
Abstract
A mouse model that recapitulates the human Ewing's sarcoma-specific chromosomal translocation was generated utilizing the Cre/loxP-mediated recombination technique. A cross between Ewsr1-loxP and Fli1-loxP mice and expression of ubiquitous Cre recombinase induced a specific translocation between Ewsr1 and Fli1 loci in systemic organs of both adult mice and embryos. As a result Ewsr1-Fli1 fusion transcripts were expressed, suggesting a functional Ews-Fli1 protein might be synthesized in vivo. However, by two years of age, none of the Ewsr1-loxP/Fli1-loxP/CAG-Cre (EFCC) mice developed any malignancies, including Ewing-like small round cell sarcoma. Unexpectedly, all the EFCC mice suffered from dilated cardiomyopathy and died of chronic cardiac failure. Genetic recombination between Ewsr1 and Fli1 was confirmed in the myocardial tissue and apoptotic cell death of cardiac myocytes was observed at significantly higher frequency in EFCC mice. Moreover, expression of Ews-Fli1 in the cultured cardiac myocytes induced apoptosis. Collectively, these results indicated that ectopic expression of the Ews-Fli1 oncogene stimulated apoptotic signals and suggested an important relationship between oncogenic signals and cellular context in the cell-of-origin of Ewing's sarcoma.
Similar content being viewed by others

Introduction
Chromosomal translocation is a common feature of malignant neoplasms1. There is growing evidence that tumor-specific translocations and inversions commonly occur among hematopoietic, mesenchymal and epithelial tumors. An increasing number of gene fusions resulting from translocation have been observed as novel technological tools have been applied. Tumor-associated chromosomal translocations include two major molecular mechanisms. One is an oncogene juxtaposition to the enhancing elements of immunoglobulin or T-cell receptor associated with lymphoid neoplasms. As a result of the juxtaposition, constitutive expression of oncogenes such as c-MYC, BCL2 or CCND1 induces abnormal cellular functions, including cell cycle progression and apoptosis suppression1. Another important outcome of translocation in cancer is gene fusion or formation of chimeric genes. Two major functional aberrations of fusion gene products are constitutive activation of signal transduction and dysregulation of transcription. Most oncogenic gene fusions in human bone and soft tissue sarcomas belong to the latter group and there is a specific relationship between tumor types and each gene fusion2.
To clarify the functional roles of sarcoma-specific chromosomal translocations and gene fusions, it would be ideal to induce chromosomal translocation in animal models in vivo. In contrast to transgenic expression of fusion genes, translocation-mediated gene fusion recapitulates gene expression levels equivalent to and splice variants similar to those in human tumors. Inducible, site-specific chromosomal translocation has been achieved using Cre-loxP-mediated recombination in murine ES cells. Using this strategy, translocations between c-myc and immunoglobulin heavy chain loci and between Dek and Can loci were successfully induced, though the efficiencies were not very high3,4. Indeed, a mouse model of Cre-loxP-mediated in vivo gene fusion between Mll and Af9 developed acute myeloid leukemia5. However, it is not known whether solid tumor-related translocation in vivo can induce malignancies of the anticipated phenotypes.
The ETS family of transcription factors includes FLI1 and ERG. They are major fusion partners for the EWSR1 gene in human Ewing's sarcoma6,7. EWS-FLI1 and EWS-ERG function as oncogenic transcription factors that dysregulate their downstream targets such as NKX2-2, NR0B1 and EZH28. It is, however, difficult to generate a good animal model by introduction of EWS-FLI1 or EWS-ERG into ES cells or mouse eggs8. Moreover, conditional EWS-FLI1 expression in hematopoietic cells induced myeloid and erythroid leukemia in mice9. Thus, it might be necessary to activate multiple target genes without activating pro-apoptosis signals for tumorigenic activity of EWS-ETS. We therefore hypothesized that EWS-ETS translocation is achieved by chance in human somatic cells of appropriate lineages and differentiation status and such in vivo translocation could properly induce Ewing's sarcoma.
In an effort to induce Ewing's sarcoma in a mouse model, we have succeeded in promoting in vivo Cre-loxP-mediated translocation between Ewsr1 and Fli1 loci on chromosomes 11 and 9, respectively. Although the Ewsr1-Fli1 fusion was confirmed at both DNA and RNA levels, no neoplastic lesion was induced in the model. Unexpectedly, the mice with systemic translocation developed dilated cardiomyopathy due to degeneration and apoptotic cell death of cardiac myocytes. The result indicates that ectopic chromosomal translocation and gene fusion activates apoptotic signals, resulting in degenerative cardiac disease.
Results
Generation of a mouse model for somatic chromosomal translocation between Ewsr1 and Fli1
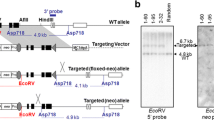
To induce locus-specific chromosomal translocation, loxP sequences were introduced into Ewsr1 intron 7 on mouse chromosome 11 and Fli1 intron 5 on chromosome 9 (Fig. 1A), since chromosomal breakpoints in human Ewing's sarcoma are most frequently observed in these loci10. Successful knock-in of loxP sequences mediated by homologous recombination was confirmed for both loci in independent ES cells by Southern blotting (Fig. 1B). Both Ewsr1fl/+ and Fli1fl/+ mice appeared normal and healthy at birth. Germline transmission of the targeted alleles was confirmed. Ewsr1fl/+ and Fli1fl/+ mice were crossed to obtain mice having both mutations.

Gene targeting for the Ewsr1-Fli1 translocation model.
(A) Physical maps of targeting alleles for Ewsr1 (top) and Fli1 (bottom) loci. Closed triangles indicate the loxP sequence. K: KpnI, X: XbaI, S: SacI, H: HindIII. (B) Southern blot analysis of ES cells. A 5.2 kb Neo-positive band and a 4.0 kb Neo-deleted band indicate homologous recombination of the Ewsr1 locus as shown by XbaI digestion (top). A 6.5 kb Neo-positive band and a 5.2 kb Neo-deleted bands for the Fli1 locus are shown by SacI digestion (bottom). Rearranged bands are indicated by arrows.
Genomic chromosomal translocation between chromosomes 9 and 11 in the Ewsr1fl/+:Fli1fl/+:CAG-Cre (EFCC) mice
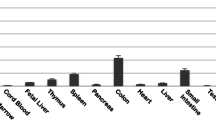
The Ewsr1fl/+ and Fli1fl/+ mice were further crossed with CAG-Cre, Mx1-Cre or Rosa26-CreER mice to induce somatic chromosomal translocation between chromosomes 9 and 11 (Fig. 2A). Dual color fluorescence in situ hybridization (FISH) analysis of embryonic fibroblasts derived from the EFCC mice showed juxtaposition of the signal on der9 of BAC clone RPCI-23 64E17 from chromosome 11 and that of 218O31 from chromosome 9 (Fig. 2B). Reciprocal genomic translocations in systemic organs were examined by genomic PCR using Ewsr1- and Fli1-specific primers and both Ewsr1-Fli1 and Fli1-Ewsr1 translocations were detected in tail skin of all the mice examined (n = 30). The translocations in systemic organs were examined in three mice and both Ewsr1-Fli1 and Fli1-Ewsr1 translocations were detected in all the organs examined (Fig. 2C). The results indicated that loxP-mediated recombination was effective at inducing somatic translocation by ubiquitous Cre recombinase expression. The frequencies of the chromosomal translocations were 1.5 × 10−5 at the highest in heart and 1 × 10−6 in bone marrow as estimated by quantitative genomic PCR comparing Ewsr1-Fli1 and Trib1 signals (Fig. 2D). The estimated translocation frequencies in the model are higher than those observed in ES cells described in the previous report3. When Cre recombinase was inducibly expressed by tamoxifen or polyIpolyC administration in a Rosa26-CreER or Mx1-Cre background, respectively, both Ewsr1-Fli1 and Fli1-Ewsr1 translocations were observed (four mice each) (Fig. 2e). However, the translocations were detected only by nested PCR in limited organs, indicating that recombination was less frequent in these Cre transgenes. In addition, inducible expression of Cre upon in the Mx1-Cre background resulted in translocations being limited to hematopoietic tissues.

Somatic chromosomal translocation between mouse chromosomes 9 and 11.
(A) A schematic diagram of the Cre-mediated translocation model. EF;wt, Ewsr1fl/+:Fli1fl/+:wild-type. Cre-Tg, Cre transgenic. EF;Cre, Ewsr1fl/+:Fli1fl/+: Cre transgenic. Illustration of mice was drawn using Microsoft PowerPoint 2011 and then converted to tif format using Adobe Photoshop CS5. (B) Metaphase FISH shows t(9;11) translocation at Ewsr1 and Fli1 loci. The green fluorescence of 64E17 shows Ewsr1 on chromosome 11 and the red fluorescence of 218O21 shows Fli1 on chromosome 9. The yellow signal indicates translocation between two loci on der9. (C) The reciprocal t(9;11) translocation was shown in systemic organs of the EFCC mouse detected as Ewsr1-Fli1 and Fli1-Ewsr1 PCR products. Ewsr1 amplification is shown as a loading control. (D) Estimated frequencies of translocation in bone marrow (BM), heart and brain calculated from the result of quantitative genomic PCR data in three independent mouse samples. (E) The reciprocal t(9;11) translocation in the organs of Rosa26-CreER and Mx1-Cre background detected by nested genomic PCR. Gel image shown is cropped and representative of gels run under the same experimental conditions.
Detection of chimeric Ewsr1-Fli1 fusion transcripts in EFCC mice
To confirm that gene fusion between Ewsr1 and Fli1 was accompanied by the anticipated transcription, RT-PCR was performed using RNA samples obtained from systemic organs of both adult and embryonic mice (three mice each) (Fig. 3A, 3B). The Ewsr1-Fli1 fusion was detected in all the embryonic organs examined and the expression of the fusion gene was decreased in bone and liver of the adult mice. Diminished Ewsr1-Fli1 expression in adult bone and liver might be related to decreased proliferative activity of osteochondrogenic tissues and disappearance of embryonic hematopoietic cells, respectively. No reciprocal Fli1-Ewsr1 fusion transcript was detected in any of the organs examined (data not shown). The cDNA sequence of the Ewsr1-Fli1 fusion transcript was analyzed by sequencing and in-frame fusion between Ewsr1 exon 7 and Fli1 exon 6 was confirmed (Fig. 3C). It is expected that the fusion product included both the EWS Q-rich repeats and the FLI1 ETS DNA binding domain11. Thus, the data strongly suggested that a functional EWS-FLI1 protein was produced by somatic chromosomal translocation in the model.

Ewsr1 is fused in-frame to Fli1.
(A, B) RT-PCR to detect Ews-Fli1 fusion transcripts in adult (A) and embryonic tissues (B). Data are representatives of three independent experiments with similar results. (C) Sequence analysis of the RT-PCR product using heart cDNA shows the in-frame fusion between Ewsr1 exon 7 and Fli1 exon 6. Deduced amino acid sequences are indicated on the nucleotide sequences.
EFCC mice died of chronic cardiac failure due to dilated cardiomyopathy
No malignant neoplasms, including Ewing's sarcoma-like lesions, were observed in EFCC mice (n = 30) for a two year period after birth. Neither sarcomas nor benign neoplasms were detect by careful examination of mice irrespective of age. Instead, most of the EFCC mice showed growth retardation and decreased motility. All the EFCC mice died by 100 weeks of age with a mean survival time of only 40 weeks (Fig. 4A). The diseased mice were carefully examined at autopsy and they showed extensive dilatation of heart (Fig. 4B). The heart weight/body weight ratio as well as heart weight itself of EFCC mice was significantly greater than that of control mice from 31 to 42 weeks (Fig 4C, Table 1). Mice of the age were selected since the severity of cardiac lesions was significantly varied in younger EFCC mice. The pathological examination further revealed the cardiac lesions and subsequent systemic congestive changes. The hearts of EFCC mice showed extensive dilatation of both the ventricles and thin ventricular wall without any signs of cardiac hypertrophy (Fig 4D). The earlier the mice became sick, the more severe the cardiac lesions were. High power views of cardiac sections indicated a disorganized arrangement of myocardial fibers with increased collagen fibers between the muscle bundles. The subendocardial area was severely affected and leukocytic infiltration was sometimes present. There was severe chronic congestion in systemic organs such as lung, liver or spleen accompanied by ischemic necrosis around the central vein of the liver (Fig. 4E).

EFCC mice died of chronic cardiac failure.
(A) Kaplan-Meier survival curve. Statistical significance was evaluated by the log-rank test. (B) Cardiac enlargement in the EFCC mouse (right) compared to EF;wild-type (left). (C) Box plot of the heart weight/body weight ratios for EF;wild-type (n = 6) and EFCC mice (n = 6). (D) Extensive ventricular dilatation of the heart in the EFCC mouse without myocardial hypertrophy (top). High power view of myocardium with H&E (middle) and Masson's trichrome staining (bottom). Extensive fibrosis is indicated as blue staining in the EFCC heart. (E) Chronic congestion of systemic organs in EFCC mice including lung, liver and spleen. Note necrotic changes around the central vein of liver.
Consistent with the pathological findings, echocardiographic analysis revealed reduced wall thickness, significant fractional shortening and decreased ejection fraction in EFCC mice (Fig. 5, Table 2). In contrast, there was no significant difference in blood pressure, heart rate or diastolic dimension between EFCC and wild-type mice (Table 2). Collectively, these findings are consistent with those of dilated cardiomyopathy.

Echocardiographic analysis of 37-week-old EFCC and EF;wild-type mice.
Analysis of cardiovascular function (top). DD, diastolic diameter of left ventricle; FS, fractional shortening; EF, ejection fraction. Representative echocardiogram for wild-type and EFCC mice (bottom). EDD, end-diastolic diameter; ESD, end-systolic diameter, IVS, interventricular septum; LV, left ventricle; PW, posterior wall.
Ewsr1-Fli1 translocation and Ewsr1-Fli1 expression induced myocardial damage
To obtain insights into the mechanisms of dilated cardiomyopathy in EFCC mice, the cardiac lesion was further investigated. Laser microdissection followed by genomic PCR to detect the Ewsr1-Fli1 translocation was carried out (Fig. 6A). Ewsr1-Fli1 was abundantly observed in the outer area of the ventricular wall, however, no signal was detected in the subendocardial area where the myocardial damage was more severe (Fig. 6A, 1 and 3). Severer damages in the subendocardial area were observed in most of mice, though the reason for such uneven distribution of cardiac lesions was unclear. The results suggested degeneration of cardiac myocytes with translocation and perhaps gradual loss due to the pathologic effects of Ewsr1-Fli1 expression. Indeed, a TUNEL assay using the cardiac sections showed significantly increased apoptosis in EFCC mice compared to wild-type (Fig. 6B).

The cardiac lesion in the EFCC mouse and Ewsr1-Fli1 translocation.
(A) Detection of Ewsr1-Fli1 translocation in the myocardium. The frozen section of the cardiac tissue from the EFCC mouse was laser microdissected for the indicated areas (1-4) (left). Genomic PCR using DNA samples obtained by laser microdissection (right). (B) A TUNEL assay showed a significantly greater increase of apoptotic cell death in the myocardium of the EFCC mouse than in that of the wild-type mouse (left). High power view of the apoptotic cell is shown in the magnified inset. Frequencies of TUNEL-positive cells per section are compared between wild type and EFCC mice (right). (C) Ewsr1-Fli1 cDNA expression induced apoptotic cell death of cardiac myocytes in vitro. The apoptotic cells were measured by positive signals in a TUNEL assay (left). Ewsr1-Fli1-induced cell death was further analyzed by Annexin V/PI staining and FACS analysis. The lower right quadrant (Annexin V+/PI-) represents early apoptosis, while the upper right quadrant (Annexin V+/PI+) and the upper left quadrant (Annexin V-/PI+) represent late apoptosis and necrosis, respectively. Data are representatives of three independent experiments with similar results (center). The expression of EWS-FLI1 protein in cardiac myocytes was detected by Western blotting using anti-FLAG M2 antibody (right). (D) Quantitative real-time RT-PCR for COL1A1 in human cardiac fibroblasts with or without Ewsr1-Fli1 (left). Expression of EWS-FLI1 protein was detected by Western blotting using anti-FLAG M2 antibody (right). (E) Expression of the Cre protein in the heart of EFCC mice and other Cre transgenic lines of variable expression levels12.
The toxic effect of Ewsr1-Fli1 was directly evaluated by its exogenous expression in cultured cardiac myocytes. The murine neonatal cardiac myocytes were infected with Ewsr1-Fli1-lentivirus and the frequencies of apoptosis were evaluated (Fig. 6C). The TUNEL assay showed that apoptosis of cardiac myocytes was significantly increased when Ewsr1-Fli1 was expressed in the cardiac myocytes. The Annexin V/PI flow cytometry analysis showed increases of both early and late apoptosis as well as necrosis in cardiac myocytes by Ewsr1-Fli1 expression (Fig. 6C). These results indicated that Ewsr1-Fli1 induced cellular apoptosis in the cardiac tissue, resulting in cellular damage and eventual dilated cardiomyopathy. In addition, Ewsr1-Fli1 expression in human cardiac fibroblasts induced increased expression of COL1A1 (Fig. 6D), suggesting that Ewsr1-Fli1 may also play some role in cardiac fibrosis.
A previous study indicated that the high level of expression of Cre recombinase itself showed cardiac toxicity12. The expression level of the Cre protein in the hearts of the EFCC mouse was therefore compared with high-expressing Cre transgenic mice (Fig. 6E). Cre expression of EFCC mice was comparable to the low Cre transgenic mice that did not show cardiac lesions. The results indicated that the cardiac lesion was caused not by Cre expression but by Ewsr1-Fli1.
Discussion
Cre/loxP-mediated chromosomal translocations in mouse models have been reported5,13,14. In those studies loxP sites were inserted into the introns of Mll or Af9 genes and the mice carrying the mutations were crossed to place loxP sites in both genes. Both ubiquitous and hematopoietic-specific expression of Cre recombinase induced in vivo chromosomal translocation and the fusion of Mll and Af9, resulting in leukemia development. In contrast, leukemia was not observed in the mice bearing chromosomal translocation between AML1 and ETO in vivo using a similar protocol15.
In the present study, Ewsr1-Fli1 fusion was successfully induced in various organs. Ewing's sarcoma, however, did not develop in the mice, suggesting that the cell-of-origin of Ewing's sarcoma might constitute a rare cellular population unlike hematopoietic neoplasms. Supporting this idea, we have recently succeeded in develo** Ewing's sarcoma-like small round cell tumors by introducing Ews-Fli1 or Ews-Erg into eSZ cells that are enriched in embryonic chondrogenic progenitors16. Therefore, when chromosomal translocation between Ewsr1 and Fli1 is efficiently induced in eSZ cells, Ewing's sarcoma can develop in a certain cohort using the current translocation model. It is likely that ubiquitous Cre expression affects most cell lineages both in develo** and adult mouse tissues including the true cell-of-origin of Ewing's sarcoma. However, the low frequency of chromosomal recombination could not induce detectable translocations in such a rare cell type. Perhaps eSZ cell-specific Cre expression may enable the induction of Ewing's sarcoma by somatic Ewsr1 and Fli1 translocation and efficient Cre expression in the specific spatiotemporal manner in the eSZ cell may be achieved using the promoter/enhancer elements of Gdf5 or Erg genes17,18.
Expression of Ews-Fli1 in the majority of primary cells induced cellular apoptosis or senescence19,20,21. Activation of the Casp3 promoter by EWS-FLI1 was reported and the activation of caspase 3-dependent signals may be responsible for apoptotic processes in mouse embryonic fibroblasts (MEFs) with ectopic Ews-Fli1 expression21. Indeed, Ews-Fli1 expression in cardiac myocytes induced apoptotic cell death, though activation of caspase 3 was not detected in cardiac myocytes unlike in MEFs (data not shown). Thus, the low capacity for cardiac myocyte regeneration after birth could not support cardiac homeostasis. This limitation, therefore, could result in gradual but irreversible cardiac damage. In support of this idea, the Ews-Fli1 fusion was not detected in the severely degenerated area but remained in relatively normal parts of the heart in EFCC mice. Moreover, introduction of Ews-Fli1 cDNA significantly induced apoptosis in primary cardiac myocytes, indicating the cardiac toxicity of the fusion gene. The cell type-specific epigenetic status may modulate growth inhibitory and tumorigenic activities of EWS-FLI1. Indeed, different chromatin modification was observed between Ewing's sarcoma-sensitive eSZ and –resistant eGP cells16. It is noted that wild-type FLI1 protein represses Col1a1 expression, inhibiting cardiac fibrosis22. Interestingly, EWS-FLI1 enhanced COL1A1 expression in human cardiac fibroblasts, suggesting that it might accelerate fibrotic processes in cardiomyopathy.
A number of transcription factors are associated with the development and maintenance of cardiac myocytes and mutations in these factors affect cardiac homeostasis, structure and functions23. Over-expression of E2F6 activates gene expression in myocardium and induces dilated cardiomyopathy in mice24. Moreover, mutations in NKX2-5 and PDRM16 were found associated with human congenital dilated cardiomyopathy25,26. It has been proposed that these proteins regulate genes involved in the ubiquitin proteasome system or proliferation of cardiomyocytes, suggesting different aspects of myocardial damage from the present model. Nevertheless, similar phenotypes shown in these models indicate the importance of cardiac-specific transcriptional regulation by transcription factors, given the low regenerative activity of adult cardiomyocytes.
Methods
Mice and gene targeting
The Ewsr1 and Fli1 targeting vectors were assembled in a pBSKSTKLoxPNeoGFP plasmid containing appropriate loxP sites, a loxP-flanked thymidine kinase (Tk) promoter-driven neo gene and a Tk promoter-driven diphtheria toxin gene. A Gfp gene was inserted immediately downstream of the 3′ loxP site for the Ewsr1 vector. The homologous regions of the Ewsr1 vector consisted of an 8.4 kb genomic fragment containing Ewsr1 exons 5 to 7 and a 1.3 kb flanking exon 8 (Fig. 1a). Similarly, the Fli1 vector included a 5.4 kb genomic fragment of Fli1 intron 5 and a 2.0 kb fragment flanking exon 6. A CMV promoter sequence was also inserted immediately upstream of the 5′ loxP site of the Fli1 vector. To establish mice carrying a single loxP allele of Ewsr1 or Fli1 genes, the linearized targeting vectors were electroporated into E14 ES cells and drug-resistant colonies were screened for homologous recombination. To remove the loxP-flanked neomycin-resistant gene cassette, the pMCCreGKPuro vector was electroporated into the ES cells and puromycin-resistant colonies were selected. Targeted clones were injected into C57BL/6 blastocysts and the resultant chimeric mice were bred to produce progeny having germ line transmission of the mutated allele. Mice harboring a targeted Ewsr1 allele (Ewsr1fl/+) and a targeted Fli1 allele (Fli1fl/+) were crossed to establish the mice that possessed loxP sites both in Ewsr1 intron 7 and in Fli1 intron 5. The resultant Ewsr1fl/+ and Fli1fl/+ mice were further crossed with CAG-Cre, Mx1-Cre or Rosa26-CreER mice27,28,29. Genoty** of the mice was performed using primers described below. Animals were handled in accordance with the guidelines of the animal care committee at the Japanese Foundation for Cancer Research, which gave ethical approval for these studies.
Southern blotting
Southern blotting was carried out using standard procedures30. Genomic DNA samples were digested with XbaI or SacI and probed with genomic DNA fragments derived from Ewsr1 or Fli1 loci (Fig. 1a).
Fluorescence in situ hybridization (FISH)
The BAC clones, RPCI-23 64E17 downstream from Ewsr1 on mouse chromosome 11 and RPCI-23 218O31 upstream from Fli1 on chromosome 9 were purchased from Invitrogen (Carlsbad, CA) for FISH analysis. The FISH analysis using metaphase spreads obtained from embryonic fibroblasts of the Ewsr1fl/+:Fli1fl/+:CAG-Cre (EFCC) mouse was performed according to the methods previously described31.
Genomic and reverse transcription-polymerase chain reaction (gPCR and RT-PCR)
Genomic DNA (100 ng) was subjected to 35 cycles of PCR amplification. The PCR primers to detect the Ewsr1-Fli1 fusions were as follows. For Ewsr1-Fli1, Ewsr1 forward primer 5′-ccccagtgcttatccttacatttg-3′ and Fli1 reverse primer 5′-cctgacccgtgtctttgtag-3′ and for Fli1-Ewsr1, Fli1 forward primer 5′-agagaacccactgcttactgg-3′ and Ewsr1 reverse primer 5′-accacgccctccaggttcac-3′ were used. To detect the rare translocation in Rosa26-CreER and Mx1-Cre transgenic mice, genomic DNA samples were pre-amplified using 35 cycles of PCR using the following primers. For Ewsr1, the 5′ primer was 5′-ccaagtaggggctctgtcag-3′ and for Fli1, the 3′ primer was 5′-ggagctgaagcagtaggaag-3′. For Fli1, the 5′ primer was 5′-gccccattgacgcaaatggg-3′ and for Ewsr1, the 3′ primer was 5′-ggggtacttggtgaaggtgc-3′. Genomic PCR for the wild-type Ewsr1 transgene, Cre recombinase or Trib1 was performed using the following primers: Ewsr1, forward, 5′-cccagtgcttatccttacatttg-3′ and GFP, reverse, 5′-accacgccctccaggttcac-3′, Cre, forward, 5′-catacctggaaaatgcttctgtcc-3′ and Cre, reverse, 5′-attgctgtcacttggtcgtggc-3′, or Trib1, forward, 5′-cagtctctccttccaagtcatc-3′ and Trib1, reverse, 5′-gattgttgctgctgttgttc-3′. The PCR products were analyzed by 2% agarose gel electrophoresis.
RT-PCR was carried out using cDNA generated from total RNA of systemic organs as previously described32. The Ewsr1-Fli1 fusion transcript was amplified using Ewsr1 exon 7 primer (5′-tcctcttcacagccgac-3′) and Fli1 exon 6 primer (5′-ctgctcagtgttcttgcc-3′). The primers for Cre recombinase (forward, 5′-cggtctggcagtaaaaactat-3′; reverse, 5′-cagggtgttataagcaatccc-3′) and Hprt (forward, 5′-gctggtgaaaaggacctct-3′; reverse, 5′-cacaggactagaacacctgc-3′) were also used. The PCR products were purified, subcloned into a plasmid and sequenced. Real-time quantitative RT-PCR was performed by using a Fast Real-Time PCR System (Applied Biosystems, Foster City, CA). The primers for human COL1A1 (forward, 5′-catgaccgagacgtgtggaa-3′; reverse, 5′-tttcttggtcggtgggtgac-3′) and GAPDH (forward, 5′-acctgacctgccgtctagaa-3′; reverse, 5′-aaagtggtcgttgagggcaa-3′) were used.
Echocardiography
Transthoracic echocardiography was performed on conscious, gently restrained mice using a 15-MHz linear probe (Power-Vision 8000, Toshiba, Tokyo, Japan), as described previously33. Parasternal long-axis view and short axis view of the left ventricle at the level of the papillary muscles were obtained. 2D-guided M-mode recordings were obtained from short axis view at the level of the papillary muscles. Measurements of interventricular septum thickness (IVST) and left ventricular posterior wall thickness (LVPWT) were made from M-mode recordings in diastole. Left ventricular internal diameter at end-diastole (LVDd) and end-systole (LVDs) were measured from M-mode recordings. Fractional shortening (FS) was calculated as 100 × [(LVDd − LVDs)/LVDd] (%). Ejection fraction (EF) was calculated using the Teichholtz method.
Cell culture and recombinant lentivirus infection
Primary neonatal ICR mouse ventricular myocytes were purchased from Cosmo Bio (Tokyo, Japan) and cells were cultured with D-MEM/F-12 medium supplemented 10% fetal bovine serum (HyClone, South Logan, UT). Human cardiac fibroblasts were purchased from PromoCell (Heidelberg, Germany) and cells were cultured with Fibroblast Medium (ScienCell, Carlsbad, CA). The human EWSR1-FLI1 cDNA (a kind gift from Susanne Baker) was FLAG-tagged and inserted into the pLVSIN-CMV-neo plasmid (Takara Bio, Tokyo, Japan) and HEK 293 cells were transfected with the plasmid using Lipofectamine 2000 (Invitrogen). Cells were harvested 48 h after lentiviral infection and subjected to further analyses.
TUNEL assay and Annexin-V analysis
Formaldehyde-fixed and paraffin-embedded cardiac tissue sections or methanol-fixed murine primary cardiac myocytes were subjected to TUNEL assays using the DeadEnd Colorimetric TUNEL System (Promega, Madison, WI) according to the manufacturer's protocol. For the Annexin V analysis cells were stained with Annexin V-FITC and propidium iodide (PI) according to the manufacturer's instruction (BD Bioscience Pharmingen, San Diego, CA). The stained cells were immediately evaluated using a FACSCalibur flow cytometer (BD Biosciences, Franklin Lakes, NJ).
Western blotting
Western blotting was performed as previously described32. A monoclonal anti-FLAG M2 antibody was purchased from Sigma (St Louis, MO), anti-Cre from Chemicon (Temecula, CA), anti-α-tubulin from Sigma and anti-GAPDH from HyTest (Turku, Finland).
Statistical analysis
Results are shown as means ± standard errors of the mean (SEM). Continuous distributions were compared with two-tailed Student's t-tests. Survival analysis was performed using the Kaplan-Meier life table method and the survival between groups was compared with the log-rank test. All P values were two-sided and a P value of less than 0.05 was considered significant.
References
Mitelman, F., Johansson, B. & Mertens, F. The impact of translocation and gene fusions on cancer causation. Nat. Rev. Cancer 7, 233–245 (2007).
Taylor, B. S. et al. Advances in sarcoma genomics and new therapeutic targets. Nat. Rev. Cancer 11, 541–557 (2011).
Smith, A. J. H. et al. A site-directed chromosomal translocation induced in embryonic stem cells by Cre-loxP recombination. Nat. Genet. 9, 376–385 (1995).
Van Deursen, J., Fornerod, M., van Rees, B. & Grosveld, G. Cre-mediated site-specific translocation between nonhomologous mouse chromosomes. Proc. Natl. Acad. Sci. USA 92, 7376–7380 (1995).
Forster, A. et al. Engineering de novo reciprocal chromosomal translocations associated with Mll to replicate primary events of human cancer. Cancer Cell 3, 449–458 (2003).
Delattre, O. et al. Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature 359, 162–165 (1992).
Sorensen, P. H. et al. A second Ewing's sarcoma translocation, t(21;22), fuses the EWS gene to another ETS-family transcription factor, ERG. Nat. Genet. 6, 146–151 (1994).
Ordonez, J. L., Osuna, D., Herrero, D., de Alava, E. & Madoz-Gurpide, J. Advances in Ewing's sarcoma research: where are we now and what lies ahead? Cancer Res. 69, 7140–7150 (2009).
Torchia, E. C., Boyd, K., Rehg, J. E., Qu, C. & Baker, S. J. EWS/FLI-1 induces rapid onset of myeloid/erythroid leukemia in mice. Mol. Cell. Biol. 27, 7918–7934 (2007).
Zucman, J. et al. Combinatorial generation of variable fusion proteins in the Ewing family of tumours. EMBO J. 12, 4481–4487 (1993).
Riggi, N., Cironi, L., Suva, M. L. & Stamenkovic, I. Sarcomas: genetics, signaling and cellular origins. Part I: The fellowship of TET. J. Pathol. 213, 4–20 (2007).
Buerger, A. et al. Dilated cardiomyopathy resulting from high-level myocardial expression of Cre-recombinase. J. Card. Fail. 12, 392–398 (2006).
Collins, E. C., Pannell, R., Simpson, E. M., Forster, A. & Rabbitts, T. H. Inter-chromosomal recombination of Mll and Af9 genes mediated by cre-loxP in mouse development. EMBO Rep. 1, 127–132 (2000).
Drynan, L. F. et al. Mll fusions generated by Cre-loxP-mediated de novo translocations can induce lineage reasignment in tumorigenesis. EMBO J. 24, 3136–3146 (2005).
Buchholz, F., Regaeli, Y., Trumpp, A. & Bishop, J. M. Inducible chromosomal translocation of AML1 and ETO genes through Cre/loxP-mediated recombination in the mouse. EMBO Rep. 11, 133–139 (2000).
Tanaka, M. et al. Ewing's sarcoma precursors are highly enriched in embryonic osteochondrogenic progenitors. J. Clin. Invest. 121, 3061–3074 (2014).
Storm, E. E. & Kingsley, D. M. GDF5 coordinates bone and joint formation during digit development. Dev. Biol. 209, 11–27 (1999).
Vijayaraj, P. et al. Erg is a crucial regulator of endocardial-mesenchymal transformation during cardiac valve morphogenesis. Development 139, 3973–3985 (2012).
Deneen, B. & Denny, C. T. Loss of p16 pathways stabilizes EWS/FLI1 expression and complements EWS/FLI1 mediated transformation. Oncogene 20, 6731–6741 (2001).
Lessnick, S. L., Dacwag, C. S. & Golub, T. R. The Ewing's sarcoma oncoprotein EWS/FLI induces a p53-dependent growth arrest in primary human fibroblasts. Cancer Cell 1, 393–401 (2002).
Sohn, E. J. et al. EWS/FLI1 oncogene activates caspase 3 transcription and triggers apoptosis in vivo. Cancer Res. 70, 1154–1163 (2010).
Elkareh, J. et al. Marinobufagenin induces increases in procollagen expression in a process involving protein kinase C and Fli-1: implications for uremic cardiomyopathy. Am. J. Physiol. Renal. Physiol. 296, F1219–F1226 (2009).
Oka, T., Xu, J. & Molkentin, J. D. Re-employment of developmental transcription factors in adult heart disease. Semin. Cell Dev. Biol. 18, 117–131 (2007).
Westendorp, B. et al. The E2F6 repressor activates gene expression in myocardium resulting in dilated cardiomyopathy. FASEB J. 26, 2569–2579 (2012).
Costa, M. W. et al. Functional characterization of a novel mutation in NKX2-5 associated with congenital heart disease and adult-onset cardiomyopathy. Circ. Cardiovasc. Genet. 6, 238–247 (2013).
Arndt, A. K. et al. Fine map** of the 1p36 deletion syndrome identifies mutation of PRDM16 as a cause of cardiomyopathy. Am. J. Hum. Genet. 93, 67–77 (2013).
Sakai, K. & Miyasaki, J. A transgenic mouse line that retains Cre recombinase activity in mature oocytes irrespective of the cre transgene transmission. Biochem. Biophys. Res. Commun. 237, 318–324 (1997).
Kuhn, R., Schwenk, F., Aguet, M. & Rajewsky, K. Inducible gene targeting in mice. Science 269, 1427–1429 (1995).
Ventura, A. et al. Restoration of p53 function leads to tumour regression in vivo. Nature 445, 661–665 (2007).
Iwasaki, M. et al. Identification of cooperative genes for NUP98-HOXA9 in myeloid leukemogenesis using a mouse model. Blood 105, 784–793 (2005).
Kawamura-Saito, M. et al. Fusion between CIC and DUX4 up-regulates PEA3 family genes in Ewing-like sarcomas with t(4;19)(q35;q13) translocation. Hum. Mol. Genet. 15, 2125–2137 (2006).
Nakamura, T. et al. Evi9 encodes a novel zinc finger protein that interacts with BCL6, a known human B-cell proto-oncogene. Mol. Cell. Biol. 20, 3178–3186 (2009).
Kuwahara, K. et al. NRSF regulates the fetal cardiac gene program and maintains normal cardiac structure and function. EMBO J. 22, 6310–6321 (2003).
Acknowledgements
We are grateful to Junichi Miyazaki for CAG-Cre, Klaus Rajewsky for MX1-Cre and Tyler Jacks for Rosa26-Cre transgenic mice. We also thank Miki Yamazaki, Yohei Kanno, Hitomi Yamanaka and Tokuichi Kawaguchi for technical assistance. This work was supported by the Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology (23791672 and 26250029) to M.T. and T.Na.
Author information
Authors and Affiliations
Contributions
M.T., T.No. and T.Na. designed the work. M.T., S.Y., Y.Y. and H.K. performed the experiments. M.T., K.K., K.N., P.Y.J., T.No. and T.Na. analyzed the data. M.T. and T.Na. wrote the paper. All co-authors contributed in the form of discussion and critical comments.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Tanaka, M., Yamaguchi, S., Yamazaki, Y. et al. Somatic chromosomal translocation between Ewsr1 and Fli1 loci leads to dilated cardiomyopathy in a mouse model. Sci Rep 5, 7826 (2015). https://doi.org/10.1038/srep07826
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep07826
- Springer Nature Limited
This article is cited by
-
RETRACTED ARTICLE: Robust method for TALEN-edited correction of pF508del in patient-specific induced pluripotent stem cells
Stem Cell Research & Therapy (2016)