier. The construct of the photovoltaic cell is a sandwich-like structure of ITO-sample-ITO. The transient photovoltage (TPV) signals of the samples were collected with by a 500 MHz digital phosphor oscilloscope (TDS 5054, Tektronix) with a preamplifier. The Mott–Schottky curves performed on an electrochemical workstation (CHI-660E, China) using a standard three electrode system. An aqueous solution containing 0.1 M Na2SO4 was employed as the electrolyte. The counter and the reference electrodes were platinum wire and Ag/AgCl (saturated KCl), respectively. The as-obtained photocatalyst film electrodes deposited on cleaned 1.5 cm × 1.0 cm fluoride-tin oxide (FTO) glass served as the working electrode. The amount of superoxide anion radicals (•O2−) during the photocatalytic process was determined by nitrotetrazolium blue chloride (NBT, 2.5 × 10−5 mol/L). NBT exhibits an absorption maximum at 265 nm, which can be detected by UV-vis spectrophotometer, however, the product of •O2− with NBT does not16. The test procedures were as follows: Firstly, NBT was dissolved in H2O with a concentration of 2.5 × 10−5 mol/L. Then, 0.20 g of the obtained catalyst was dispersed in 200 mL of the NBT solution. Irradiation experiments were carried out under visible light for 20 min and sampled at an interval of 5min. Finally, the suspension was centrifuged, filtered and measured on Thermofisher Evolution 220 spectrophotometer. Methyl orange (MO, anionic dye, 10 mg/L) was selected as the objective pollutants to evaluate the activity of the photocatalysts. 0.20 g of photocatalyst powder was dispersed in 200 mL of dye aqueous solution in a 500 ml photo-catalytic reactor. The suspensions were stirred in darkness at 300 rpm for 30 min in order to reach adsorption equilibrium. Irradiation experiments were carried out under a 300 W xenon arc lamp with a 400 nm cutoff filter. The total light intensity was 0.44 W/cm2 in the range of 400–1064 nm measured by a Newport 842-PE optical power/energy meter. For kinetic studies, samples were taken at regular time intervals and were analyzed by a UV–vis spectrophotometer. To investigate the stability of the as-prepared photocatalyst, the nanopowders were separated from solution and used for the next run experiment. How to cite this article: Wang, X.-j. et al. A Chelation Strategy for In-situ Constructing Surface Oxygen Vacancy on {001} Facets Exposed BiOBr Nanosheets. Sci. Rep. 6, 24918; doi: 10.1038/srep24918 (2016).Methods
Preparation of samples
Characterization
Detection of superoxide anion radicals
Photocatalytic activity evaluation
Additional Information
References
Li, L. D. et al. Sub-10 nm rutile titanium dioxide nanoparticles for efficient visible-light-driven photocatalytic hydrogen production. Nat. Commun. 6, 5881 (2015).
Shah, M. W. et al. Facile Synthesis of Defective TiO2−x Nanocrystals with High Surface Area and Tailoring Bandgap for Visible-light Photocatalysis. Sci. Rep. 5, 15804 (2015).
Kisch, H. Semiconductor Photocatalysis-Mechanistic and Synthetic Aspects. Angew. Chem. Int. Edit. 52, 812−847 (2013).
Wang, L. et al. A dye-sensitized visible light photocatalyst-Bi24O31Cl10 . Sci. Rep. 4, 7384 (2014).
Bai, S., Jiang, W. Y., Li, Z. Q. & **ong, Y. J. Surface and Interface Engineering in Photocatalysis. ChemNanoMat 1, 223−239 (2015).
Iwashina, K., Iwaseab, A. & Kudo, A. Sensitization of wide band gap photocatalysts to visible light by molten CuCl treatment. Chem. Sci. 6, 687−692 (2015).
Osterloh, F. E. Inorganic nanostructures for photoelectrochemical and photocatalytic water splitting. Chem. Soc. Rev. 42, 2294−2320 (2013).
Ding, J., Huang, Z. N., Zhu, J. H., Kou, S. Z. & Zhang, X. B. Low-temperature synthesis of high-ordered anatase TiO2 nanotube array films coated with exposed {001} nanofacets. Sci. Rep. 5, 17773 (2015).
Liu, S. W., Yu, J. G. & Jaroniec, M. Tunable photocatalytic selectivity of hollow TiO2 microspheres composed of anatase polyhedra with exposed {001} facets. J. Am. Chem. Soc. 132, 11914−11916 (2010).
Zhang, D., Li, J., Wang, Q. G. & Wu, Q. S. High {001} facets dominated BiOBr lamellas: facile hydrolysis preparation and selective visible-light photocatalytic activity. J. Mater. Chem. A 1, 8622−8629 (2013).
Li, H., Shang, J., Ai, Z. H. & Zhang, L. Z. Efficient Visible Light Nitrogen Fixation with BiOBr Nanosheets of Oxygen Vacancies on the Exposed {001} Facets. J. Am. Chem. Soc. 137, 6393−6399 (2015).
Petrik, N. G. & Kimmel, G. A. Reaction Kinetics of Water Molecules with Oxygen Vacancies on Rutile TiO2 (110). J. Phys. Chem. C119, 23059−23067 (2015).
Pihosh, Y. et al. Photocatalytic generation of hydrogen by core-shell WO3/BiVO4 nanorods with ultimate water splitting efficiency. Sci. Rep. 5, 11141 (2015).
Kong, L. et al. Unusual reactivity of visible-light-responsive AgBr–BiOBr heterojunction photocatalysts. J. Catal. 293, 116−125 (2012).
Li, F. T. et al. Ionic liquid self-combustion synthesis of BiOBr/Bi24O31Br10 heterojunctions with exceptional visible-light photocatalytic performances. Nanoscale 7, 1116−1126 (2015).
Sathasivam, S. et al. Tungsten Doped TiO2 with Enhanced Photocatalytic and Optoelectrical Properties via Aerosol Assisted Chemical Vapor Deposition. Sci. Rep. 5, 10952 (2015).
Ran, J. R., Ma, T. Y., Gao, G. P., Du, X. W. & Qiao, S. Z. Porous P-doped graphitic carbon nitride nanosheets for synergistically enhanced visible-light photocatalytic H2 production. Energy Environ. Sci. 8, 3708−3717 (2015).
Jiang, G. H. et al. Novel highly active visible-light-induced photocatalysts based on BiOBr with Ti do** and Ag decorating. ACS Appl. Mater. Interfaces 4, 4440−4444 (2012).
Cheng, H. F. et al. In situ ion exchange synthesis of the novel Ag/AgBr/BiOBr hybrid with highly efficient decontamination of pollutants. Chem. Commun. 47, 7054−7056 (2011).
Cheng, H. F., Huang, B. B. & Dai, Y. Engineering BiOX (X = Cl, Br, I) nanostructures for highly efficient photocatalytic applications. Nanoscale 6, 2009−2026 (2014).
Zhao, Z. W. et al. Mass-Controlled Direct Synthesis of Graphene-like Carbon Nitride Nanosheets with Exceptional High Visible Light Activity. Less is Better. Sci. Rep. 5, 14643 (2015).
Boltersdorf, J. & Maggard, P. A. Silver Exchange of Layered Metal Oxides and Their Photocatalytic Activities. ACS. Catal. 3, 2547−2555 (2013).
Wu, Y. H. et al. Well-defined BiOCl colloidal ultrathin nanosheets: synthesis, characterization and application in photocatalytic aerobic oxidation of secondary amines. Chem. Sci. 6, 1873−1878 (2015).
Xu, J. et al. Layered C3N3S3 Polymer/Graphene Hybrids as Metal-Free Catalysts for Selective Photocatalytic Oxidation of Benzylic Alcohols under Visible Light. ACS. Catal. 4, 3302−3306 (2014).
Kim, K. C. et al. Enhancement of Initial Growth of ZnO Films on Layer-Structure0d Bi2Te3 by Atomic Layer Deposition. Chem. Mater. 26, 6448−6453 (2014).
Cao, S. W., Low, J. X., Yu, J. G. & Jaroniec, M. Polymeric Photocatalysts Based on Graphitic Carbon Nitride. Adv. Mater. 27, 2150−2176 (2015).
Li, J., Zhao, K., Yu, Y. & Zhang, L. Z. Facet-Level Mechanistic Insights into General Homogeneous Carbon Do** for Enhanced Solar-to-Hydrogen Conversion. Adv. Funct. Mater. 25, 2189−2201 (2015).
Ye, L. Q., Zan, L., Tian, L. H., Peng, T. Y. & Zhang, J. J. The {001} facets-dependent high photoactivity of BiOCl nanosheets. Chem. Commun. 47, 6951−6953 (2011).
Jiang, J., Zhao, K., **ao, X. Y. & Zhang, L. Z. Synthesis and facet-dependent photoreactivity of BiOCl single-crystalline nanosheets. J. Am. Chem. Soc. 134, 4473−4476 (2012).
**g, L. Q., Zhou, W., Tian, G. H. & Fu, H. G. Surface tuning for oxide-based nanomaterials as efficient photocatalysts. Chem. Soc. Rev. 42, 9509−9549 (2013).
Kuang, P. Y. et al. Enhanced Photoelectrocatalytic Activity of BiOI Nanoplate–Zinc Oxide Nanorod p–n Heterojunction. Chem. Eur. J. 21, 15360−15368 (2015).
Huo, Y. N. et al. BiOBr visible-light photocatalytic films in a rotating disk reactor for the degradation of organics. J. Mater. Chem. A 3, 14801−14808 (2015).
Li, L. L., Ai, L. H., Zhang, C. H. & Jiang, J. Hierarchical {001}-faceted BiOBr microspheres as a novel biomimetic catalyst: dark catalysis towards colorimetric biosensing and pollutant degradation. Nanoscale 6, 4627−4634 (2014).
Xu, J., Meng, W., Zhang, Y., Li, J. & Guo, C. S. Photocatalytic degradation of tetrabromobisphenol A by mesoporous BiOBr: efficacy, products and pathway. Appl. Catal. B 107, 355−362 (2011).
Huo, Y., Zhang, J., Miao, M. & **, Y. Solvothermal synthesis of flower-like BiOBr microspheres with highly visible-light photocatalytic performances. Appl. Catal. B 111–112, 334−341 (2012).
Fang, Y. F., Ma, W. H., Huang, Y. P. & Cheng, G. W. Exploring the reactivity of multicomponent photocatalysts: insight into the complex valence band of BiOBr. Chem. Eur. J. 19, 3224−3229 (2013).
Cheng, H. F. et al. One-Pot Miniemulsion-Mediated Route to BiOBr Hollow Microspheres with Highly Efficient Photocatalytic Activity. Chem. Eur. J. 17, 8039−8043 (2011).
Lv, Y. H., Liu, Y. F., Zhu, Y. Y. & Zhu, Y. F. Surface oxygen vacancy induced photocatalytic performance enhancement of a BiPO4 nanorod. J. Mater. Chem. A 2, 1174−1182 (2014).
Tan, S. J. et al. Molecular oxygen adsorption behaviors on the rutile TiO2 (110)-1 × 1 surface: an in situ study with low-temperature scanning tunneling microscopy. J. Am. Chem. Soc. 133, 2002−2009 (2011).
**ao, X. Y., **g, J. & Zhang, L. Z. Selective oxidation of benzyl alcohol into benzaldehyde over semiconductors under visible light: The case of Bi12O17Cl2 nanobelts. Appl. Catal. B 142–143, 487−493 (2013).
Zhang, G. et al. Formation of Bi2WO6 Bipyramids with Vacancy Pairs for Enhanced Solar-Driven Photoactivity. Adv. Funct. Mater. 25, 3726−3734 (2015).
Zhao, K. et al. Surface structure-dependent molecular oxygen activation of BiOCl single-crystalline nanosheets. J. Am. Chem. Soc. 135, 15750−15753 (2013).
Li, H., Shi, J. G., Zhao, K. & Zhang, L. Z. Sustainable molecular oxygen activation with oxygen vacancies on the {001} facets of BiOCl nanosheets under solar light. Nanoscale 6, 14168−14173 (2014).
**, X. L. et al. Bismuth-rich strategy induced photocatalytic molecular oxygen activation properties of bismuth oxyhalogen: The case of Bi24O31Cl10 . Appl. Catal. B. 165, 668–675 (2015).
Yu, L. H. et al. Highly efficient Bi2O2CO3/BiOCl photocatalyst based on heterojunction with enhanced dye-sensitization under visible light. Appl. Catal. B. 187, 301–309 (2016).
Huang, Y. C. et al. Poly(3-hexylthiophene) Nanotubes with Tunable Aspect Ratios and Charge Transport Properties. ACS Appl. Mater. Interfaces 6, 22920−22927 (2014).
Wang, G. et al. Computational and photoelectrochemical study of hydrogenated bismuth vanadate. J. Phys. Chem. C 117, 10957−10964 (2013).
Lv, Y. H., Zhu, Y. Y. & Zhu, Y. F. Enhanced Photocatalytic Performance for the BiPO4–x Nanorod Induced by Surface Oxygen Vacancy. J. Phys. Chem. C 117, 18520−18528 (2013).
Zhou, L. J. et al. Porous nanoplate-assembled CdO/ZnO composite microstructures: A highly sensitive material for ethanol detection. Sensor. Actuat. B 197, 370−375 (2014).
Zhang, L., Li, S., Liu, B., Wang, D. & **e, T. Highly Efficient CdS/WO3 Photocatalysts: Z-Scheme Photocatalytic Mechanism for Their Enhanced Photocatalytic H2 Evolution under Visible Light. ACS. Catal. 4, 3724−3729 (2014).
Kronik, L. & Shapira, Y. Surface photovoltage phenomena: theory, experiment and applications. Surf. Sci. Rep. 37, 1−206 (1999).
Jiang, J., Zhang, X., Sun, P. B. & Zhang, L. Z. ZnO/BiOI heterostructures: photoinduced charge-transfer property and enhanced visible-light photocatalytic activity. J. Phys. Chem. C 115, 20555−20564 (2011).
Mahrov, B., Boschloo, G., Hagfeldt, A., Dloczik, L. & Dittrich, T. Photovoltage study of charge injection from dye molecules into transparent hole and electron conductors. Appl. Phys. Lett. 84, 5455 (2004).
Wang, Y. J., Shi, R., Lin, J. & Zhu, Y. F. Enhancement of photocurrent and photocatalytic activity of ZnO hybridized with graphite-like C3N4 . Energy Environ. Sci. 4, 2922−2929 (2011).
Acknowledgements
We are grateful for the financial support from the National Natural Science Foundation of China (20140654, 21376061), the Program for New Century Excellent Talents in University (NCET-12-0686), Natural Science Foundation of Hebei Province (B2015208005, B2015208010), Scientific Research Foundation for High-Level Talent in University of Hebei Province (GCC2014057), Young Talents Program in University of Hebei Province (No. BJ2016022) and the Foundation of Hebei University of Science and Technology (2013YY17).
Author information
Authors and Affiliations
Contributions
F.t.L. and X.j.W. conceived the project. X.j.W. and L.j.D. carried out experiments and data analyses. Y.Z. performed the natural bond orbital calculation. X.j.W. and F.t.L. wrote the manuscript. Y.p.L., J.Z. and Y.j.H. performed the characterization of samples and analyzed the results. All authors contributed to polishing paper and mechanism discussion.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Wang, Xj., Zhao, Y., Li, Ft. et al. A Chelation Strategy for In-situ Constructing Surface Oxygen Vacancy on {001} Facets Exposed BiOBr Nanosheets. Sci Rep 6, 24918 (2016). https://doi.org/10.1038/srep24918
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep24918
- Springer Nature Limited
This article is cited by
-
Electronic structure regulation and built-in electric field synergistically strengthen photocatalytic nitrogen fixation performance on Ti-BiOBr/TiO2 heterostructure
Rare Metals (2024)
-
Photocatalytic nitrogen fixation over flower-like molecular cobalt phthalocyanine@ZnIn2S4 heterojunctions under visible-light irradiation
Journal of Materials Science (2023)
-
Built-in electric field enhanced BiFeO3 photo-Fenton degradation Rhodamine B solution
Journal of Materials Science (2022)
-
The synthesis of high photocatalytic activity BiOBr nanosheets with dominant exposed (010) facets
Journal of Materials Science: Materials in Electronics (2020)
-
Fabrication of oxygen-vacancy-rich black-BiOBr/BiOBr heterojunction with enhanced photocatalytic activity
Journal of Materials Science (2020)