
Abstract
Populus wulianensis is an endangered species endemic to Shandong Province, China. Despite the economic and ornamental value of this species, few genomics and genetic studies have been performed. In this study, we performed a relevant analysis of the full-length transcriptome sequencing data of P. wulianensis and obtained expressed sequence tag (EST)-simple sequence repeat (SSR) markers with polymorphisms that can be used for further genetic research. In total, 8.18 Gb (3,521,665) clean reads with an average GC content of 42.12% were obtained. From the corrected 64,737 high-quality isoforms, 42,323 transcript sequences were obtained after redundancy analysis with CD-HIT. Among these transcript sequences, 41,876 sequences were annotated successfully. A total of 23,539 potential EST-SSRs were identified from 16,057 sequences. Excluding mononucleotides, the most abundant motifs were trinucleotide SSRs (47.80%), followed by di- (46.80%), tetra- (2.98%), hexa- (1.58%) and pentanucleotide SSRs (0.84%). Among the 100 designed EST-SSRs, 18 were polymorphic with high PIC values (0.721 and 0.683) and could be used for analyses of the genetic diversity and population structure of P. wulianensis. These full-length transcriptome sequencing data will facilitate gene discovery and functional genomics research in P. wulianensis, and the novel EST-SSRs developed in our study will promote molecular-assisted breeding, genetic diversity and conservation biology research in this species.
Similar content being viewed by others


Introduction
Populus wulianensis is an endangered species endemic to Shandong Province, China, with high economic and ornamental value1,2, and similar to other species of the genus Populus, it is an important source of timber. The effective number of wild survivors of P. wulianensis is less than 1000, and thus, this species should be considered among China’s plant species with extremely small populations (PSESP)3. There are different views on the taxonomic status of P. wulianensis. Liang and Li reported P. wulianensis as an independent species in 1986 and thought that it might have originated from the cross between Populus adenopoda and Populus davidiana4. However, Zhang et al. suggested that P. wulianensis is an intraspecific variation of P. adenopoda5. At present, taxonomists generally recognize the taxonomic independence of the species1,2,6,7. Very few studies have investigated this species, and the previous studies have only focused on its taxonomy, the establishment of regeneration systems and the control of vitrification in test-tube seedlings5,8. As a result, few studies have provided molecular data for this endangered species.
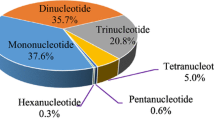
In recent years, a large number of molecular markers, such as amplified fragment length polymorphisms (AFLPs), random amplified polymorphic DNA (RAPD), single primer amplification reaction (SRAP), and simple sequence repeats (SSRs), have been developed and applied in the fields of genomic map**, molecular-assisted breeding, DNA fingerprinting, genetic diversity and population structure analysis, and conservation biology9,10,11,12. Among these molecular markers, SSRs are more reliable and widely used in the above-mentioned research fields due to their abundance in the genome, high polymorphism rate, high information content and codominance13,14. SSR markers are divided into two types, namely, expressed sequence tag (EST)-SSRs and genomic SSRs (gSSRs), which originate from RNA transcriptome sequencing and arbitrary genome sequencing, respectively. Compared with than gSSRs, EST-SSRs are functional molecular markers with the advantages of easier and more efficient development, lower cost and more interspecific transferability15,16, The transcriptome is an essential tool for understanding life processes. In this study, the full-length transcriptome sequence of P. wulianensis was obtained based on SMRT sequencing. In total, 8.18 Gb (3,521,665) of clean reads with an average GC content of 42.12% were obtained (Table 1). The mean clean read length was 2177 (Table 1). We screened a total of 289,128 reads of insert (ROIs) from the original sequence data (Table 1). The mean quality of the ROIs in each library was greater than 0.9 (Table 1). As shown in Table 1, increases in the size of the inserted fragment were associated with gradual decreases in the percentage of full-length sequences to ROI sequences (FLP). The Iso-Seq module of the SMRT Analysis software was used to perform a cluster analysis of the above-mentioned full-length sequences. A total of 87,004 consensus isoforms with an average length of 17,642 were obtained (Table 2). Combined with the non-full-length sequences, the quiver program was used to correct the consensus isoforms in each cluster, and 64,737 corrected high-quality isoforms with an accuracy higher than 99% were obtained (Table 2). As shown in Table 2, the highest percentage of polished high-quality isoforms (84.61%) was obtained with a sequence length of 0–1 kb. In contrast, if the sequence size was higher than 6 kb, the percentage of polished high-quality isoforms was only 5.52%. From the corrected 64,737 high-quality isoforms, 42,323 transcript sequences suitable for further analysis were obtained through redundancy analysis with CD-HIT (Supplementary Table S2). The obtained 42,323 nonredundant transcript sequences were aligned to the following databases using BLAST software (version 2.2.26) (Supplementary Table S2): RefSeq nonredundant proteins (NR), Swiss-Prot Protein Sequence (Swiss-Prot), Gene Ontology (GO), Cluster of Orthologous Groups of proteins (COG), Clusters of orthologous groups for eukaryotic complete genomes (KOG), evolutionary genealogy of genes: Nonsupervised Orthologous Groups (eggNOG), Pfam protein families (Pfam), and Kyoto Encyclopedia of Genes and Genomes (KEGG). A total of 41,876 nonredundant transcript sequences were favorably annotated (Supplementary Table S2). In total, 41,851 transcript sequences were aligned with the NR database (Supplementary Table S2), and among these, 35,070 (83.80%) transcript sequences showed higher than 90% similarity (Supplementary Fig. S1), whereas only 0.45% of the transcript sequences showed less than 50% similarity (Supplementary Fig. S1). Sequence alignment was used to identify homologous species. As shown in Supplementary Fig. S2, 22,776 (54.42%) transcript sequences were annotated to Populus trichocarpa, whereas 14,486 (34.61%) transcript sequences were annotated to Populus euphratica. Notably, only 4.03% of the transcript sequences were annotated to Populus tomentosa, and 6.03% of the transcript sequences were annotated to plants outside the genus Populus. The GO database provides a set of dynamically updated standard vocabularies to comprehensively describe the functional attributes of genes and gene products in organisms. In our study, a total of 35,580 transcript sequences were annotated in the GO database and assigned to 51 subcategories within the cellular component (84,429, 41.38%), molecular function (45,154, 21.13%) and biological process (80,133, 37.50%) categories in the GO database (Supplementary Table S2, Fig. 1). In the cellular component category, cell (18,706, 8.75%) constituted the largest group of transcript sequences, followed by cell part (18,613, 8.71%), membrane (13,424, 6.28%), and organelle (12,611, 5.90%). Only 40 (0.02%) transcript sequences were assigned to nucleoids (Supplementary Table S2, Fig. 1). Similarly, in the molecular function ontology, transcript sequences involved in catalytic activity (19,420, 9.09%) formed the largest group, followed by binding (19,135, 8.95%) and transporter activity (2,490, 1.17%) (Supplementary Table S2, Fig. 1). Fewer than 10 transcript sequences were found to be involved in metallochaperone activity (4), protein tag (3), and translation regulator activity (2) (Supplementary Table S2, Fig. 1). In the biological process category, the largest group of transcript sequences was assigned to metabolic processes (18,826, 8.81%), followed by cellular processes (18,132, 8.48%) and single-organism processes (13,626, 6.38%) (Supplementary Table S2, Fig. 1). In contrast, subclasses such as developmental process (1717, 0.80%) and multicellular organismal process (1566, 0.73%) in the biological process category were assigned to less than 1% of the total transcript sequences (Supplementary Table S2, Fig. 1). GO annotations of P. wulianensis transcript sequences. The KOG, COG and eggNOG databases are all databases for orthologous gene function annotation. The KOG database is based on orthologous relationships among genes and uses evolutionary relationships to divide homologous genes from different species into different ortholog clusters. The COG database is an earlier database that was used to identify orthologous genes and to classify gene products by homology. The eggNOG database is mainly used for functional description annotation and the functional classification of orthologous groups. In our study, a total of 29,851, 20,657, and 41,710 unique transcript sequences were assigned to 25 KOG categories, 26 COG categories, and 25 eggNOG categories, respectively (Supplementary Table S2). Among the 25 KOG categories, the greatest number of transcript sequences was assigned to general function prediction only (4743, 15.89%), followed by posttranslational modification, protein turnover, chaperones (3293, 11.03%) and signal transduction mechanisms (3121, 10.64%) (Supplementary Table S2, Fig. 2). Extracellular structures and cell motility formed the two smallest groups, with 96 (0.32%) and 10 (0.03%) transcript sequences, respectively (Supplementary Table S2, Fig. 2). KOG functional classification of P. wulianensis transcript sequences. Among the 26 COG categories, approximately 30% of the transcript sequences were annotated as general function prediction only (2166, 10.49%), signal transduction mechanisms (2097, 10.15%), and carbohydrate transport and metabolism (2079, 10.06%) (Supplementary Table S2, Supplementary Fig. S3). The results showed that no transcript sequences were annotated as nuclear structure (Supplementary Table S2, Supplementary Fig. S3). Unlike the KOG and COG categories, no transcript sequence was annotated to general function prediction only when using the eggNOG categories (Supplementary Table S2, Supplementary Fig. S4). The largest number of transcript sequences were annotated to function unknown (18,793, 45.06%), followed by posttranslational modification, protein turnover, chaperones (3280, 7.86%) and transcription (2889, 6.93%) (Supplementary Table S2, Supplementary Fig. S4). With the exception of unknown function, no class contained more than 10% of the transcripts (Supplementary Table S2, Supplementary Fig. S4). The pathway annotation analysis of expressed genes helps increase the understanding of gene functions, and KEGG is a powerful tool for in vivo metabolic analysis and metabolic network research. In this study, a total of 19,686 transcript sequences were associated with 128 KEGG pathways in six categories, namely, cellular processes, environmental information processing, genetic information processing, human diseases, metabolism and organismal systems (Supplementary Table S2, Fig. 3). Among the six categories, the category involving the most unigenes was metabolism (13,075, 66.4%) with 96 KEGG pathways, followed by genetic information processing (4230, 21.49%) with 21 KEGG pathways (Supplementary Table S2, Fig. 3). The remaining four categories included only 2381 unigenes, approximately 12% of the total (Supplementary Table S2, Fig. 3). KEGG metabolic categories in the P. wulianensis transcriptome. Transcripts longer than 500 bp were screened using MIcroSAtellite identification tool (MISA). Among the 42,311 evaluated sequences, we identified 16,057 transcript sequences that contained EST-SSRs. As shown in Table 3, a total of 23,539 SSRs (including 12,520 mononucleotide repeats) were identified from the abovementioned 16,057 SSR-containing sequences. One SSR site was found per 2.64 kb (23,539 SSR loci within 89,101,859 bp). The identification results showed that a total of 5,050 transcript sequences contained more than one EST-SSR locus (Table 3), and a total of 2506 SSRs were present in compound form. In this study, the highest number of identified SSRs (excluding mononucleotide repeats) were trinucleotide repeats (5267, 47.80%), followed by dinucleotide repeats (5157, 46.80%) and tetranucleotide repeats (328, 2.98%) (Table 3). The rarest type of EST-SSR was the pentanucleotide type (93, 0.84%) and not the hexanucleotide type (174, 1.58%) (Table 3). The SSR density results showed that the repeat type with the highest distribution density was trinucleotide (excluding mononucleotide repeats and compound SSRs), with an average of 47.34 SSR loci per Mb, followed by dinucleotide, tetranucleotide, hexanucleotide, and pentanucleotide repeats, with averages of 44.01, 2.93, 1.66, and 0.83 SSR loci per Mb, respectively (Table 3, Supplementary Fig. S5). Among all the nucleotide repeats (excluding mononucleotide repeats), hexanucleotide repeats (74, 56.92%) included the most repeat types, and the main repeat types included AACAGC/CTGTTG (12, 0.11%), AAAAAC/ATTTGT (8, 0.07%), and ACCGCC/CGGTGG (7, 0.06%) (Table 4). The tetranucleotide repeats (23, 17.69%) and pentanucleotide repeats (19, 14.62%) also included multiple repeat types (Table 4). Although the four types of dinucleotide repeats, AG/CT (3788, 34.38%), AT/AT (774, 7.02%), AC/GT (570, 5.17%), and CG/CG (25, 0.23%), only accounted for 3.08% of all repeat types, the proportion of SSRs that were dinucleotide sequences was as high as 46.80% (Tables 3 and 4). Among all SSR repeat types (excluding mononucleotide repeats), the highest number was obtained for AG/CT dinucleotide repeats, accounting for 34.38% of all repeat motifs, followed by AAG/CTT (1138, 10.33%) and AGC/CTG (1078, 9.78%) (Table 4). Among all SSR tandem repeats (excluding mononucleotide repeats), the most common tandem repeat number was 6 (3055, 27.72%), followed by 5 (2980, 27.04%) and 7 (1622, 14.72%) (Supplementary Table S4). The number of loci with at least 16 tandem repeats was 197, accounting for 1.79% of all SSR tandem repeats (Supplementary Table S4). The development of primers constitutes the basis for further research on the genetic structure and diversity of species. Twelve samples, including ten P. wulianensis samples, one P. adenopoda sample and one P. davidiana sample, were subjected to PCR amplification using 100 pairs of newly developed EST-SSRs. Among the 100 EST-SSRs, 12 failed to generate a product, and the other 88 primer pairs successfully resulted in amplification (Supplementary Table S5). Of these 88 primer pairs, 31 exhibited poor universal applicability, eight produced multiple bands, and 12 were monomorphic (Supplementary Table S5). Of the remaining 37 primer pairs capable of generating polymorphic amplification products, 19 primer pairs generated unstable and unclear amplification, and the remaining 18 produced stable and clear amplification products (Supplementary Table S5). Details of these 18 primer pairs can be found in Supplementary Table S6. To further verify the polymorphism of these 18 primer pairs, we performed amplification experiments using 30 samples from six populations, including 24 P. wulianensis samples, three P. adenopoda samples and three P. davidiana samples, and a more accurate high-performance capillary electrophoresis method. The amplification results obtained for all the samples showed that a total of 150 alleles were observed, and the number of alleles (NA) ranged from 5 to 15 per locus, with an average allele number of 8.333 (Supplementary Table S7). Their polymorphism information content (PIC) values ranged from 0.591 to 0.865, with an average of 0.721 (Fig. 4). The average values of observed heterozygosity (HO) and expected heterozygosity (HE) were 0.772 and 0.775, respectively, and these values ranged from 0.261 to 0.966 and from 0.637 to 0.877, respectively (Supplementary Table S7). The amplification results from only the 24 samples of P. wulianensis detected a total of 117 alleles, and the number of alleles ranged from 3 to 13, with an average NA of 6.50 (Supplementary Table S7). The PIC values ranged from 0.528 to 0.857, with an average of 0.683 (Fig. 4). The average HO and HE values were 0.772 and 0.775, respectively (Supplementary Table S7). The amplification results obtained from the samples of the six populations are shown in Supplementary Table S7. PIC values of 18 polymorphic EST-SSR markers. Transcriptome research is one of the essential tools for understanding the life processes of species38. Based on next-generation sequencing (NGS), researchers have performed a large number of gene expression and quantitative studies of Populus species39,40,41. Zhang et al. characterized cDNA libraries of mature xylem from tension wood (72.17%), opposite wood (70.13%), and normal wood (73.58%) of P. tomentosa, and their results showed that the length of most of the transcripts (more than 70%) was approximately 500–1000 bp42. In our study, a total of 8.18 Gb (3,521,665) of clean reads was obtained. The length of high-quality transcripts (approximately 97.16%) obtained in this study was almost greater than 1000 bp (Table 2), and this transcript length was clearly significantly higher than that obtained previously with second-generation sequencing42, which suggests that third-generation full-length transcriptome sequencing based on PacBio SMRT sequencing technology can compensate for the limitation of the short reading length observed with NGS and promote further in-depth study of P. wulianensis and its related species without reference genomes. In our study, a total of 41,876 nonredundant transcript sequences were successfully annotated with the functional databases (Supplementary Table S2). These annotated sequences lay a foundation for further studies of genetic differentiation in P. wulianensis. It has been previously reported that P. tomentosa best matches P. trichocarpa in the NR database41, and the species-based annotation performed in the present study also showed that P. wulianensis was most closely related to P. trichocarpa. However, in contrast to P. trichocarpa (sect. Tacamahaca), P. wulianensis and P. tomentosa both belong to sect. Populus, which indicates that the homology between P. wulianensis and P. tomentosa is higher than that between P. wulianensis and P. trichocarpa or P. tomentosa and P. trichocarpa. Notably, only 4.03% of the transcript sequences were annotated to P. tomentosa in our study (Supplementary Fig. S2). This contradiction is most likely due to the scarcity of reports on the transcriptome of species that belong to sect. Populus. Furthermore, the results of the present study showed that the number of annotations of P. wulianensis transcripts obtained with the GO and KEGG databases was significantly higher than that obtained for P. tomentosa (16,774 and 11,670, respectively)41. This finding is mainly attributed to the fact that the PacBio SMRT sequencing technology was used to obtain third-generation full-length transcriptomes in this study. It has been reported that the isoform sequencing (Iso-Seq) of transcriptomes by PacBio is advantageous for genome annotation43. Overall, the annotation information provided in our study is more sufficient than that obtained based on NGS and thus increases our understanding of the active biological and metabolic processes in P. wulianensis. Obviously, the transcript data obtained in our study solved the problem of the lack of a reference genome for this species and will provide rich annotation information for the more convenient identification of specific expression and the more precise quantification of gene and transcript expression in P. wulianensis and its related species. SSRs are well known and widely used in studies of the genetic diversity and population structure of species44,45,46. Compared with gSSRs, EST-SSRs are functional molecular markers with the advantages of easier and more efficient development, lower cost and more interspecific transferability15,16, The plant materials used in this study were obtained from the wild. All the samples were collected with the approval and permission of the local authorities. Based on the protection of wild plant resources, particularly endangered plant resources, we were only collected a small number of plant specimens. During the sample collection and experiment, we strictly abided by China’s laws and regulations regarding the protection of endangered wild plant resources and complied with the Convention on the Trade in Endangered Species of Wild Fauna and Flora. Prof. Dekui Zang formally identified all the samples, and information on the samples and specimens used in this study can be found in Supplementary Table S1. For transcriptomic analysis, samples from five tissue types (root tips, stems, leaves, buds and male flowers) were collected from vigorously growing and healthy P. wulianensis male specimens growing in Kunyu Mountain National Nature Reserve (Supplementary Table S1). Similarly, samples from six tissue types (root tips, stems, leaves, buds, female flowers and fruits) were collected from vigorously growing and healthy P. wulianensis female specimens (Supplementary Table S1). All the samples were frozen in liquid nitrogen immediately after collection and stored at − 80 °C until the experiment. The extraction of total DNA and the isolation of total RNA were performed according to Wiland-Szymańskas57 and Ghawana58, respectively. To ensure the accuracy of the data, the purity, concentration, and nucleic acid absorption peaks of the isolated RNA were detected using a Nanodrop spectrophotometer, and the RNA integrity was accurately tested with an Agilent 2100 instrument. We used electrophoresis to assess the contamination of the RNA samples with genomic DNA. For the polymorphism analysis of the developed EST-SSRs, the young leaves of ten P. wulianensis individuals at Kunyu Mountain, Zhaohu Mountain, Jiuxian Mountain and Juyu Mountain were collected and stored in silica gel (Supplementary Table S1). Similarly, samples of Populus adenopoda and Populus davidiana from Tianmu Mountain and Culai Mountain, respectively, were collected and stored using the above method (Supplementary Table S1). The modified cetyltrimethylammonium bromide (CTAB) method was used for total genomic DNA extraction59. A cDNA library was constructed using qualified samples. The SMARTer™ PCR cDNA Synthesis Kit was used for the synthesis of full-length cDNA from mRNA. The full-length cDNA fragments were screened using BluePippin and then amplified again by PCR. Subsequently, end repair of the full-length cDNA was performed, and the dumbbell-shaped SMRT adapter was connected. After exonuclease digestion, BluePippin was used for secondary screening to obtain a cDNA library. Qubit 2.0 and Agilent 2100 were used to accurately quantify and detect the library size, respectively. After the libraries passed quality control, full-length transcriptome sequencing was performed using PacBio RSII according to the target offline data volume. The ROI sequences were extracted from the original sequences according to the following criteria: full passes ≥ 0 and sequence accuracy > 0.75. The ROI sequences were divided into full-length and non-full-length sequences based on the presence of the 3′ primer, 5′ primer, and PolyA (optional). The ROI sequences from the same transcript were clustered using the iterative isoform-clustering (ICE) algorithm. ROIs with similar sequences were clustered, and each cluster yielded a consensus sequence. High-quality sequences (accuracy > 99%) were obtained by polishing the consensus sequences using the non-full-length sequences and used for subsequent analysis. CD-HIT was used to remove redundant sequences from the high-quality transcripts to obtain nonredundant sequences (identity > 0.99)60. To obtain annotation information for the transcripts, the obtained nonredundant transcript sequences were aligned to the NR, Swiss-Prot, GO, COG, KOG, EggNOG, Pfam, and KEGG databases using BLAST software (version 2.2.26)61,62,63,64,65,66,67,68. R version 3.6.1 was used for data analysis69. Potential EST-SSRs included in transcript sequences longer than 500 bp were searched and analyzed using the MIcroSAtellite identification tool (MISA). In our study, the SSR loci were identify based on the following criteria: repeat numbers of mono-, di-, tri-, tetra-, penta-, hexa- repeat motifs greater than or equal to 10, 6, 5, 5, 5, and 5, respectively. The maximum number of bases for two SSRs in an interrupted composite microsatellite was 100. EST-SSR primers were designed using Primer 3.0 software. The primer design was performed based on the following principles: (1) the primer length was 18–27 bp, usually 20 bp; (2) the annealing temperature was 57–63 °C, the optimal temperature was 60 °C, and the difference between the Tm values of the upstream and downstream primers did not exceed 5 °C; (3) the GC content was 20–80%, and the optimal GC content was 50%; and (4) the PCR amplification products were expected to have a length of 100–280 bp. The EST-SSR primers were synthesized by Shanghai Biological Engineering (Shanghai) Company. To screen out primers with polymorphisms, the abovementioned 100 primer pairs were synthesized for amplification. PCR was performed in a 20-μL reaction volume, which included 1 μL of template DNA (40 ng/µL), 1 μL of the forward primer (10 μmol/L), 1 μL of the reverse primer (10 μmol/L), 10 μL of 2× EasyTaq PCR SuperMix, and 7 μL of ddH2O. PCR amplification was performed using the following temperature program: predenaturation at 94 °C for 5 min; nine cycles of denaturation at 94 °C for 30 s, annealing at 59 °C for 30 s, and extension at 72 °C for 30 s; 21 cycles of denaturation at 94 °C for 30 s, annealing at 55 °C for 30 s, and extension at 72 °C for 30 s; extension at 72 °C for 3 min; and preservation at 4 °C49. During the selection of polymorphic primers, the PCR amplification products were run on a vertical plate electrophoresis apparatus using a 6% nondenaturing polyacrylamide gel. In the primer polymorphism verification experiments, high-performance capillary electrophoresis, which has higher accuracy than a nondenaturing polyacrylamide gel, was performed. Wu, Z. Y., Raven, P. H. & Hong, D. Y. Flora of China Vol. 4 (Science Press & Missouri Botanical Garden Press, Bei**g & St. Louis, 1999). Zang, D. K. Rare and Endangered Plants in Shandong (China Forestry Press, Bei**g, 2017). Wade, E. M. et al. Plant species with extremely small populations (PSESP) in China: A seed and spore biology perspective. Plant Diversity 38, 209–220. https://doi.org/10.1016/j.pld.2016.09.002 (2016). Liang, S. B. & Li, X. W. A new species of Populus from Shandong. Bull. Bot. Res. 6, 135–137 (1986). Zhang, L., Wang, M., Ma, T. & Liu, J. Taxonomic status of Populus wulianensis and P. ningshanica (Salicaceae). PhytoKeys 108, 117–129. https://doi.org/10.3897/phytokeys.108.25600 (2018). Qin, H. et al. China Checklist of Higher Plants, In the Biodiversity Committee of Chinese Academy of Sciences ed., Catalogue of Life China: 2020 Annual Checklist(Bei**g, China, 2020) Li, F., Li, W. & Fan, S. Arboraceous Flora of Shandong Vol. 1 (Science Press, Bei**g, 2016). Wang, Y. et al. Establishment of generation system for Populus wulianensis and control of vitrification of its test-tube seedlings. Mol. Plant Breeding 17, 6434–6446 (2019). Li, S. et al. Development and comparative genomic map** of Dasypyrum villosum 6V#4S-specific PCR markers using transcriptome data. Theor. Appl. Genet. 130, 2057–2068. https://doi.org/10.1007/s00122-017-2942-0 (2017). de Oliveira, E. J. et al. Polymorphic microsatellite marker set for Carica papaya L. and its use in molecular-assisted selection. Euphytica 173, 279–287. https://doi.org/10.1007/s10681-010-0150-y (2010). Zarini, H. N., Jafari, H., Ramandi, H. D., Bolandi, A. R. & Karimishahri, M. R. A comparative assessment of DNA fingerprinting assays of ISSR and RAPD markers for molecular diversity of Saffron and other Crocus spp. in Iran. Nucleus 62(1), 39–50. https://doi.org/10.1007/s13237-018-0261-8 (2019). Tamaki, I., Setsuko, S. & Tomaru, N. Genetic diversity and structure of remnant Magnolia stellata populations affected by anthropogenic pressures and a conservation strategy for maintaining their current genetic diversity. Conserv. Genet. 17, 715–725. https://doi.org/10.1007/s10592-016-0817-6 (2016). Powell, W., Machray, G. C. & Provan, J. Polymorphism revealed by simple sequence repeat. Trends Plant Sci. 1, 215–222. https://doi.org/10.1016/1360-1385(96)86898-1 (1996). Li, W. et al. De Novo transcriptomic analysis and development of EST–SSRs for Styrax japonicus. Forests. 9, 1–14. https://doi.org/10.3390/f9120748 (2018). Bouck, A. & Vision, T. The molecular ecologist’s guide to expressed sequence tags. Mol. Ecol. 16, 907–924. https://doi.org/10.1111/j.1365-294X.2006.03195.x (2010). Ling, P. et al. EST-SSR marker characterization based on RNA-sequencing of Lolium multiflorum and cross transferability to related species. Mol. Breeding 38, 80. https://doi.org/10.1007/s11032-018-0775-4 (2018). **ng, W. et al. De novo assembly of transcriptome from Rhododendron latoucheae Franch. using Illumina sequencing and development of new EST-SSR markers for genetic diversity analysis in Rhododendron. Tree Genet. Genomes 13, 53. https://doi.org/10.1007/s11295-017-1135-y (2017). Bazzo, B. R. et al. Development of novel EST-SSR markers in the macaúba palm (Acrocomia aculeata) using transcriptome sequencing and cross-species transferability in Arecaceae species. BMC Plant Biol. 18, 276. https://doi.org/10.1186/s12870-018-1509-9 (2018). Jeong, S. W. et al. Development of EST-SSR markers through de novo RNA sequencing and application for biomass productivity in kenaf (Hibiscus cannabinus L.). Genes Genomics 39, 1139–1156. https://doi.org/10.1007/s13258-017-0582-z (2017). Tang, D. Q. et al. De novo sequencing of the Freesia hybrida petal transcriptome to discover putative anthocyanin biosynthetic genes and develop EST-SSR markers. Acta Physiol. Plant. 40, 168 (2018). Xu, Q. S. et al. Transcriptome profiling using single-molecule direct RNA sequencing approach for in-depth understanding of genes in secondary metabolism pathways of Camellia sinensis. Front. Plant Sci. 8, 1205. https://doi.org/10.3389/fpls.2017.01205 (2017). Jo, I. H. et al. Isoform sequencing provides a more comprehensive view of the panax ginseng transcriptome. Genes 8, 228 (2017). Bhati, M., Kadri, N. K., Crysnanto, D. & Hubert, P. Assessing genomic diversity and signatures of selection in Original Braunvieh cattle using whole-genome sequencing data. BMC Genomics. 21, 27 (2020). Shi, J. S., Wang, Z. H. & Chen, J. H. Progress on whole genome sequencing in woody plants. Hereditas 34, 145–156 (2012). **ang, X., Zhang, Z., Wang, Z., Zhang, X. & Wu, G. Transcriptome sequencing and development of EST-SSRmarkers in Pinus dabeshanensis, an endangered conifer endemic to China. Mol. Breeding 35, 158. https://doi.org/10.1007/s11032-015-0351-0 (2015). Zulkapli, M. M. et al. Iso-Seq analysis of Nepenthes ampullaria, Nepenthes rafflesiana andNepenthes × hookeriana for hybridisation study in pitcher plants. Genomics Data 12, 130–131. https://doi.org/10.1016/j.gdata.2017.05.003 (2017). Thomas, S., Underwood, J. G., Tseng, E. & Holloway, A. K. Long-read sequencing of chicken transcripts and identification of new transcript isoforms. PLoS ONE 9, e94650 (2014). Zhang, D. Y., Zhang, T. X. & Wang, G. X. Development and application of second-generation sequencing technology. Environ. Sci. Technol. 39, 96–102 (2016). Abdel-Ghany, S. E. et al. A survey of the sorghum transcriptome using single-molecule long reads. Nat. Commun. 7, 11706. https://doi.org/10.1038/ncomms11706 (2016). Hoang, N. V. et al. A survey of the complex transcriptome from the highly polyploid sugarcane genome using full-length isoform sequencing and de novo assembly from short read sequencing. BMC Genomics 18, 395. https://doi.org/10.1186/s12864-017-3757-8 (2017). Kuang, X., Sun, S., Wei, J., Li, Y. & Sun, C. Iso-Seq analysis of the Taxus cuspidata transcriptome reveals the complexity of Taxol biosynthesis. BMC Plant Biol. 19, 210. https://doi.org/10.1186/s12870-019-1809-8 (2019). He, L. et al. Hybrid sequencing of full-length cDNA transcripts of stems and leaves in Dendrobium officinale. Genes 8, 257 (2017). Hovde, B. T. et al. Detection of abrin-like and prepropulchellin-like toxin genes and transcripts using whole genome sequencing and full-length transcript sequencing of Abrus precatorius. Toxins 11, 691. https://doi.org/10.3390/toxins11120691 (2019). Chen, X. et al. Full-length transcriptome sequencing and methyl jasmonate-induced expression profile analysis of genes related to patchoulol biosynthesis and regulation in Pogostemon cablin. BMC Plant Biol. 19, 266 (2019). Tian, X. et al. Transcriptome sequencing and EST-SSR marker development in Salix babylonica and S. suchowensis. Tree Genet. Genomes 15, 9. https://doi.org/10.1007/s11295-018-1315-4 (2019). **a, H. et al. Distinguishing upland and lowland rice ecotypes by selective SSRs and their applications in molecular-assisted selection of rice drought resistance. Euphytica 206, 11–20. https://doi.org/10.1007/s10681-015-1446-8 (2015). Wang, P. et al. Characterization and development of EST-SSR markers from a cold-stressed transcriptome of centipedegrass by illumina paired-end sequencing. Plant Mol. Biol. Rep. 35, 215–223. https://doi.org/10.1007/s11105-016-1017-8 (2017). Gordon, S. P. et al. Widespread polycistronic transcripts in fungi revealed by single-molecule mRNA sequencing. PLoS ONE 10, e0132628. https://doi.org/10.1371/journal.pone.0132628 (2015). Iori, V. et al. Physiology and genetic architecture of traits associated with cadmium tolerance and accumulation in Populus nigra L. Trees 30, 125–139. https://doi.org/10.1007/s00468-015-1281-5 (2016). Ariani, A., Romeo, S., Groover, A. T. & Sebastiani, L. Comparative epigenomic and transcriptomic analysis of Populus roots under excess Zn. Environ. Exp. Bot. 132, 16–27. https://doi.org/10.1016/j.envexpbot.2016.08.005 (2016). Yang, X. et al. Transcriptome profiling of Populus tomentosa under cold stress. Ind. Crops Products 135, 283–293. https://doi.org/10.1016/j.indcrop.2019.04.056 (2019). Chen, J., Chen, B. & Zhang, D. Q. Transcript profiling of Populus tomentosagenes in normal, tension, and opposite wood by RNA-seq. BMC Genomics 16, 164. https://doi.org/10.1186/s12864-015-1390-y (2015). An, D., Cao, H. X., Li, C., Humbeck, K. & Wang, P. Isoform sequencing and state-of-art applications for unravelling complexity of plant transcriptomes. Genes 9, 43. https://doi.org/10.3390/genes9010043 (2018). Zhang, Y. et al. Genetic variation, population structure and linkage disequilibrium in Switchgrass with ISSR, SCoT and EST-SSR markers. Hereditas 153, 4. https://doi.org/10.1186/s41065-016-0007-z (2016). Shahabzadeh, Z., Mohammadi, R., Darvishzadeh, R. & Jaffari, M. Genetic structure and diversity analysis of tall fescue populations by EST-SSR and ISSR markers. Mol. Biol. Rep. 47, 655–669. https://doi.org/10.1007/s11033-019-05173-z (2020). Zavinon, F. et al. Genetic diversity and population structure in Beninese pigeon pea [Cajanus cajan (L.) Huth] landraces collection revealed by SSR and genome wide SNP markers. Genet. Resour. Crop Evol. 67, 191. https://doi.org/10.1007/s10722-019-00864-9 (2020). Kim, J. H. et al. Characterization and development of EST-SSR markers in sweet potato (Ipomoea batatas (L.) Lam). 3 Biotech 6, 243. https://doi.org/10.1007/s13205-016-0565-9 (2016). Yan, L. et al. De novo transcriptome analysis of Fraxinus velutina using Illumina platform and development of EST-SSR markers. Biol. Plant 61, 210–218. https://doi.org/10.1007/s10535-016-0681-8 (2017). Zhang, X., Song, C., Zhang, Y., Yang, Y. & Huang, M. Development of EST-SSR in Populus deltoides and P. euramericana. Sci. Silvae Sin. 12, 53–59. https://doi.org/10.11707/j.1001-7488.20090910 (2009). Chen, L. Y. et al. Characterization of transcriptome and development of novel EST-SSR makers based on next-generation sequencing technology in Neolitsea sericea(Lauraceae) endemic to East Asian land-bridge islands. Mol. Breeding 35, 187. https://doi.org/10.1007/s11032-015-0379-1 (2015). Wang, J., Li, Z., Guo, Q., Ren, Q. & Wu, Y. Genetic variation within and between populations of a desert poplar (Populus euphratica) revealed by SSR markers. Ann. For. Sci. 68, 1143. https://doi.org/10.1007/s13595-011-0119-6 (2011). Ciftci, A. & Kaya, Z. Genetic diversity and structure of Populus nigra populations in two highly fragmented river ecosystems from Turkey. Tree Genet. Genomes 15, 66. https://doi.org/10.1007/s11295-019-1370-5 (2019). Wei, Z., Du, Q., Zhang, J., Li, B. & Zhang, D. Genetic diversity and population structure in chinese indigenous poplar (Populus simonii) populations using microsatellite markers. Plant Mol. Biol. Rep. 31, 620–632. https://doi.org/10.1007/s11105-012-0527-2 (2013). Saito, Y. et al. Genetic diversity of Populus euphratica populations in northwestern China determined by RAPD DNA analysis. New Forest. 23, 97–103. https://doi.org/10.1023/A:1015605928414 (2002). Mutegi, S. M. et al. Genetic diversity of the African poplar (Populus ilicifolia) populations in Kenya. Tree Genet. Genomes 12, 66. https://doi.org/10.1007/s11295-016-1013-z (2016). Shen, D.F., Bo, W.H., Xu, F. & Wu, R. Genetic diversity and population structure of the Tibetan poplar (Populus szechuanica var. tibetica) along an altitude gradient. BMC Genet.15, S11. https://www.biomedcentral.com/1471-2156/15/S1/S11(2014) Li, J., Wang, S., Yu, J., Wang, L. & Zhou, S. A modified CTAB protocol for plant DNA extraction. Chin. Bull. Bot. 48, 72–78 (2013). Ghawana, S. et al. An RNA isolation system for plant tissues rich in secondary metabolites. BMC Res. Notes.4, 85. https://www.biomedcentral.com/1756-0500/4/85 (2011) Hewitt, G. M. & Johnston, A. Molecular Techniques in Taxonomy 283–293 (Springer, Berlin, 1991). Li, W. & Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659 (2006). Deng, Y. Y. et al. Integrated NR database in protein annotation system and its localization. Computer Eng. 32, 71–74 (2006). Apweiler, R. et al. UniProt: The Universal Protein knowledgebase. Nucleic Acids Res. 32, D115–D119 (2004). Ashburner, M. et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 25, 25–29 (2000). Tatusov, R. L., Galperin, M. Y., Natale, D. A. & Koonin, E. V. The COG database: A tool for genome scale analysis of protein functions and evolution. Nucleic Acids Res. 28, 33–36 (2000). Koonin, E. V. et al. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biol. 5, R7 (2004). Finn, R. D. et al. Pfam: The protein families database. Nucleic Acids Res. 42, D222–D230 (2013). Kanehisa, M., Goto, S., Kawashima, S., Okuno, Y. & Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 32, D277–D280 (2004). Altschul, S. F. et al. Gapped BLAST and PSIBLAST: A new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. https://doi.org/10.1093/nar/25.17.3389 (1997). R Core Team. R: A language and environment for statisticalcomputing. R Foundation for Statistical Computing, Vienna,Austria. https://www.R-project.org/.(2019) This study was supported by the Shandong Agricultural Seeds Engineering Project (2019LZGC01802) and the National Forest Germplasm Resources Platform Project 2019-2020. We would like to thank AJE (https://www.aje.cn/) for providing linguistic assistance during the preparation of this manuscript. Q.W. and F.Z. jointly performed the bioinformatics analyses and jointly performed the screening and verification experiments of polymorphic EST-SSR markers; X.X. and Y.Z. provided guidance for the bioinformatics analysis and performed the data mining analysis; D.Z. and Y.M. designed and coordinated the study; and Q.W. wrote the manuscript. Q.W., F.Z. and Y.M. revised the manuscript. D.Z. is the recipient of the above fundings. Based on their contribution to this study, Q.W. and F.Z. are co-first authors. Y.M. is the corresponding author. All the authors read and approved the final manuscript. The authors declare no competing interests. Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. Wu, Q., Zang, F., **e, X. et al. Full-length transcriptome sequencing analysis and development of EST-SSR markers for the endangered species Populus wulianensis.
Sci Rep 10, 16249 (2020). https://doi.org/10.1038/s41598-020-73289-5 Received: Accepted: Published: DOI: https://doi.org/10.1038/s41598-020-73289-5 Genetic Resources and Crop Evolution (2024) Scientific Reports (2023) Parasites & Vectors (2023) BMC Genomics (2022) BMC Plant Biology (2022)Results
Full-length transcriptome sequencing
Functional annotation of transcript sequences



Characteristics of SSRs in the transcriptome
Verification of novel and polymorphic EST-SSRs

Discussion
Methods
Plant materials, DNA and RNA isolation
cDNA library construction and online sequencing
Analysis of transcriptome sequencing and annotation
EST-SSR detection and primer design
Amplification and validation of EST-SSRs
References
Acknowledgements
Author information
Authors and Affiliations
Contributions
Corresponding author
Ethics declarations
Competing interests
Additional information
Publisher's note
Supplementary information
Rights and permissions
About this article
Cite this article
This article is cited by
Genetic diversity and population structure analysis in wood-apple (Limonia acidissima L.) using simple sequence repeat (SSR) markers
Genetic diversity, population structure, and DNA fingerprinting of Ailanthus altissima var. erythrocarpa based on EST-SSR markers
Transcriptome analysis of Haemaphysalis flava female using Illumina HiSeq 4000 sequencing: de novo assembly, functional annotation and discovery of SSR markers
Full-length fruit transcriptomes of southern highbush (Vaccinium sp.) and rabbiteye (V. virgatum Ait.) blueberry
De novo transcriptome assembly and analysis of gene expression in different tissues of moth bean (Vigna aconitifolia) (Jacq.) Marechal
