
Abstract
Scale-up of production is needed for industrial applications and clinical translation of human induced pluripotent stem cells (hiPSCs). However, in cryopreservation of hiPSCs, successful rewarming of vitrified cells can only be achieved by convective warming of small volumes (generally 0.2 mL). Here, we present a scalable nano-warming technology for hiPSC cryopreservation employing inductive heating of magnetic nanoparticles under an alternating magnetic field. The conventional method by water bath heating at 37 °C resulted in a decrease of cell viability owing to devitrification caused by slow warming of samples with large volumes (≥ 20 mL). Nano-warming showed uniform and rapid rewarming of vitrified samples and improved viability of hiPSCs in the 20-mL system. In addition to single cells, hiPSC aggregates prepared using a bioreactor-based approach were successfully cryopreserved by the nano-warming technique. These results demonstrate that nano-warming is a promising methodology for cryopreservation in mass production of hiPSCs.
Similar content being viewed by others

Introduction
Pluripotent stem cells, including human embryonic stem cells (hESCs) and induced pluripotent stem cells (hiPSCs), are a promising cell source for regenerative medicine because of their unlimited proliferation potential and differentiation capability1,2. The first patient was treated with an hESC‐based cellular therapy product in a clinical trial for spinal cord injury in 20103. In 2014, the first patient received hiPSC-derived retinal pigment epithelial cells for macular degeneration4. Although pluripotent stem cells have been generated at laboratory scale, large scale production processes by standardized and economically viable procedures and technologies will be required5. One of the challenges in large scale expansion of pluripotent stem cells is suspension culture of cell aggregates in stirred bioreactors6,7 in which single cells form cell aggregates in the presence of the small molecule Y27632 (Rho-associated coiled-coil kinase inhibitor)8. Furthermore, bioreactor-based suspension culture of pluripotent stem cells can be used for large scale induction of functional cells such as iPSC-derived cardiomyocytes9. In addition, a recent report suggests that cell aggregates can be used as building blocks for tissue engineering10.
One bottleneck in manufacturing pluripotent stem cells is robust cryopreservation, and large-scale cell cryopreservation will be mandatory for industrial applications and clinical translation. Cryopreservation is classified into two distinct methods: slow freezing and vitrification. In vitrification methods, cryoprotectant solutions with high cryoprotectant concentrations are used, and cells are rapidly frozen by direct immersion of the container in liquid nitrogen. Originally, vitrification methods were developed for cryopreservation of oocytes and embryos11. Fujioka et al. developed DAP213, a cryoprotectant solution containing dimethyl sulfoxide (DMSO) for primate ESCs, and succeeded in cryopreservation of ESCs by vitrification of a 0.2-mL scale12. However, Katkov et al. reported that cryopreservation with DMSO diminished expression of the pluripotency marker Oct3/4 in ESCs13. More recently, Ota et al. developed a DMSO-free cryoprotectant solution called StemCell Keep14 in which carboxylated ε-poly-l-lysine was added as a cryoprotectant. StemCell Keep showed successful cryopreservation of mesenchymal stem cells15, hESCs14 and hiPSCs16. However, the size limitation remains and the 0.2-mL scale (1 × 106 cells/mL) is the gold standard for vitrification of hiPSCs.
In the cryopreservation process, both cooling and warming rates should exceed the critical cooling rate (CCR)17 and critical warming rate (CWR)18 to avoid failure of vitrification, resulting in cell damage caused by ice crystal growth19. In large scale cryopreservation, one of the major technological barriers is achieving the CWR to avoid devitrification, because the conventional method of rewarming is performed by immersing vitrified cells in a water bath at 37 °C to avoid cellular damage caused by overheating. This leads to a low warming rate at the centre of large volume samples with large diameters. Additionally, high temperature gradients between the centre and edge of large volume samples may cause cracking and devitrification. Therefore, technological breakthroughs to overcome slow and uneven rewarming are required for large-scale cryopreservation of human iPSCs.
Because magnetic nanoparticles have unique features including magnetic attraction, they have been used for medical applications27. In this study, iPSC aggregates that underwent slow freezing showed a clear cell–cell boundary, poor E-cadherin expression, and low cell viability (Supplementary Fig. S2). These results suggest that slow freezing is unsuitable for cryopreservation of multicellular biomaterials such as cell aggregates, tissues, and organs. In the present study, we succeeded in cryopreservation of iPSC aggregates by vitrification, which is considered an important finding for the large-scale production of iPSCs using stirred bioreactors. Here, we focused on scale-up of cryopreservation volumes for iPSC production, whereas cryopreservation of tissues and organs will also require large-scale vitrification. Manuchehrabadi et al. demonstrated that nano-warming is effective for tissue cryopreservation in which porcine arteries with a wall thickness of 1–2 mm were evaluated26. To achieve successful cryopreservation of more complex and thicker tissues and organs, continuous breakthroughs including perfusion of cryoprotectant solutions in tissues and organs will be necessary.
Toxicology of magnetite nanoparticles is an important issue, and the main requirements for cryopreservation are maintaining cell viability and preserving normal cell behaviours. In the present study, for both single (Figs. 1c, 4b) and aggregated (Fig. 5b) iPSCs, cell viabilities in magnetite-loaded StemCell Keep were comparable to that in StemCell Keep without magnetite nanoparticles. Moreover, iPSCs after nano-warming showed expression of pluripotency markers (Figs. 4c, 5c), and trilineage differentiation was induced successfully (Fig. 4d and Supplementary Fig. S1). Ota et al. reported that the teratoma assay showed that StemCell Keep-cryopreserved human ESCs differentiated into three germ layers14. The effects of magnetite nanoparticles on in vivo differentiation of iPSCs remain to be fully elucidated. In our preliminary study of biodistribution, magnetite nanoparticles (90 mg, i.p.) were injected into mice, and none of the 10 mice died during the study. Transient accumulation of magnetite was observed in the liver of the mice, but the magnetite nanoparticles had been cleared from circulation at 10 days after administration28. In addition to their high biocompatibility, magnetite nanoparticles can be easily separated by a magnet. In the present study, magnetite nanoparticles were separated with a magnet, and iPSCs after nano-warming were cultured for AP staining (Fig. 4b) and trilineage differentiation (Fig. 4c). We found no detectable magnetite nanoparticles (< 0.02 mg Fe per mL by the potassium thiocyanate method29) in iPSCs. Toxicity of magnetic nanoparticles used in nano-warming should be investigated in more detail before entering clinical trials.
In the present study, we demonstrated the success of nano-warming and convective failure for hiPSCs vitrified in a 20-mL system, which corresponded to a 100 times larger volume than the gold standard (0.2 mL). Here, the volume of the cryoprotectant solution was scaled up at the concentration of 1 × 106 cells/mL, and we succeeded in cryopreservation of 2 × 107 cells per vial in a batch. Studies suggest that 1 × 109 functional cells per patient are required for regenerative medicine using hepatocytes, pancreatic β-cells, or cardiomyocytes5, and a 1-L cryopreservation system will be required in the future. In the present study, uniform and rapid rewarming rates beyond the CWR were achieved in 1- to 30-mL systems (Fig. 3b), suggesting that nano-warming is a scalable technology. For further scale-up of nano-warming, larger coils of an alternating magnetic field applicator will be required. We previously developed a coil system of 30 cm in diameter for MFH. In the 30-cm coil system, non-specific heating in agarose gel phantoms occurs by eddy current loss at 360 kHz, but we succeeded in reducing non-specific heat generation by lowering the frequency of the alternating magnetic field irradiation to 100–200 kHz (unpublished results). However, achieving the CCR may be more challenging for large-scale vitrification. Rough estimation suggests that StemCell Keep in a vial with a radius of ≤ 2 cm can achieve a CCR of ≥ 4.9 °C/min. Therefore, to vitrify 1 L of solution, the height of the container must be ≥ 80 cm. In the present study, we used glass vials, but their thermal conductivity is known to be low. Most metals cannot be used for nano-warming because they generate heat under an alternating magnetic field. A potential approach includes the use of a container with relatively high thermal conductivity, such as aluminium oxide.
In the current work, we focussed on hiPSC cryopreservation by nano-worming. To confirm the efficacy of this novel method, we conducted nano-warming using another hiPSC line, 253G130 (Supplementary Fig. S3): For 20-mL samples, the cell viability after convective warming was 34.5 ± 3.6%, whereas the cell viability after nano-warming was 67.7 ± 7.8%, which was comparable to that of the gold standard (70.6 ± 3.5%). Also, we believe that nano-worming is applicable to other stem cells, tissue cells and tissue-engineered cell constructs. Recently, Liu et al. used adipose-derived stem cells and showed that nano-warming of cell–alginate hydrogel constructs improved cell survival after cryopreservation31. In our preliminary study, cell viability of pancreatic beta cell line MIN6 was improved by nano-warming. Moreover, cell viability of primary mouse islets was improved by nano-warming. In addition to the cell viability, glucose-stimulated insulin secretion was observed at the similar level to the non-frozen control after nano-warming. These results suggest that nano-warming is a potent approach for cryopreservation.
In conclusion, we propose novel methodology for iPSC cryopreservation. Nano-warming is a scalable technology that may facilitate the industrialization of iPSC production and its clinical translation for regenerative medicine.
Methods
Preparation of the magnetite-loaded cryopreservation solution
StemCell Keep is a DMSO-free cryopreservation solution containing 6.5 M ethylene glycol, 10% carboxylated poly-l-lysine, and 0.5 M sucrose (BioVerde, Kyoto, Japan)14. The magnetite nanoparticles (Fe3O4; average particle size: 10 nm) were obtained from Dai-ichi High Frequency (Tokyo, Japan). The magnetic characteristics at 796 kA/m (room temperature) were as follows: 2.0 kA/m coercivity, 63.9 Am2/kg saturation flux density, and 2.6 Am2/kg remanent flux density. To prepare magnetite-loaded cryoprotectant solutions, the magnetite nanoparticles were added to StemCell Keep in cylindrical glass vials (Supplementary Table S1) at the indicated concentrations. The vials were vortexed for 15 min before use.
Cell culture and preparation for cryopreservation
hiPSCs (clone 201B71) were provided by the RIKEN BRC (Ibaraki, Japan). The cells were cultured on a surface coated with recombinant laminin-511 E8 fragments (iMatrix-511; Nippi, Tokyo, Japan) in culture medium to maintain the undifferentiated state (StemFit AK02N; A**omoto, Tokyo, Japan). Cell culture was carried out at 37 °C with 5% CO2 in a humidified atmosphere. To cryopreserve single hiPSCs, hiPSC colonies were dissociated into single cells by treatment with Accutase (Innovative Cell Technologies, San Diego, CA) containing 10 μM ROCK inhibitor (Y-27632; Nacalai Tesque, Kyoto, Japan). Cells were counted with a cell counter (Countess II FL; Thermo Fischer Scientific, Waltham, MA) by the trypan blue exclusion method, and added to the cryoprotectant solution at 1 × 106 cells/mL in the glass vials.
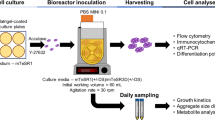
For bioreactor experiments, 6 × 106 cells were suspended in medium containing 10 μM ROCK inhibitor and applied to a 30-mL stirred bioreactor (Cat# BMV-S03A; Able, Tokyo, Japan). The agitation speed was 55 rpm. Cells were cultured at 37 °C with 5% CO2 in a humidified atmosphere. To cryopreserve hiPSC aggregates, hiPSC aggregates at day 2 of culture were collected from the bioreactor. To determine the number of cells in the aggregates, triplicate 1-mL samples of the cell culture were collected and dissociated into single cells using Accumax (Innovative Cell Technologies). The cells were then counted with the Countess II FL by the trypan blue exclusion method. Cell aggregates corresponding to 1 × 106 cells/mL were added to the cryoprotectant solution in the glass vials.
Convective cooling and warming
Convective cooling for freezing and convective warming for thawing were carried out by immersing the glass vials or 2-mL polypropylene cryovials (solution volume: 0.2 mL; Corning, New York, NY) as a control (gold standard) in liquid nitrogen and a 37 °C water bath, respectively. Temperatures were measured using optical fibre probes (FX-9020; Anritsu Meter, Tokyo, Japan).
Nano-warming
An alternating magnetic field was created using a vertical coil (inner diameter 7 cm; length 8 cm) with a transistor inverter (HI-HEATER6020; Dai-ichi High Frequency) operating at 108, 163, or 208 kHz. The maximum levels of power and magnetic field intensity were 10 kW and 31.9 kA/m, respectively. The vial was placed inside the coil, so that the vial was positioned at the centre of the coil. Temperatures during application of the alternating magnetic field were measured by the optical fibre probes. After nano-warming, cells were washed with the culture medium and remaining magnetite nanoparticles were separated with a magnet (diameter 5 cm; height 1 cm; magnetic induction 370 mT).
Cell viability
Convective cooling for freezing was carried out by immersing the samples in liquid nitrogen for 15 min, and the samples were then warming for thawing. For cell thawing, a water bath for convective warming or alternating magnetic field applicator for nano-warming were turned off when the temperature reached 0 °C to prevent any toxicity from StemCell Keep. After thawing, cells in a 1 mL suspension were collected and their viability was assayed using a ReadyProbes Cell Viability Imaging Kit (Blue/Green) based on Hoechst 33342 for live cells and SYTOX green nucleic acid stain for dead cells (Thermo Fischer Scientific). Cell viability was determined as the percentage of live cells among the total cells counted in five fluorescence images of each sample under a fluorescence microscope (BZ-X810; Keyence, Tokyo, Japan).
Alkaline phosphate (AP) staining
Cells were seeded in a 6-well culture plate coated with recombinant laminin-511 E8 fragments at 1.3 × 104 cells/well and cultured for 5 days to induce colony formation. After washing with phosphate buffered saline (PBS), the colonies were fixed with 4% paraformaldehyde in PBS for 3 min at room temperature and exposed to a solution containing naphthol AS-MX phosphate (Nacalai Tesque) as a substrate and Fast red TR salt hemi (zinc chloride) salt (Santa Cruz Biotechnology, Dallas, TX) as a coupler for 30 min at 37 °C. AP-positive colonies were determined as the percentage of positive colonies to total colonies counted in five images of each sample under the BZ-9000 microscope.
Induction of differentiation
After nano-warming, cells were washed with the culture medium and remaining magnetite nanoparticles were separated with the magnet. hiPSCs were induced to differentiate into the three germ layers, ectoderm, mesoderm, and endoderm, using a STEMdiff Trilineage Differentiation Kit (STEMCELL Technologies, Vancouver, Canada), according to the manufacturer’s instructions.
RT-PCR
For semiquantitative RT-PCR, total RNA was extracted from 1 × 106 cells using an RNAiso Plus Kit (Takara Bio, Shiga, Japan) and reverse transcribed using reverse transcriptase (ReverTra Ace; Toyobo, Osaka, Japan). PCR was performed using the primers for lineage-specific genes listed in Supplementary Table S2. PCR was initiated using a DNA polymerase (G-Taq; Cosmo Genetech, Seoul, Korea) at 94 °C for 2 min, followed by 35 cycles of amplification at 98 °C for 10 s, 57 °C for 30 s, and 68 °C for 30 s, and final extension at 30 °C for 5 min. The PCR products were subjected to electrophoresis on a 2% agarose gel. DNA fragments were visualized by ethidium bromide staining.
Immunostaining of Oct3/4
hiPSC aggregates were embedded in optical cutting temperature compound (Tissue-Tek, Tokyo, Japan), and thin sections (20 μm thick) were fixed with 4% paraformaldehyde for 10 min at room temperature. The specimens were permeabilized with PBS containing 0.5% Triton X-100 (FUJIFILM Wako Pure Laboratory Chemicals, Osaka, Japan) for 5 min and blocked in Block Ace (Dainippon Sumitomo Pharma, Osaka, Japan) at 4 °C overnight. The specimens were then probed with primary antibodies against Oct3/4 (cat# sc-9081, Santa Cruz Biotechnology) at 4 °C overnight. After washing with Tris-buffered saline, the specimens were immersed in PBS containing 10% Block Ace and an Alexa Fluor 488-conjugated secondary antibody (Thermo Fischer Scientific) for 60 min at room temperature. Cell nuclei were stained with 4′,6-diamidino-2-phenylindol (DAPI, Thermo Fischer Scientific) for 20 min. After washing with PBS, the specimens were observed under a confocal laser-scanning microscope (Olympus, Tokyo, Japan).
Statistical analysis
Statistical comparisons were made using the Mann–Whitney rank sum test. Values of P < 0.05 were considered to be significantly different.
Data availability
The datasets generated during and/or analysed during current study are available in this article and its Supplementary Information files, or from the corresponding author on request.
References
Takahashi, K. et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell131, 861–872 (2007).
Shi, Y., Inoue, H., Wu, J. C. & Yamanaka, S. Induced pluripotent stem cell technology: a decade of progress. Nat. Rev. Drug Discov.16, 115–130 (2017).
Ilic, D., Devito, L., Miere, C. & Codognotto, S. Human embryonic and induced pluripotent stem cells in clinical trials. Br. Med. Bull.116, 19–27 (2015).
Mandai, M. et al. Autologous induced stem-cell-derived retinal cells for macular degeneration. N. Engl. J. Med.376, 1038–1046 (2017).
Kropp, C., Massai, D. & Zweigerdt, R. Progress and challenges in large-scale expansion of human pluripotent stem cells. Process Biochem.59, 244–254 (2017).
Zweigerdt, R., Olmer, R., Singh, H., Haverich, A. & Martin, U. Scalable expansion of human pluripotent stem cells in suspension culture. Nat. Protoc.6, 689–700 (2011).
Galvanauskas, V., Grincas, V., Simutis, R., Kagawa, Y. & Kino-Oka, M. Current state and perspectives in modeling and control of human pluripotent stem cell expansion processes in stirred-tank bioreactors. Biotechnol. Prog.33, 355–364 (2017).
Watanabe, K. et al. A ROCK inhibitor permits survival of dissociated human embryonic stem cells. Nat. Biotechnol.25, 681–686 (2007).
Matsuura, K. et al. Creation of human cardiac cell sheets using pluripotent stem cells. Biochem. Biophys. Res. Commun.425, 321–327 (2012).
Arai, K. et al. Fabrication of scaffold-free tubular cardiac constructs using a Bio-3D printer. PLoS ONE13, e0209162 (2018).
Rall, W. F. & Fahy, G. M. Ice-free cryopreservation of mouse embryos at − 196 degrees C by vitrification. Nature313, 573–575 (1985).
Fujioka, T., Yasuchika, K., Nakamura, Y., Nakatsuji, N. & Suemori, H. A simple and efficient cryopreservation method for primate embryonic stem cells. Int. J. Dev. Biol.48, 1149–1154 (2004).
Katkov, I. I. et al. Cryopreservation by slow cooling with DMSO diminished production of Oct-4 pluripotency marker in human embryonic stem cells. Cryobiology53, 194–205 (2006).
Ota, A., Matsumura, K., Lee, J. J., Sumi, S. & Hyon, S. H. StemCell Keep™ is effective for cryopreservation of human embryonic stem cells by vitrification. Cell Transpl.26, 773–787 (2017).
Matsumura, K. et al. Cryopreservation of a two-dimensional monolayer using a slow vitrification method with polyampholyte to inhibit ice crystal formation. ACS Biomater. Sci. Eng.2, 1023–1029 (2016).
Matsumura, K., Bae, J. Y., Kim, H. H. & Hyon, S. H. Effective vitrification of human induced pluripotent stem cells using carboxylated ε-poly-l-lysine. Cryobiology63, 76–83 (2011).
Canova, C. & Carvalho, B. M. Modified method for obtaining the critical cooling rate for vitrification of polymers materials research. Mater. Res.10, 449–452 (2007).
Hopkins, J. B., Badeau, R., Warkentin, M. & Thorne, R. E. Effect of common cryoprotectants on critical warming rates and ice formation in aqueous solutions. Cryobiology65, 169–178 (2012).
Fehy, G. M. & Wowk, B. Principles of cryopreservation by vitrification. In Cryopreservation and Freeze-Drying Protocols (eds Wolkers, W. F. & Oldenhof, H.) 21–82 (Springer Science + Business Media, New York, 2015).
**e, W. et al. Shape-, size- and structure-controlled synthesis and biocompatibility of iron oxide nanoparticles for magnetic theranostics. Theranostics8, 3284–3307 (2018).
Ito, A., Shinkai, M., Honda, H. & Kobayashi, T. Medical application of functionalized magnetic nanoparticles. J. Biosci. Bioeng.100, 1–11 (2005).
Dennis, C. L. & Ivkov, R. Physics of heat generation using magnetic nanoparticles for hyperthermia. Int. J. Hyperthermia29, 715–729 (2013).
Hergt, R., Dutz, S. & Roder, M. Effects of size distribution on hysteresis losses of magnetic nanoparticles for hyperthermia. J. Phys. Condens. Matter20, 385214 (2008).
Johannsen, M., Thiesen, B., Wust, P. & Jordan, A. Magnetic nanoparticle hyperthermia for prostate cancer. Int. J. Hyperthermia26, 790–795 (2010).
Ito, A. & Kobayashi, T. Intracellular hyperthermia using magnetic nanoparticles: a novel method for hyperthermia clinical applications. Therm. Med.24, 113–129 (2008).
Manuchehrabadi, N. et al. Improved tissue cryopreservation using inductive heating of magnetic nanoparticles. Sci. Transl. Med.9, eaah4586 (2017).
Mandumpal, J. B., Kreck, C. A. & Mancera, R. L. A molecular mechanism of solvent cryoprotection in aqueous DMSO solutions. Phys. Chem. Chem. Phys.13, 3839–3842 (2011).
Ito, A. et al. Time course of biodistribution and heat generation of magnetite cationic liposomes in mouse model. Jpn. J. Hyperthermic Oncol.19, 151–159 (2003).
Owen, C. S. & Sykes, N. L. Magnetic labelling and cell sorting. J. Immunol. Methods73, 41–48 (1984).
Nakagawa, M. et al. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat. Biotechnol.26, 101–106 (2008).
Liu, X., Zhao, G., Chen, Z., Panhwar, F. & He, X. Dual suppression effect of magnetic induction heating and microencapsulation on ice crystallization enables low cryoprotectant vitrification of stem cell–alginate hydrogel constructs. ACS Appl. Mater. Interfaces10, 16822–16835 (2018).
Acknowledgements
This work was supported in part by JST PRESTO (No. JPMJPR17I1), a Japan Society for the Promotion of Science Grant-in-Aid for Scientific Research (Grant no. 17H03469), the AJINOMOTO CO. INNOVATION ALLIANCE AMINO ACID RESEARCH PROGRAM, and Kyoto University Nano Technology Hub in “Nanotechnology Platform Project” sponsored by the Ministry of Education, Cultures, Sports, Science and Technology (MEXT) of Japan. We thank Mitchell Arico from Edanz Group (http://www.edanzediting.com/ac) for editing a draft of this manuscript.
Author information
Authors and Affiliations
Contributions
A.I., M.T., S.T., and M.H. were involved in the experimental design. A.I., K.Y., S.M., K.S., Y.H., T.N., T.Y., and M.H. conducted experiments and analysed data. A.I., M.T., S.T., and M.H. wrote the manuscript. All authors discussed the results and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ito, A., Yoshioka, K., Masumoto, S. et al. Magnetic heating of nanoparticles as a scalable cryopreservation technology for human induced pluripotent stem cells. Sci Rep 10, 13605 (2020). https://doi.org/10.1038/s41598-020-70707-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-70707-6
- Springer Nature Limited
This article is cited by
-
De novo design of a nanoregulator for the dynamic restoration of ovarian tissue in cryopreservation and transplantation
Journal of Nanobiotechnology (2024)
-
Advanced cryopreservation engineering strategies: the critical step to utilize stem cell products
Cell Regeneration (2023)
-
Nanowarming and ice-free cryopreservation of large sized, intact porcine articular cartilage
Communications Biology (2023)
-
The Beneficial Effects of Static Magnetic Field and Iron Oxide Nanoparticles on the Vitrification of Mature Mice Oocytes
Reproductive Sciences (2023)
-
Chemical approaches to cryopreservation
Nature Reviews Chemistry (2022)