
Abstract
Chronic obstructive pulmonary disease (COPD) is the 3rd leading cause of death worldwide. Cigarette smoke which has approximately 2–3 µg of Cadmium (Cd) per cigarette contributes to the environmental exposure and development and severity of COPD. With the lack of a cadmium elimination mechanism in humans, the contribution of cadmium induced stress to lung epithelial cells remains unclear. Studies on cadmium responsive miRNAs suggest regulation of target genes with an emphasis on the critical role of miRNA–mRNA interaction for cellular homeostasis. Mir-381, the target miRNA in this study is negatively regulated by cadmium in airway epithelial cells. miR-381 is reported to also regulate ANO1 (Anoctamin 1) expression negatively and in this study low dose cadmium exposure to airway epithelial cells was observed to upregulate ANO1 mRNA expression via mir-381 inhibition. ANO1 which is a Ca2+-activated chloride channel has multiple effects on cellular functions such as proliferation, mucus hypersecretion and fibroblast differentiation in inflamed airways in chronic respiratory diseases. In vitro studies with cadmium at a high concentration range of 100–500 µM is reported to activate chloride channel, ANO1. The secretory epithelial cells are regulated by chloride channels like CFTR, ANO1 and SLC26A9. We examined “ever” smokers with COPD (n = 13) lung tissue sections compared to “never” smoker without COPD (n = 9). We found that “ever” smokers with COPD had higher ANO1 expression. Using mir-381 mimic to inhibit ANO1, we demonstrate here that ANO1 expression is significantly (p < 0.001) downregulated in COPD derived airway epithelial cells exposed to cadmium. Exposure to environmental cadmium contributes significantly to ANO1 expression.
Similar content being viewed by others


Introduction
COPD is associated with airflow limitation as a consequence of the combined effects of emphysema, airway wall thickening, mucus obstruction, and peribronchiolar fibrosis which presents clinically as cough, sputum production, and progressive dyspnea. The severity of these abnormalities varies between patients but a subset is particularly impacted by mucus dysfunction including hypersecretion and impaired mucociliary clearance which can lead to mucus plugging. The mechanisms underlying this mucus dysfunction are complex and incompletely understood though exposure to cigarette smoke (CS), aeroallergens, and particulate and gas pollution can contribute.
Cadmium (Cd) is a toxicant that humans are exposed to through air, food and water. Combustion releases cadmium oxide which can be easily adsorbed on the particulate matter (PM) of < 2.5 including diesel exhaust particles or CS. CS which has approximately 2–3 µg of Cd per cigarette is associated with an increased number of mucus producing cells, impaired mucus clearance and increased mucosal permeability to allergens1. But the mechanisms contributing to this mucosal dysfunction have many facets.
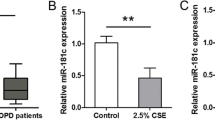
The evolutionarily conserved small non-coding RNAs (miRNAs) have been studied to determine their role in the regulation of protein translation and their contribution to disease pathophysiology2. miRNA or lncRNAs may be positively or negatively regulated by epigenetic regulators like EZH2. Heavy metals are not only known to induce changes in protein expression profiles but also have an effect on microRNA through epigenetic marker regulation3. Prediction of Cd-responsive miRNAs suggested regulation of 214 target genes4. ANO1 is a mechano-sensitive chloride channel, preferentially Ca2+ activated, encoded by the ANO1 gene located on human chromosome 11q13. A 960 amino acid protein with 10 membrane spanning segments it is expressed on the apical region of a variety of epithelial cells such as gastrointestinal, bronchial and pulmonary vessels smooth muscle cells and endothelial cells of arteries. Studies have reported regulation of ANO1 by miR-9, miR-144, miR-181a-2-3p and miR-3815,6,7,8. miR-9 in bronchial epithelial cells negatively regulates ANO1 expression in cystic fibrosis (CF)9. In a study of CF subjects, expression of miR-9 is higher which suppresses the expression of ANO1 by directly binding to the 3ʹ UTR of ANO1 mRNA. This study proposed miR-9 as a potential target for anti-CF therapy where its downregulation increases ANO1 associated chloride efflux to potentiate mucociliary clearance5. Also, knockout of ANO1 was shown to reduce Cl− conductance and inhibit mucus secretion. Interestingly, activation of ANO1 by denufosol induced cough and failed to provide any benefit to CF patients10. In chronic lung diseases such as asthma and CF which have high susceptibly to environmental pollutants/allergens expression of ANO1 in airway epithelial cells is upregulated in the presence of mucus expression inducing interleukins- IL-4, IL-8 and IL-135,11,12. In asthma increased ANO1 is shown to effectively modulate MUC5AC expression and mucus production. However, in COPD expression of ANO1 is not well studied. Since ANO1 supports fluid secretion and airway smooth muscle contraction, inhibition rather than activation is suggested as an appropriate treatment mechanism for inflammatory airway diseases. miR-381 was our target miRNA for this study because its association is closely related to barrier dysfunction and cell proliferation and ANO1 is most recognized for cellular functions such as cell proliferation, attachment and a controversial role in the secretion of mucus5,13,4c).
TGF-β signaling which has been extensively studied and reported to highly expressed and promote airway remodeling in asthma is also suppressed by an increased expression of miR-381. TGF-β promotes goblet cell transformation resulting in increased mucus production and secretion28. Knockdown of miR-381 increases epithelial cell proliferation with increased Ki-67 stained cells and intensity13. In gastric cancer induction of miR-381 expression suppresses differentiation of epithelial cells and reduces cell proliferation29. Our observations from experiments involving low-dose Cd-exposure of epithelial cells suggested a similar understanding that ANO1 is a direct target for miR-381 which is downregulated upon low-dose Cd-exposure (Fig. 4c). Thus, CS-induced Cd-toxicity may alter cellular homeostasis mechanisms at very low concentrations. Thus Cd-exposure in a person with an existing pulmonary condition can have an additive or adverse effect with increased susceptibility towards infections and environmental allergens and that miRNAs may act as potential therapeutic targets to be explored further in Cd-exposure and subsequent lung injury.
Data availability
The datasets generated during the current study are available from the corresponding author on reasonable request.
Abbreviations
- CACCs:
-
Calcium activated chloride channels
- COPD:
-
Chronic obstructive pulmonary disease
- ANO1:
-
Anoctamin 1
- miR:
-
Micro RNA
References
Polosa, R. & Thomson, N. C. Smoking and asthma: Dangerous liaisons. Eur. Respir. J. 41(3), 716–726 (2013).
Bartel, S. et al. Human airway epithelial extracellular vesicle miRNA signature is altered upon asthma development. Allergy 75(2), 346–356 (2020).
Heffler, E. et al. MicroRNA profiling in asthma: Potential biomarkers and therapeutic targets. Am. J. Respir. Cell Mol. Biol. 57(6), 642–650 (2017).
Liu, Q. et al. MicroRNAs–mRNAs expression profile and their potential role in malignant transformation of human bronchial epithelial cells induced by cadmium. BioMed Res. Int. 2015, 902025 (2015).
Huang, F. et al. Calcium-activated chloride channel TMEM16A modulates mucin secretion and airway smooth muscle contraction. Proc. Natl. Acad. Sci. U.S.A. 109(40), 16354–16359 (2012).
Jiang, Y. et al. MicroRNA-144 suppresses aggressive phenotypes of tumor cells by targeting ANO1 in colorectal cancer. Oncol. Rep. 41(4), 2361–2370 (2019).
Kim, J. et al. Role of miRNA-181a-2-3p in cadmium-induced inflammatory responses of human bronchial epithelial cells. J. Thorac. Dis. 11(7), 3055–3069 (2019).
Kukoyi, A. T. et al. MiR-144 mediates Nrf2 inhibition and alveolar epithelial dysfunction in HIV-1 transgenic rats. Am. J. Physiol. Cell Physiol. 317(2), C390–C397 (2019).
Sonneville, F. et al. MicroRNA-9 downregulates the ANO1 chloride channel and contributes to cystic fibrosis lung pathology. Nat. Commun. 8(1), 710 (2017).
Danahay, H. & Gosling, M. TMEM16A: An alternative approach to restoring airway anion secretion in cystic fibrosis? Int. J. Mol. Sci. 21(7), 2386 (2020).
Kunzelmann, K. et al. TMEM16A in cystic fibrosis: Activating or inhibiting? Front. Pharmacol. 10, 3 (2019).
Kondo, M. et al. Chloride ion transport and overexpression of TMEM16A in a guinea-pig asthma model. Clin. Exp. Allergy 47(6), 795–804 (2017).
Benedetto, R. et al. TMEM16A is indispensable for basal mucus secretion in airways and intestine. FASEB J. 33(3), 4502–4512 (2019).
Bai, W., Liu, M. & **ao, Q. The diverse roles of TMEM16A Ca2+-activated Cl− channels in inflammation. J. Adv. Res. 33, 53 (2021).
Tien, J. et al. A comprehensive search for calcium binding sites critical for TMEM16A calcium-activated chloride channel activity. eLife 3, e02772 (2014).
Vogelmeier, C. F. et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive lung disease 2017 report. GOLD executive summary. Am. J. Respir. Crit. Care Med. 195(5), 557–582 (2017).
Cao, X. et al. Tight junction disruption by cadmium in an in vitro human airway tissue model. Respir. Res. 16(1), 30 (2015).
Forti, E. et al. Characterisation of cadmium chloride induced molecular and functional alterations in airway epithelial cells. Cell. Physiol. Biochem. 25(1), 159–168 (2009).
Li, F. J. et al. Citrullinated vimentin mediates development and progression of lung fibrosis. Sci. Transl. Med. 13(585), 2927 (2021).
Hu, X. et al. Environmental cadmium enhances lung injury by respiratory syncytial virus infection. Am. J. Pathol. 189(8), 1513–1525 (2019).
Ghosh, B. et al. Strong correlation between air–liquid interface cultures and in vivo transcriptomics of nasal brush biopsy. Am. J. Physiol. Lung Cell Mol. Physiol. 318(5), 1056–1062 (2020).
Dulin, N. O. Calcium-activated chloride channel ANO1/TMEM16A: Regulation of expression and signaling. Front. Physiol. 11, 1428 (2020).
Le, S. C. & Yang, H. An additional Ca2+ binding site allosterically controls TMEM16A activation. Cell Rep. 33(13), 108570 (2020).
Cerveri, I. et al. The impact of cigarette smoking on asthma: A population-based international cohort study. Int. Arch. Allergy Immunol. 158(2), 175–183 (2012).
Langhammer, A. et al. Cigarette smoking gives more respiratory symptoms among women than among men The Nord–Trøndelag Health Study (HUNT). J. Epidemiol. Community Health 54(12), 917–922 (2000).
Piipari, R. et al. Smoking and asthma in adults. Eur. Respir. J. 24(5), 734–739 (2004).
Hei**k, I. H. et al. Epithelial cell dysfunction, a major driver of asthma development. Allergy 75(8), 1902–1917 (2020).
Halwani, R. et al. Role of transforming growth factor-β in airway remodeling in asthma. Am. J. Respir. Cell Mol. Biol. 44(2), 127–133 (2011).
Cao, Q. et al. MicroRNA-381 inhibits the metastasis of gastric cancer by targeting TMEM16A expression. J. Exp. Clin. Cancer Res. 36(1), 29 (2017).
Funding
The funding was provided by National Institute of Environmental Health Sciences (P42 ES027723).
Author information
Authors and Affiliations
Contributions
P.S. and V.A. drew the hypothesis and experimental plan. P.S., F.J.L., K.D., C.S. and H.Z. performed the experiment. P.S., F.J.L. and V.A. performed data interpretation. P.S. and V.A. wrote the manuscript. P.S., A.K., M.T.D. and V.A. edited and reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Singh, P., Li, F.J., Dsouza, K. et al. Low dose cadmium exposure regulates miR-381–ANO1 interaction in airway epithelial cells. Sci Rep 14, 246 (2024). https://doi.org/10.1038/s41598-023-50471-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-50471-z
- Springer Nature Limited