
Abstract
Circulating miRNAs can be found in extracellular vesicles (EV) and could be involved in intercellular communication. Here, we report the biodistribution of EV associated miR-155 using miR-155 KO mouse model. Administration of exosomes loaded with synthetic miR-155 mimic into miR-155 KO mice resulted in a rapid accumulation and clearance of miR-155 in the plasma with subsequent distribution in the liver, adipose tissue, lung, muscle and kidney (highest to lowest, respectively). miR-155 expression was detected in isolated hepatocytes and liver mononuclear cells of recipient KO mice suggesting its cellular uptake. In vitro, exosome-mediated restoration of miR-155 in Kupffer cells from miR-155 deficient mice augmented their LPS-induced MCP1 mRNA increase. The systemic delivery of wild type plasma to miR-155 KO mice also resulted in a rapid accumulation of miR-155 in the circulation and distribution to the liver and adipose tissue. In summary, our results demonstrate tissue biodistribution and biologic function of EV-associated miR-155.
Similar content being viewed by others


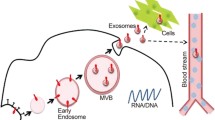
Introduction
MicroRNAs (miRNAs) are a class of non-coding RNAs that regulate gene expression1. miRNAs are highly stable and can be detected in various human body fluids, including peripheral blood, plasma and serum2,3,4. Circulating/extracellular miRNAs are present either in extracellular vesicles (EV) (including exosomes, microvesicles and apoptotic bodies) or associated with non-membranous particles that include RNA inducing silencing complex (RISC) proteins (AGO proteins)5,6,7. Exosomes are of multivesicular bodies origin with a size range from 40–100 nm and express exosomal markers, including CD9, CD63, CD81, Alix, Flotillin-1 and Tsg1018. Microvesicles directly bud from the plasma membrane with a size range from 50–1000 nm and have no unique markers8. Currently consensus is still under development regarding the origin and nomenclature of exosomes and microvesicles.
Multiple lines of evidence indicate that circulating miRNAs have the potential to be distributed into organs via the circulation9,10,11. miRNAs are found to be stable in the circulation and this property makes them attractive for biomarker discovery. miRNAs are also receiving interest given their potential as therapeutic tools and targets in various diseases. Evidences suggest that extracellular vesicles (EV)/exosomes carry various cargos including nucleic acids (mRNA and non-coding RNA), proteins and lipids and serve as vehicles to transfer material between cells/organs6,12,13,14.
Exosomal meditated transfer of miRNA has been proposed to be a mode of intercellular communication11, All methods were carried out in accordance with the approved guidelines. Mice deficient in miRNA-155 (miR-155 KO) were obtained from Jackson Laboratory and the colony was maintained in the animal facility at the University of Massachusetts Medical School. Eight to ten week old C57/Bl6J wild type (WT) male or female mice were obtained from Jackson Laboratory. All experimental protocols were approved by the Institutional Animal Use and Care Committee of the University of Massachusetts Medical School (Worcester, MA). To induce the miR-155 induction in the circulation, wild type female mice were either injected with saline or 2.5 mg/kg CpG (i.p) for three days as described53. On day 4, CpG treated mice received 0.5 mg/kg LPS (i.p.) 3 h prior to sacrifice. At the end of treatment, mice were cheek bled and plasma was separated and stored at −80 °C for further analyses. Liver and other tissue was washed with PBS and immediately either snap frozen in liquid nitrogen (protein analyses) or in RNA later (Qiagen) (RNA analyses) respectively. To separate the plasma, blood was collected in EDTA containing microtainer tubes (BD Biosciences) and centrifuged at 5000 g for 10 mins at room temperature. The centrifugation step was repeated twice to minimize platelet contamination and the clear plasma fraction was aliquoted and stored at −80 0C. Exosomes were generated from the murine B cell line (M12.4.1). B cells were cultured in RPMI medium supplemented with 10% FBS and 1% penicillin/streptomycin. To induce exosome release, the cells were cultured in exosome-depleted FBS (System Biosciences, USA) medium stimulated with IL-4 (50 ng/mL)+CD40 (5 μg/mL) for 3 days as described54. IL-4 and CD40 treatment was used to increase exosome production as reported previously54. For the isolation of exosomes, culture medium was centrifuged at 500 g for 10 mins to deplete cells and then at 10,000 g for 20 mins to eliminate residual cellular debris as described previously22. The resulting supernatant was passed through 0.4 μm and 0.22 μm filters and concentrated using the Amicon Ultra-15 Centrifugal Filter Unit with Ultracel-100 membrane (Millipore, MA). CD63 enriched exosomes were isolated using anti-CD63 immuno-magnetic positive selection as described32. Anti-CD63 antibody (Abcam cat. #ab8219 and Santa Cruz cat. #15363) was used as primary antibody followed by corresponding secondary antibody coupled with magnetic beads (Miltenyl biotech cat. #130-048-602). Exosomes were eluted from anti-CD63 beads using elution buffer from Invitrogen and finally exosomes were resuspended in PBS. The Miltenyl Biotec midi-MACS separator along side LS columns (cat. #130-042-901) was used. The exosomes were quantified using NanoSight LM10 system (NanoSight, UK) equipped with a fast video capture and Nanoparticle Tracking Analysis system (NTA), according to the manufacturer’s instructions. The samples were measured for 60s at room temperature with manual shutter and gain adjustments. NTA was used to measure particle size (measured in nanometers) and concentration of particles (particles/ml). Each measurement was repeated for three times. For electron microscopy, exosomes were re-suspended in PBS and placed on a formvar-coated copper grid and incubated for 30 mins as described22. The grid was washed with PBS and sample was fixed by placing the grid on the top of the 2% paraformaldehyde placed on the parafilm for 10 mins. Fixation was followed by several washes with deionized water and samples contrasted by adding 2% uranyl acetate for 15 mins. Samples were embedded by adding a drop of 0.13% methyl cellulose and 0.4% uranyl acetate for 10 mins. The grid was examined in a Philips CM10 transmission electron microscope and images were captured using a Gatan CCD digital camera. Western blot analysis was carried out for exosomal and non-exosomal markers. Exosomes or B cells were lysed with RIPA buffer and 30 μg of protein from each fraction was run on 10% SDS-PAGE gel. Proteins were transferred to nitrocellulose membrane and were blocked in TBS containing 5% non-fat dry milk and 0.1% Tween-20 for 1 hour followed by incubation with primary CD63 or CD81 or GRP78 antibody at 4°C for overnight. Next day, membranes were washed 3 times with TBST and then incubated with corresponding horseradish peroxidase-conjugated secondary antibodies for 1 hour at room temperature (Santa Cruz Biotechnology). The target proteins were visualized on blot using a Clarity™ Western ECL substrate kit (BioRad) according to the manufacturer’s protocol and analyzed via Fujifilm LAS-4000 luminescent image analyzer. The loading of exosomes with miRNA-155 mimic or scrambled mimic was performed based on our previously optimized protocol22. Briefly, resuspended exosomes were diluted in the Gene Pulser electroporation buffer (Bio-Rad Laboratories, CA) in 1:1 ratio. Mouse miRNA-155 mimic (Ambion, NY) or scrambled miRNA mimic control#1 (scrambled mimic) at final concentration of 5 μmol/ml were added to 200 μl of exosome sample containing 1 μg/μl exosomal protein using standardized conditions as we previously described22. Approximately 1 μ mole of miRNA is loaded into 200 μl of exosome fractions, containing about 4 × 108 particles (exosomes). The mixtures were transferred into cold 0.2cm electroporation cuvettes and electroporated at 150 V/100 μF capacitance using a Gene pulser II System (Bio-Rad Laboratories, CA). Immediately after electroporation, the mixture was treated with one unit of RNase A (Qiagen, Germany) for 30 mins to remove the free-floating miRNA mimic. Deactivation of RNase A was achieved by adding 2 μl of RNase inhibitor (Life technologies) and exosomes were re-isolated using ExoQuick-TC™ reagent as described by the manufacturer. The final pellet (exosome) was resuspended in PBS, aliquoted in 100 μl volume to avoid freeze and thaw cycles and stored at −80 °C. Of note, freeze and thaw of exosomes (2 times) did not influence miR-155 levels significantly. Prior to injecting into recipient miR-155 KO mice, WT plasma (miR-155 enriched) or miR-155 mimic loaded exosomes (exo-miR-155 mimic) or scrambled loaded exosomes were brought to room temperature (thaw) and vortexed gently to make a homogenous suspension. 150 μl of WT plasma or 100 μl exosomes either containing miR-155 mimic or scrambled mimic was injected into recipient miR-155 KO mice (intraperitoneal [i.p.] or intravenous [i.v.]) for various times. Blood was drawn and animals were perfused using our laboratory standardized protocol55. To rule out any blood contamination red blood lysis buffer (Sigma, USA) was used as per manufacture’s instructions. At the conclusion of the experiment, various organs were removed, immediately washed in PBS, blotted on tissue and stored in RNA later at −80 °C. To isolate the different liver cell populations, some mice were perfused using the laboratory protocol55. Briefly, mice were anesthetized with ketamine and the livers were perfused with saline solution for 10 mins followed by in vivo digestion with HBSS solution containing collagenase for 5 mins. The perfused liver was placed into petri dish containing HBSS and collagenase and cells were released by separating the liver lobes. To separate hepatocytes, cell suspension was centrifuged at 200 g for 5 mins at room temperature. The pellet containing hepatocytes was washed 2 times with PBS and cultured in low DMEM medium (10% FBS+antibiotics) containing insulin as described55. To isolate mononuclear cells (MNCs), supernatants were centrifuged at 1200 g for 10 mins and pellet was suspended in PBS and washed two times. Red blood lysis buffer (Sigma, USA) was used to remove blood contamination as per manufacture’s instructions. The resulting pellet was suspended in 40% percoll and layered on 70% percoll and centrifuged at 2000 g for 25 mins at room temperature. The interphase containing mononuclear cells were collected and washed with PBS 2 times and cultured in 10% low glucose DMEM containing antibiotics. Kupffer cells (KCs) were isolated as described previously using an established protocol28. Briefly, after 10 mins perfusion of liver with saline solution, in vivo digestion was done using liberase enzyme for 5 mins and followed by in vitro digestion for 30 mins. The non-hepatocyte fraction was separated by Percoll gradient and centrifuged at 1600 g for 30 mins. The inter-cushion layer was collected, washed with PBS two times and cultured in low glucose DMEM supplemented with 10% FBS and antibiotics. The non-adherent cells were removed after 3–4 h of plating with PBS and new medium was added. The KCs were used for subsequent experiments. To understand the biological effect of exo-miR-155 mimic, we performed in vitro studies. Primary hepatocytes and Kupffer cells (KCs) were isolated from miR-155 KO mice and cultured with either control mimic or miR-155 mimic loaded exosomes (~2 × 108 particles/ml) for 6 h. After incubation, cells were washed twice with PBS to remove free-floating exosomes. KCs were counted and plated onto 96 well plates (@1 × 106 cells/ml) and stimulated with or without LPS (100 ng/ml) for 6 h. The supernatants were collected and centrifuged to remove the cell debris and used for MCP1 ELISA. 10–20 mg of each organ was homogenized in QIAzol Lysis reagent (Qiagen, Germany) using stainless steel beads in TissueLyser II (Qiagen, USA). Total RNA was isolated using Direct-zol RNA MiniPrep kit with on column DNA digestion (Zymo Research Corp., USA). For mRNA analysis, cDNA synthesis was carried out using iScript reverse transcription system kit (BioRad, USA) and quantitative analyses of genes were performed using gene-specific primers on a Bio-Rad iCycler real time machine. Primer sequences for TNFα, MCP1, IL-1β and 18S were same as described28. Cq value was normalized to 18S and fold change was calculated using the delta-delta Ct method. For the detection of miRNAs, TaqMan miRNA Assays (Applied Biosystems) were employed as described previously28. SnoRNA202 (tissue/cells) or synthetic cel-miR-39 (exosomes and plasma) was used to normalize the technical variations between the samples. To enhance miR-155 signal in recipient miR-155 KO mice after the administration of WT plasma (miR-155 enriched) a pre-amplification step was introduced. The cDNA was pre-amplified using pre-amplification kit as described by the supplier (Applied Biosystem, USA) and diluted pre-amplified product was used for real time PCR. Supernatants were collected from cultured KCs after co-culture and centrifuged at 2000 g for 10 mins to remove cellular debris and were frozen at −80 °C until use. Protein levels of TNFα, IL-1β and MCP1 were measured from cell-free culture supernatant by ELISA. The quantification of TNFα (BioLegends, San Diego, CA), MCP1 (BioLegend Inc., San Diego, CA) and IL-1β (R&D Systems, Inc., Minneapolis, MN) were carried out based on manufacturers’ recommendations using an ELISA reader. Statistical analysis was performed using non-parametric Mann-Whitney test. Data is shown as average ± standard error of the mean (SEM) and differences were considered statistically significant at p ≤ 0.05. How to cite this article: Bala, S. et al. Biodistribution and function of extracellular miRNA-155 in mice. Sci. Rep. 5, 10721; doi: 10.1038/srep10721 (2015).Methods
Animal studies
Exosome isolation from tissue culture media
Electron Microscopy
Western Blotting
Loading of miR-155 mimic into exosomes
Delivery of WT plasma or miR-155 mimic loaded exosomes
Tissue collection, perfusion and cell isolation
In vitro co-culture of exosomes
RNA isolation and PCR analysis
Enzyme-linked Immunosorbent Assay (ELISA)
Statistical analysis
Additional Information
References
Ambros, V. The functions of animal microRNAs. Nature 431, 350–355 (2004).
Chen, X. et al. Characterization of microRNAs in serum: a novel class of biomarkers for diagnosis of cancer and other diseases. Cell Res. 18, 997–1006 (2008).
Mitchell, P. S. et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc. Natl. Acad. Sci. USA. 105, 10513–10518 (2008).
Weber, J.A. et al. The microRNA spectrum in 12 body fluids. Clin. Chem. 56, 1733–1741 (2010).
Arroyo, J.D. et al. Argonaute2 complexes carry a population of circulating microRNAs independent of vesicles in human plasma. Proc. Natl. Acad. Sci. USA. 108, 5003–5008 (2011).
Vickers, K.C., Palmisano, B.T., Shoucri, B.M., Shamburek, R.D. & Remaley, A.T. MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nat. Cell Biol. 13, 423–433 (2011).
Szabo, G. & Bala, S. MicroRNAs in liver disease. Nat. Rev. Gastroenterol. Hepatol. 10, 542–52 (2013).
Lee, Y., El Andaloussi, S. & Wood, M.J. Exosomes and microvesicles: extracellular vesicles for genetic information transfer and gene therapy. Hum. Mol. Genet. 21, R125–34 (2012).
Kosaka, N. et al. Secretory mechanisms and intercellular transfer of microRNAs in living cells. J. Biol. Chem. 285, 17442–17452 (2010).
Mittelbrunn, M. et al. Unidirectional transfer of microRNA-loaded exosomes from T cells to antigen-presenting cells. Nat. Commun. 2, 282 (2011).
Valadi, H. et al. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 9, 654–659 (2007).
Turchinovich, A., Samatov, T.R., Tonevitsky, A.G. & Burwinkel, B. Circulating miRNAs: cell-cell communication function? Front. Genet. 4, 119 (2013).
Rana, S., Malinowska, K. & Zoller, M. Exosomal tumor microRNA modulates premetastatic organ cells. Neoplasia 15, 281–295 (2013).
Zernecke, A. et al. Delivery of microRNA-126 by apoptotic bodies induces CXCL12-dependent vascular protection. Sci. Signal. 2, ra81 (2009).
**n, H. et al. Exosome-mediated transfer of miR-133b from multipotent mesenchymal stromal cells to neural cells contributes to neurite outgrowth. Stem Cells 30, 1556–1564 (2012).
Skog, J. et al. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 10, 1470–1476 (2008).
Li, C.C. et al. Glioma microvesicles carry selectively packaged coding and non-coding RNAs which alter gene expression in recipient cells. RNA Biol. 10, 1333–1344 (2013).
Nolte-'t Hoen, E.N., Buschow, S. I., Anderton, S.M., Stoorvogel, W. & Wauben, M.H. Activated T cells recruit exosomes secreted by dendritic cells via LFA-1. Blood 113, 1977–1981 (2009).
Camussi, G., Deregibus, M.C., Bruno, S., Cantaluppi, V. & Biancone, L. Exosomes/microvesicles as a mechanism of cell-to-cell communication. Kidney Int. 78, 838–848 (2010).
Anand, P. K., Anand, E., Bleck, C. K., Anes, E. & Griffiths, G. Exosomal Hsp70 induces a pro-inflammatory response to foreign particles including mycobacteria. PLoS One 5, e10136 (2010).
Kooijmans, S. A., Vader, P., van Dommelen, S. M., van Solinge, W. W. & Schiffelers, R. M. Exosome mimetics: a novel class of drug delivery systems. Int. J. Nanomedicine 7, 1525–1541 (2012).
Momen-Heravi, F., Bala, S., Bukong, T. & Szabo, G. Exosome-mediated delivery of functionally active miRNA-155 inhibitor to macrophages. Nanomedicine 10, 1517–27 (2014).
Hagiwara, K., Ochiya, T. & Kosaka, N. A paradigm shift for extracellular vesicles as small RNA carriers: from cellular waste elimination to therapeutic applications. Drug Deliv. Transl. Res. 4, 31–37 (2014).
Pegtel, D. M. et al. Functional delivery of viral miRNAs via exosomes. Proc. Natl. Acad. Sci. USA. 107, 6328–6333 (2010).
Alvarez-Erviti, L. et al. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 29, 341–345 (2011).
Ohno, S. et al. Systemically injected exosomes targeted to EGFR deliver antitumor microRNA to breast cancer cells. Mol. Ther. 21, 185–191 (2013).
O’Connell, R.M., Rao, D.S. & Baltimore, D. MicroRNA Regulation of Inflammatory Responses. Annu. Rev. Immunol. . 30, 295–312 (2011).
Bala, S. et al. Up-regulation of microRNA-155 in macrophages contributes to increased tumor necrosis factor {alpha} (TNF{alpha}) production via increased mRNA half-life in alcoholic liver disease. J. Biol. Chem. 286, 1436–1444 (2011).
Bala, S. et al. Circulating microRNAs in exosomes indicate hepatocyte injury and inflammation in alcoholic, drug-induced and inflammatory liver diseases. Hepatology 56, 1946–57 (2012).
Williams, Z. et al. Comprehensive profiling of circulating microRNA via small RNA sequencing of cDNA libraries reveals biomarker potential and limitations. Proc. Natl. Acad. Sci. USA. 110, 4255–4260 (2013).
Gallo, A., Tandon, M., Alevizos, I. & Illei, G.G. The majority of microRNAs detectable in serum and saliva is concentrated in exosomes. PLoS One 7, e30679 (2012).
Bukong, T.N., Momen-Heravi, F., Kodys, K., Bala, S. & Szabo, G. Exosomes from Hepatitis C Infected Patients Transmit HCV Infection and Contain Replication Competent Viral RNA in Complex with Ago2-miR122-HSP90. PLoS Pathog. 10, e1004424 (2014).
Rottiers, V. & Naar, A.M. MicroRNAs in metabolism and metabolic disorders. Nat. Rev. Mol. Cell Biol. 13, 239–250 (2012).
Vickers, K.C. & Remaley, A.T. Lipid-based carriers of microRNAs and intercellular communication. Curr. Opin. Lipidol. 23, 91–97 (2012).
Creemers, E. E., Tijsen, A.J. & Pinto, Y.M. Circulating microRNAs: novel biomarkers and extracellular communicators in cardiovascular disease? Circ. Res. 110, 483–495 (2012).
Sun, D. et al. A novel nanoparticle drug delivery system: the anti-inflammatory activity of curcumin is enhanced when encapsulated in exosomes. Mol. Ther. 18, 1606–1614 (2010).
Zhuang, X. et al. Treatment of brain inflammatory diseases by delivering exosome encapsulated anti-inflammatory drugs from the nasal region to the brain. Mol. Ther. 19, 1769–1779 (2011).
Imai, T. et al. Macrophage-dependent clearance of systemically administered B16BL6-derived exosomes from the blood circulation in mice. J. Extracell Vesicles 4, 26238 (2015).
Takahashi, Y. et al. Visualization and in vivo tracking of the exosomes of murine melanoma B16-BL6 cells in mice after intravenous injection. J. Biotechnol. 165, 77–84 (2013).
Peinado, H. et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med. 18, 883–891 (2012).
Feng, D. et al. Cellular internalization of exosomes occurs through phagocytosis. Traffic 11, 675–687 (2010).
Zech, D., Rana, S., Buchler, M. W. & Zoller, M. Tumor-exosomes and leukocyte activation: an ambivalent crosstalk. Cell. Commun. Signal. 10, 37-811X–10-37 (2012).
Kooijmans, S. A. et al. Electroporation-induced siRNA precipitation obscures the efficiency of siRNA loading into extracellular vesicles. J. Control. Release 172, 229–238 (2013).
Han, J., Wang, Q. W. & Wang, S. Q. Fluorescent tag is not a reliable marker for small RNA transfection in the presence of serum. J. Biosci. 38, 471–478 (2013).
Chevillet, J.R. et al. Quantitative and stoichiometric analysis of the microRNA content of exosomes. Proc. Natl. Acad. Sci. USA. 111, 14888–14893 (2014).
Aswendt, M., Adamczak, J., Couillard-Despres, S. & Hoehn, M. Boosting bioluminescence neuroimaging: an optimized protocol for brain studies. PLoS One 8, e55662 (2013).
Koberle, V. et al. Differential stability of cell-free circulating microRNAs: implications for their utilization as biomarkers. PLoS One 8, e75184 (2013).
Zhang, X. et al. Regulation of Toll-like receptor-mediated inflammatory response by complement in vivo. Blood 110, 228–236 (2007).
Champion, J.A. & Mitragotri, S. Role of target geometry in phagocytosis. Proc. Natl. Acad. Sci. USA. 103, 4930–4934 (2006).
Bala, S. & Szabo, G. MicroRNA Signature in Alcoholic Liver Disease. Int. J. Hepatol. 2012, 498232 (2012).
Piper, R. C. & Katzmann, D. J. Biogenesis and function of multivesicular bodies. Annu. Rev. Cell Dev. Biol. 23, 519–547 (2007).
Melo, S. A. et al. Cancer Exosomes Perform Cell-Independent MicroRNA Biogenesis and Promote Tumorigenesis. Cancer. Cell. 26, 707–721 (2014).
Petrasek, J., Dolganiuc, A., Csak, T., Kurt-Jones, E. A. & Szabo, G. Type I interferons protect from Toll-like receptor 9-associated liver injury and regulate IL-1 receptor antagonist in mice. Gastroenterology 140, 697–708.e4 (2011).
Saunderson, S.C. et al. Induction of exosome release in primary B cells stimulated via CD40 and the IL-4 receptor. J. Immunol. 180, 8146–8152 (2008).
Csak, T. et al. Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger signals to stimulate immune cells. Hepatology 54, 133–144 (2011).
Acknowledgements
We thank Anna Cerny for her technical assistance in conducting mouse experiments. The work was supported by NIAAA grant-1R01AA020744 (GS). Electron microscopy was carried out using the core electron microscopy facility at the University of Massachusetts Medical School.
Author information
Authors and Affiliations
Contributions
S.B., G.S. and V.A. conceived the project, designed the experiments and analyzed the data. S.B., T.C., F.M.H., D.L., K.K., D.C. and A.S conducted the experiments. S.B. and G.S. wrote the manuscript and all authors read, made critical suggestions and approved the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Bala, S., Csak, T., Momen-Heravi, F. et al. Biodistribution and function of extracellular miRNA-155 in mice. Sci Rep 5, 10721 (2015). https://doi.org/10.1038/srep10721
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep10721
- Springer Nature Limited