
Abstract
Sepsis-induced acute respiratory distress syndrome (ARDS) is one of the leading causes of death in critically ill patients, and macrophages play very important roles in the pathogenesis and treatment of sepsis-induced ARDS. The aim of this study was to screen macrophage-related biomarkers for the diagnosis and treatment of sepsis-induced ARDS by bioinformatics and machine learning algorithms. A dataset including gene expression profiles of sepsis-induced ARDS patients and healthy controls was downloaded from the gene expression omnibus database. The limma package was used to screen 325 differentially expressed genes, and enrichment analysis suggested enrichment mainly in immune-related pathways and reactive oxygen metabolism pathways. The level of immune cell infiltration was analysed using the ssGSEA method, and then 506 macrophage-related genes were screened using WGCNA; 48 showed differential expression. PPI analysis was also performed. SVM-RFE and random forest map analysis were used to screen 10 genes. Three key genes, SGK1, DYSF and MSRB1, were obtained after validation with external datasets. ROC curves suggested that all three genes had good diagnostic efficacy. The nomogram model consisting of the three genes also had good diagnostic efficacy. This study provides new targets for the early diagnosis of sepsis-induced ARDS.
Similar content being viewed by others


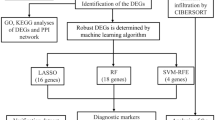
Introduction
Sepsis is defined as life-threatening organ dysfunction caused by a dysregulated host response to infection1. Despite significant advances in treatment, sepsis affects approximately 19–48.9 million people worldwide each year and remains one of the leading causes of death in critically ill patients worldwide2. Sepsis causes various complications, such as lung injury, liver injury, kidney injury, myocardial injury and brain injury3,4,5. Acute respiratory distress syndrome (ARDS) is a common complication in patients with sepsis and is characterized by diffuse alveolar injury; patients present with clinical symptoms such as acute respiratory distress, hypoxemia and pulmonary oedema6. The mortality rate of sepsis-induced ARDS is 30–40%, which is higher than that of other types of ARDS7,8. Moreover, when patients progress to severe ARDS, the mortality rate increases to more than 40%5E).

Random forest plot and SVM analysis. (A)–(B). SVM-RFE analysis plot; the horizontal axis represents the number of feature genes. The best fivefold cross-validation precision and error are 0.842 and 0.158, respectively. (Software: R (4.0.2) version, R packet: e1071 (1.7–13). URL: https://cran.rstudio.com/web/packages/e1071/index.html). (C) Random forest tree. (D) The precision and Gini coefficient of random forest plot analysis of macrophage-related differentially expressed genes to determine gene importance. ((C, D) Software: R (4.0.2) version, R packet: randomForestSRC (3.2.1). URL: https://www.randomforestsrc.org/https://ishwaran.org/). (E) SVM-RFE method and random forest plot method to screen genes in the intersection of the Venn diagram.
Differential expression validation in the validation dataset
The differential expression analysis of the genes obtained after random forest plot and SVM analyses in the GSE154918, GSE28750, and GSE185263 datasets showed that, in GSE154918, the expression of all 10 genes was significantly different (P < 0.05) (Fig. 6A). In the GSE28750 dataset, the expression levels of SGK1, ANPEP, DYSF and MSRB1 were significantly different between groups (P < 0.05) (Fig. 6B). In the GSE185263 dataset, the expression levels of SGK1, MYD88, DYSF, PLEKHO2, CYTH4 and MSRB1 were significantly different between groups (P < 0.05) (Fig. 6C). Only SGK1, DYSF and MSRB1 showed differential expression in all three validation sets.

Gene expression validation. (A) Gene expression validation in the GSE154918 dataset. (B) Gene expression validation in the GSE28750 dataset. (C) Gene expression validation in the GSE185263 dataset. (Software: R (4.0.2) version, R packet: ggplot2 (3.4.1). URL: https://ggplot2.tidyverse.org/). (D)–(M). ROC analysis of the value of these genes in diabetes mellitus diagnosis. (Software: R (4.0.2) version, R packet: pROC (1.18.0). URL: http://expasy.org/tools/pROC/).
ROC curve analysis of the diagnostic value of marker genes
To further understand the diagnostic value of marker genes in ARDS, ROC curve analysis was performed using samples from the control and sepsis-induced ARDS groups in the GSE32707 dataset as study samples. The results suggested that all 10 marker genes had a good predictive effect in terms of the diagnosis of ARDS, with an area under the curve greater than 0.65; SGK1 (AUC = 0.791) had the best diagnostic effect, followed by LST1 (AUC = 0.743), MSRB1 (AUC = 0.740) and DYSF (AUC = 0.721) (Fig. 6D–M). All three validated differentially expressed genes showed good diagnostic value for ARDS.
Construction of the nomogram
Based on the validation results of external datasets and ROC analysis, SGK1, DYSF and MSRB1 were used as the basic genes for subsequent analysis. To further understand the diagnostic value of SGK1, DYSF and MSRB1 in ARDS, nomogram analysis was performed, and a model was constructed. The calibration curves suggested that the nomogram model curves had high overlap with the ideal model, indicating that the nomogram model composed of three genes, SGK1, DYSF and MSRB1, showed good diagnostic prediction for ARDS. Moreover, the area under the curve (AUC) was 0.809, demonstrating that the prediction effect of the nomogram model was better than that of each of the three genes (SGK1, DYSF and MSRB1) alone (Fig. 7A,B).

Nomogram and cluster analysis. (A) Nomogram plot. (B) Nomogram model prediction effect plot. (Software: R (4.0.2) version, R packet: rms (6.5–0). URL: https://hbiostat.org/R/rms/, https://github.com/harrelfe/rms). (C) Matrix heatmap of key gene clustering analysis. (D) Cluster analysis delta area plot. (E) PCA of different clusters after cluster analysis. (Software: R (4.0.2) version, R packet: ConsensusClusterPlus [1.62.0]. URL: https://bioconductor.org/packages/ConsensusClusterPlus/;). (F) Cluster analysis subtype grou** and sample consistency of sample diagnostic grou** Sankey plot. (G) Analysis of the expression differences of key genes in different clusters.
Clustering analysis
To understand the effect of SGK1, DYSF and MSRB1 on sample clustering, cluster analysis was performed. The results suggested that the samples could be divided into 2 clusters according to the expression values of SGK1, DYSF and MSRB1 (Fig. 7C,D). PCA showed that the cluster analysis had a good clustering effect (Fig. 7E). Sankey plots showed good agreement between cluster and disease grou** for cluster analysis, with Cluster 1 having a high proportion of samples in the control group and Cluster 2 having a high proportion of samples in the ARDS group (Fig. 7F). Wilcox analysis suggested that SGK1, DYSF and MSRB1 were differentially expressed in Cluster 1 and Cluster 2 (Fig. 7G) (P < 0.05), and the trend of difference was consistent with the trend of difference between the control and ARDS groups. These results further demonstrate the diagnostic value of SGK1, DYSF and MSRB1 for ARDS.
Transcription factors and miRNA prediction of SGK1, DYSF and MSRB1
Based on the prediction results in the JASPAR, HumanTFDB, and GTRD databases, a total of 173 transcription factors of SGK1 (Fig. 8A), 140 transcription factors of MSRB1 (Fig. 8B), and 172 transcription factors of DYSF (Fig. 8C,D) were obtained. Based on the prediction results in six databases, miRWalk, RNA22, RNAInter, TargetMiner, TargetScan and miRDB, a total of 28 miRNAs for SGK1 were obtained (see Supplementary Fig. S3A online). miRNAs for DYSF and MSRB1 were not predicted in the TargetMiner database, and the prediction results in the remaining five databases suggested that a total of 12 miRNAs of MSRB1 (see Supplementary Fig. S3B online) and 21 miRNAs of DYSF (see Supplementary Fig. S3C,D online) were obtained.

Transcription factor prediction. (A) Transcription factors of DYSF were predicted by three databases, as shown in Venn diagram. (B) Transcription factors of MSRB1 were predicted by three databases, as shown in Venn diagram. (C) Transcription factors of DYSF were predicted by three databases, as shown in Venn diagram. (Software: R (4.0.2) version, R packet: VennDiagram (1.7.3). URL: https://cran.rstudio.com/web/packages/VennDiagram/index.html). (D) Interaction of SGK1, MSRB1 and DYSF with predicted transcription factors.
Discussion
Sepsis affects more than 30 million people worldwide each year and is one of the leading causes of death in critically ill patients22. Sepsis-induced ARDS is a type of acute progressive respiratory failure caused by sepsis, and its pathogenesis is mostly focused on the inflammatory response, oxidative stress, and abnormal coagulation23. As innate immune cells, macrophages play a key role in the inflammatory response. It was found that secretory autophagy of alveolar macrophages (AMs) promotes the inflammatory response and lung injury through secreted IL-1β24. Impaired phagocytosis of alveolar macrophages in advanced sepsis further increases the severity of ARDS, and IFN-β treatment reverses the impairment of AM function induced by IL-10 and reduces the severity and mortality of ARDS in a dose-dependent manner25.
The results of the enrichment analysis in this study suggest significant enrichment of multiple pathways. Immune cells play a substantial role in the development and treatment of ARDS. Neutrophils play a major role in the process of leukocyte migration. Neutrophils are the first immune cells to migrate to the site of inflammation after stimulation by chemokines released from damaged lung tissue, and massive activation of neutrophils leads to peripheral tissue damage and lung dysfunction26,27. Eliav et al.28 found that the leukocyte migration inhibitor GT-73 significantly reduced the number of infiltrating leukocytes in LPS-induced ARDS in mice, decreased the levels of cytokines and was protective against ARDS. The reactive oxygen species-related pathway was also significantly enriched, and in ARDS, the production of reactive oxygen species (ROS) is detrimental to lung tissue, and ROS can damage lung endothelial cells and lead to impaired alveolar-capillary barrier function29. NF-κB is an important transcription factor that controls the release of proinflammatory mediators, and ROS can also cause activation of NF-κB, which can exacerbate inflammatory lung injury30. Studies have shown that suppressing the production of ROS is beneficial in reducing inflammatory lung injury31.
GSVA suggests significant differences in cytokine/cytokine receptor and Toll-like receptor pathway enrichment levels. In ARDS, multiple receptors of innate immune cells (macrophages, dendritic cells or monocytes), such as Toll-like receptors, recognize PAMPs and DAMPs to induce cytokine release syndrome32. It has been shown that the inflammatory response to LPS-induced ARDS can be attenuated by inhibiting TLR4 expression33. AM is a major source of cytokines and chemokines that initiate the immune response, and the overproduction of these proinflammatory cytokines (IL-1β, IL-6, TNF-α, IL-8, etc.) leads to the development of acute lung injury34. Additionally, AM is a major source of anti-inflammatory cytokines, such as IL-10 and TGF-β, which can suppress the inflammatory response to acute lung injury.
The three key genes screened in this study have been partially studied in lung injury and other diseases. SGK1 is a member of the protein kinase subfamily, a serine/threonine protein kinase with high homology to second messengers such as protein kinase B. As a hub for multiple signal transduction pathways and cellular phosphorylation events, SGK1 plays an important role in cell proliferation, ion channel regulation, signal transduction and other physiological processes and is thought to have an essential function in inflammation35. Studies have shown that SGK1 enhances the function of sodium channels to promote clearance of alveolar oedema fluid in a mouse model of lung injury36,37. Michalick et al. found that SGK1 may provide promising new targets for the prevention or treatment of ventilator-associated lung injury38. ** of cluster analysis with sample grou**. Wilcox test was used to analyse the differences in gene expression between different clustered grou**s.
Transcription factors and miRNA prediction of key genes
The promoter sequences of the core genes (including the transcription start site up to 2000 bp upstream of it) were obtained from the NCBI database (https://www.ncbi.nlm.nih.gov/gene/). After that, transcription factor prediction was performed in the JASPAR database (https://jaspar.genereg.net/), HumanTFDB database (http://bioinfo.life.hust.edu.cn/HumanTFDB#!/) and GTRD database (http://gtrd.biouml.org/#!) based on the obtained promoter sequences. The transcription factors obtained from the three databases were intersected to obtain transcription factors that existed in all three databases at the same time and then visualized using Cytoscape software. miRNAs of key genes were predicted in the miRWalk, RNA22, RNAInter, TargetMiner, TargetScan and miRDB databases, and then the miRNAs in each database were overlapped to identify the intersecting miRNAs; the Venn diagram was visualized using Cytoscape software.
Data availability
The datasets analysed during the current research are all available in the GEO database (GSE32707, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE32707; GSE154918, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE154918; GSE28750, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE28750; GSE185263,https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE185263) and MsigDB (http://www.gsea-msigdb.org/gsea/index.jsp).
References
David, S. & Brunkhorst, F. Sepsis-3: What has been confirmed in therapy?. Der Internist 58, 1264–1271. https://doi.org/10.1007/s00108-017-0338-5 (2017).
Chiu, C. & Legrand, M. Epidemiology of sepsis and septic shock. Curr. Opin. Anaesthesiol. 34, 71–76. https://doi.org/10.1097/aco.0000000000000958 (2021).
L’Heureux, M., Sternberg, M., Brath, L., Turlington, J. & Kashiouris, M. Sepsis-induced cardiomyopathy: A comprehensive review. Curr. Cardiol. Rep. 22, 35. https://doi.org/10.1007/s11886-020-01277-2 (2020).
Ren, C., Yao, R., Zhang, H., Feng, Y. & Yao, Y. Sepsis-associated encephalopathy: A vicious cycle of immunosuppression. J. Neuroinflamm. 17, 14. https://doi.org/10.1186/s12974-020-1701-3 (2020).
Petejova, N. et al. Acute kidney injury in septic patients treated by selected nephrotoxic antibiotic agents-pathophysiology and biomarkers-a review. Int. J. Mol. Sci. https://doi.org/10.3390/ijms21197115 (2020).
Matthay, M. et al. Acute respiratory distress syndrome. Nat. Rev. Dis. Primers. 5, 18. https://doi.org/10.1038/s41572-019-0069-0 (2019).
Englert, J., Bobba, C. & Baron, R. Integrating molecular pathogenesis and clinical translation in sepsis-induced acute respiratory distress syndrome. JCI Insight https://doi.org/10.1172/jci.insight.124061 (2019).
Bellani, G. et al. Epidemiology, patterns of care, and mortality for patients with acute respiratory distress syndrome in intensive care units in 50 countries. JAMA 315, 788–800. https://doi.org/10.1001/jama.2016.0291 (2016).
Pan, C., Liu, L., **e, J. & Qiu, H. Acute respiratory distress syndrome: Challenge for diagnosis and therapy. Chin. Med. J. 131, 1220–1224. https://doi.org/10.4103/0366-6999.228765 (2018).
Kaku, S. et al. Acute respiratory distress syndrome: Etiology, pathogenesis, and summary on management. J. Intensive Care Med. 35, 723–737. https://doi.org/10.1177/0885066619855021 (2020).
Kumar, V. Pulmonary innate immune response determines the outcome of inflammation during pneumonia and sepsis-associated acute lung injury. Front. Immunol. 11, 1722. https://doi.org/10.3389/fimmu.2020.01722 (2020).
Dang, W. et al. The role of lung macrophages in acute respiratory distress syndrome. Inflamm Res. Off. J. Eur. Histamine Res. Soc. 71, 1417–1432. https://doi.org/10.1007/s00011-022-01645-4 (2022).
Locati, M., Curtale, G. & Mantovani, A. Diversity, mechanisms, and significance of macrophage plasticity. Annu. Rev. Pathol. 15, 123–147. https://doi.org/10.1146/annurev-pathmechdis-012418-012718 (2020).
Chen, X. et al. Macrophage polarization and its role in the pathogenesis of acute lung injury/acute respiratory distress syndrome. Inflamm. Res. Off. J. Eur. Histamine Res. Soc. 69, 883–895. https://doi.org/10.1007/s00011-020-01378-2 (2020).
Shapouri-Moghaddam, A. et al. Macrophage plasticity, polarization, and function in health and disease. J. Cell. Physiol. 233, 6425–6440. https://doi.org/10.1002/jcp.26429 (2018).
Reilly, J., Christie, J. & Meyer, N. Fifty years of research in ARDS. Genomic contributions and opportunities. Am. J. Respir. Crit. Care Med. 196, 1113–1121. https://doi.org/10.1164/rccm.201702-0405CP (2017).
Sun, M., Yang, Q., Hu, C., Zhang, H. & **ng, L. Identification and validation of autophagy-related genes in sepsis-induced acute respiratory distress syndrome and immune infiltration. J. Inflamm. Res. 15, 2199–2212. https://doi.org/10.2147/jir.S355225 (2022).
Giassa, I. & Alexiou, P. Bioinformatics and machine learning approaches to understand the regulation of mobile genetic elements. Biology https://doi.org/10.3390/biology10090896 (2021).
Huang, S. et al. Applications of support vector machine (SVM) learning in cancer genomics. Cancer Genom. Proteomics 15, 41–51. https://doi.org/10.21873/cgp.20063 (2018).
Libbrecht, M. & Noble, W. Machine learning applications in genetics and genomics. Nat. Rev. Genet. 16, 321–332. https://doi.org/10.1038/nrg3920 (2015).
Deo, R. Machine learning in medicine. Circulation 132, 1920–1930. https://doi.org/10.1161/circulationaha.115.001593 (2015).
Huang, M., Cai, S. & Su, J. The pathogenesis of sepsis and potential therapeutic targets. Int. J. Mol. Sci. 20, 1. https://doi.org/10.3390/ijms20215376 (2019).
Huppert, L., Matthay, M. & Ware, L. Pathogenesis of acute respiratory distress syndrome. Semin. Respir. Crit. Care Med. 40, 31–39. https://doi.org/10.1055/s-0039-1683996 (2019).
Xu, X. et al. Secretory autophagosomes from alveolar macrophages exacerbate acute respiratory distress syndrome by releasing IL-1β. J. Inflamm. Res. 15, 127–140. https://doi.org/10.2147/jir.S344857 (2022).
Hiruma, T. et al. IFN-β improves sepsis-related alveolar macrophage dysfunction and postseptic acute respiratory distress syndrome-related mortality. Am. J. Respir. Cell Mol. Biol. 59, 45–55. https://doi.org/10.1165/rcmb.2017-0261OC (2018).
Aulakh, G. Neutrophils in the lung: “The first responders”. Cell Tissue Res. 371, 577–588. https://doi.org/10.1007/s00441-017-2748-z (2018).
Yang, S., Tsai, Y., Pan, Y. & Hwang, T. Understanding the role of neutrophils in acute respiratory distress syndrome. Biomed. J. 44, 439–446. https://doi.org/10.1016/j.bj.2020.09.001 (2021).
Qu, M. et al. Neutrophil extracellular traps-triggered impaired autophagic flux via METTL3 underlies sepsis-associated acute lung injury. Cell Death Discov. 8, 375. https://doi.org/10.1038/s41420-022-01166-3 (2022).
Liu, X. et al. Neferine protects endothelial glycocalyx via mitochondrial ROS in lipopolysaccharide-induced acute respiratory distress syndrome. Front. Physiol. 9, 102. https://doi.org/10.3389/fphys.2018.00102 (2018).
Jiang, K. et al. Barbaloin protects against lipopolysaccharide (LPS)-induced acute lung injury by inhibiting the ROS-mediated PI3K/AKT/NF-κB pathway. Int. Immunopharmacol. 64, 140–150. https://doi.org/10.1016/j.intimp.2018.08.023 (2018).
Wagner, J. et al. Sevoflurane posttreatment prevents oxidative and inflammatory injury in ventilator-induced lung injury. PLoS ONE 13, e0192896. https://doi.org/10.1371/journal.pone.0192896 (2018).
Root-Bernstein, R. Innate receptor activation patterns involving TLR and NLR synergisms in COVID-19, ALI/ARDS and sepsis cytokine storms: A review and model making novel predictions and therapeutic suggestions. Int. J. Mol. Sci.. https://doi.org/10.3390/ijms22042108 (2021).
Tang, S. et al. Pre-treatment with ten-minute carbon dioxide inhalation prevents lipopolysaccharide-induced lung injury in mice via down-regulation of toll-like receptor 4 expression. Int. J. Mol. Sci. https://doi.org/10.3390/ijms20246293 (2019).
Bissonnette, E., Lauzon-Joset, J., Debley, J. & Ziegler, S. Cross-talk between alveolar macrophages and lung epithelial cells is essential to maintain lung homeostasis. Front. Immunol. 11, 583042. https://doi.org/10.3389/fimmu.2020.583042 (2020).
Lu, R. et al. SGK1, a critical regulator of immune modulation and fibrosis and a potential therapeutic target in chronic graft-versus-host disease. Front. Immunol. 13, 822303. https://doi.org/10.3389/fimmu.2022.822303 (2022).
Fei, X. et al. Aldosterone alleviates lipopolysaccharide-induced acute lung injury by regulating epithelial sodium channel through PI3K/Akt/SGK1 signaling pathway. Mol. Cell. Probes 57, 101709. https://doi.org/10.1016/j.mcp.2021.101709 (2021).
Li, J. et al. Melatonin attenuates sepsis-induced acute lung injury through improvement of epithelial sodium channel-mediated alveolar fluid clearance via activation of SIRT1/SGK1/Nedd4-2 signaling pathway. Front. Pharmacol. 11, 590652. https://doi.org/10.3389/fphar.2020.590652 (2020).
Michalick, L. et al. Transient receptor potential vanilloid 4 and serum glucocorticoid-regulated kinase 1 are critical mediators of lung injury in overventilated mice in vivo. Anesthesiology 126, 300–311. https://doi.org/10.1097/aln.0000000000001443 (2017).
**, X. et al. SGK1 mediates hypoxic pulmonary hypertension through promoting macrophage infiltration and activation. Anal. Cell. Pathol. (Amst.) 3013765, 2019. https://doi.org/10.1155/2019/3013765 (2019).
White, Z., Milad, N., Sellers, S. & Bernatchez, P. Effect of dysferlin deficiency on atherosclerosis and plasma lipoprotein composition under normal and hyperlipidemic conditions. Front. Physiol. 12, 675322. https://doi.org/10.3389/fphys.2021.675322 (2021).
Zhang, X. et al. DYSF promotes monocyte activation in atherosclerotic cardiovascular disease as a DNA methylation-driven gene. Transl. Res. J. Lab. Clin. Med. 247, 19–38. https://doi.org/10.1016/j.trsl.2022.04.001 (2022).
Tarrago, L. et al. The selenoprotein methionine sulfoxide reductase B1 (MSRB1). Free Radical Biol. Med. 191, 228–240. https://doi.org/10.1016/j.freeradbiomed.2022.08.043 (2022).
Lee, B. et al. Selenoprotein MsrB1 promotes anti-inflammatory cytokine gene expression in macrophages and controls immune response in vivo. Sci. Rep. 7, 5119. https://doi.org/10.1038/s41598-017-05230-2 (2017).
Lee, H. et al. The selenoprotein MsrB1 instructs dendritic cells to induce T-Helper 1 immune responses. Antioxidants (Basel Switz.) https://doi.org/10.3390/antiox9101021 (2020).
Ritchie, M. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. https://doi.org/10.1093/nar/gkv007 (2015).
Yu, G., Wang, L., Han, Y. & He, Q. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 16, 284–287. https://doi.org/10.1089/omi.2011.0118 (2012).
Hänzelmann, S., Castelo, R. & Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 14, 7. https://doi.org/10.1186/1471-2105-14-7 (2013).
Langfelder, P. & Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 9, 559. https://doi.org/10.1186/1471-2105-9-559 (2008).
Szklarczyk, D. et al. The STRING database in 2017: Quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 45, D362–D368. https://doi.org/10.1093/nar/gkw937 (2017).
Taylor, J. Random survival forests. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 6, 1974–1975. https://doi.org/10.1097/JTO.0b013e318233d835 (2011).
Liu, Q. et al. Gene selection and classification for cancer microarray data based on machine learning and similarity measures. BMC Genom. https://doi.org/10.1186/1471-2164-12-s5-s1 (2011).
Robin, X. et al. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 12, 77. https://doi.org/10.1186/1471-2105-12-77 (2011).
Wilkerson, M. & Hayes, D. ConsensusClusterPlus: A class discovery tool with confidence assessments and item tracking. Bioinformatics (Oxf. Engl.) 26, 1572–1573. https://doi.org/10.1093/bioinformatics/btq170 (2010).
Acknowledgements
We appreciate the works by the Gene Expression Omnibus (GEO) datasets for the source of data used in our study.
Author information
Authors and Affiliations
Contributions
B.C. conceived and designed the whole project and revised and proofread the manuscript. Q.L. and H.Z. analysed the data and edited and wrote the manuscript. All authors contributed to manuscript revision and read and approved the submitted version. Q.L. and H.Z. contributed equally to this work.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, Q., Zheng, H. & Chen, B. Identification of macrophage-related genes in sepsis-induced ARDS using bioinformatics and machine learning. Sci Rep 13, 9876 (2023). https://doi.org/10.1038/s41598-023-37162-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-37162-5
- Springer Nature Limited