
Abstract
Treatment of plant seeds with electromagnetic fields or non-thermal plasmas aims to take advantage of plant functional plasticity towards stimulation of plant agricultural performance. In this study, the effects of pre-sowing seed treatment using 200 Pa vacuum (7 min), 5.28 MHz radio-frequency cold plasma (CP −2, 5, and 7 min) and electromagnetic field (EMF −5, 10, 15 min) on seed germination kinetics, content of phytohormones, morphometric parameters of seedlings and leaf proteome were assessed. CP 7 min and EMF 15 min treatments caused 19–24% faster germination in vitro; germination in the substrate was accelerated by vacuum (9%) and EMF 15 min (17%). The stressors did not change the seed germination percentage, with exception of EMF 5 min treatment that caused a decrease by 7.5%. Meanwhile both CP 7 min and EMF 15 min treatments stimulated germination, but the EMF treatment resulted in higher weight of leaves. Stressor-specific changes in phytohormone balance were detected in seeds: vacuum treatment decreased zeatin amount by 39%; CP treatments substantially increased gibberellin content, but other effects strongly varied with the treatment duration; the abscisic acid content was reduced by 55–60% after the EMF treatment. Analysis of the proteome showed that short exposure of seeds to the EMF or CP induced a similar long-term effect on gene expression in leaves, mostly stimulating expression of proteins involved in photosynthetic processes and their regulation.
Similar content being viewed by others
Introduction
An interdisciplinary field of research on low temperature non-equilibrium plasma, also termed cold plasma (CP) and electromagnetic field (EMF) applications for agriculture1,2 is directed towards exploiting the potential of plant functional plasticity. Seed treatment with CP or EMF is a modern eco-agricultural technology for increasing plant agricultural performance. Numerous studies have demonstrated that such treatments are effective for enhancing agronomic seed quality and have potential to be used for seed decontamination, activation of germination and seedling growth.
The majority of studies in this area are focused on assessment of CP and EMF effects on physiological (germination), structural (changes in seed coat surface) and morphometric (early seedling growth) estimates (reviewed by3,4,5). A few reports have also considered changes in biochemical characteristics, such as amount of pigments and secondary metabolites, enzymatic activities or antioxidative capacity6,7,8,9,10,11,12. Stressor-induced changes in physiological or biochemical activities are associated with selective modulation of protein expression in the growing seedling, e.g., activation of photosynthesis is expected to be related to changes in the leaf proteome13,14. Although several studies described morphological, genotoxic or biochemical changes induced by the CP and EMF treatments14,15,16,17,18,19, the proteomic profiles of plant response to CP and EMF treatments have not been reported so far.
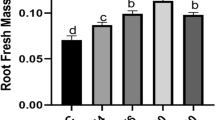
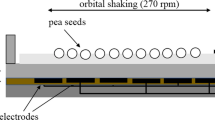
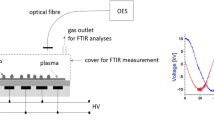
On the other hand, although germination tests are commonly used as a hallmark of the response to treatments, until now there has been no attempt to estimate CP and EMF effects on the content of seed phytohormones, which are known to be key regulators of germinationMeasurement of seed germination and seedling morphological parameters Germination tests were started four days after the treatment both in vitro and in the substrate. For germination test in vitro seeds were evenly distributed on three layers of filter paper in 13.5 mm diameter plastic Petri dishes (three replicates of 30 seeds each) and watered with 6 mL distilled water. Petri dishes with seeds were placed in a climatic chamber KK 750 (Pol-Eko-Aparatura, Poland) with automatic control of moisture (60%), light, and temperature. Alternating light and temperature regimes were maintained in the chamber (darkness: 14 °C for 8 h; light: 21 °C for 16 h). Seeds were provided with additional water in a Petri dish, if necessary, to prevent drying. Germinated seeds (judged by the appearance of a visible 1-mm radicle) were counted daily until their number stopped increasing. For the germination tests in the substrate, the seeds were sown into plastic containers (12 × 18 × 30 cm) filled with peat substrate, placing the seeds in 0.5 cm depth from the substrate surface. Germination tests were replicated three times for all experimental conditions including control seeds (3 × 30 seeds, n = 30 for one replicate). Germinated seeds were counted daily as judged by the appearance of the top of green sprout from the surface of the substrate. The germination results of each experimental replicate were analyzed using the application of Richards’ function66 for the analysis of germinating seed population67. The indices of germination kinetics derived from Richards plots were: Vi (%) – final germination percentage indicating seed viability, Me (days) – median germination time (t50%) indicating the germination halftime of a seed lot or germination rate67. The containers with grown seedlings were kept for 2 weeks in the climatic chamber with constant humidity (60%) and alternating light and temperature regimes (darkness: 14 °C for 8 h; light: 21 °C for 16 h). For morphometric analysis seedlings were carefully removed from containers, their roots washed to remove the substrate and wiped well with a moisture absorbent paper. Fresh weight and length of all seedlings and their parts (roots, shoots and leaves) was estimated. For the extraction of plant hormones 1 g of seeds was ground and extracted in 5 mL of 85% methanol for 24 hours at 4 °C. The homogenate was centrifuged at 13500 × g for 5 min, the supernatant was collected and kept at −80 °C until HPLC analysis. Extractions were performed in triplicates. Seed extracts were treated and analysed by a modified method of Bendokas et al.68 Plant hormones were separated and quantified using high performance liquid chromatography (HPLC). Agilent 1200 series HPLC system (Agilent Technologies Inc., USA) with a diode array detector and a reversed phase column (Spherisorb ODS2, 4 × 125 mm, Waters Corporation, USA) were used. Quaternary solvent (A 50% methanol, B 50% methanol, 1.2% acetic acid, C water, D methanol) gradient elution was used as follows: initial conditions 10% B, 60% C; 10.5 min 50% B, 15.75 min 50% B; 23 min 40% B, 60% D, 30 min 40% B, 60% D, and 32 min 10% B, 60% C. Gibberellins (GA3 and GA7) and ABA were detected at a wavelength of 254 nm, while auxins (IAA and IBA), Z and SA − at 280 nm. Peak positions of analytes were identified by the retention time, peak spiking and spectral properties. Hormone concentrations were valued via a linear regression equation of standard calibration curves. The analyses were performed in triplicate and the results were presented as mean ± standard error of mean. Seedlings grown from the control and vacuum, CP and EMF treated seeds were maintained in a climatic chamber as described above and the analysis was performed after 2 weeks of cultivation. Four and three biological repeats of protein samples from shoots and roots, respectively were prepared using phenol extraction and ammonium acetate precipitation, as described previously69. Internal standards were prepared from a pooled mixture of all protein extracts. Protein separation and detection was performed using a differential gel electrophoresis procedure as described previously70. Sample aliquots of 50 µg were labeled with Cy3 and Cy5 fluorescent dyes, and the internal standard was labeled with Cy2 dye (Lumiprobe, USA). For the preparative gel, 500 µg of unlabeled internal standard was mixed with 50 µg of Cy2 labeled internal standard. Isoelectric focusing was performed on 24 cm IPG strips with a linear gradient of pH 4–7 using Ettan IPGphor (GE Healthcare, USA). Further, the proteins were separated on 1-mm thick 10–16% polyacrylamide gradient gels using Ettan DALTsix (GE Healthcare, USA). Gels were scanned using a fluorescence scanner FLA 9000 (GE Healthcare, USA). Relative protein quantification was performed using DeCyder 2-D Differential Analysis Software, v.7.0 (GE Healthcare, USA). Preparative gel was fixed in 50% methanol and 10% acetic acid. Protein spots were excised manually and subjected to protein digestion with trypsin, according to a method described previously71. Protein digests were loaded and desalted on a 100 μm × 20 mm Acclaim PepMap C18 trap column and separated on a 75 μm × 150 mm Acclaim PepMap C18 column using an Ultimate3000 RSLC system (Thermo-Scientific, USA), coupled to a Maxis G4 Q-TOF mass spectrometer detector with a Captive Spray nano-electrospray ionization source (Bruker Daltonics, Germany). Peptide identification was performed using the MASCOT server (Matrix Science, USA) against Helianthus annuus L., genome database v.1.025. Threshold value for the identification of proteins was a Mascot score of >50 and at least 2 peptides. Blast2GO software72 was used for the annotation and gene ontology analysis of the protein sequences identified with the NCBI Protein database. The obtained GO terms were summarized using the REVIGO server73, the A. thaliana database and the SimRel semantic similarity method with the level set at 0.7 value. A. thaliana homologues of the identified proteins were obtained by a search against TAIR10 gene models using the BLAST tool at the Sunflower Genome Database (https://sunflowergenome.org/blast/) and interactions were assessed using the String database with default settings74. The Biological Variation Analysis module of the DeCyder software was used to match protein spots in biological repeats across different gels and ANOVA analysis was used to identify statistically significant (p ≤ 0.01) differences in protein abundance. Additionally, a threshold value of at least a 1.5-fold difference in protein abundance was used. Since CP treatments require application of a vacuum, the effect of CP treatment was compared to control and vacuum treated experimental groups. Means of various parameters between the control and treatment groups were compared using Student’s t-tests for independent samples, as there was no reason for comparing different conditions of affected groups. The differences were considered to be statistically significant at p ≤ 0.05. The number of measured seeds or plants in the control and treatment groups varied from 17–24 (analysis of morphometric parameters) to 30 (germination tests and estimation of phytohormones content) for one replicate. Data are presented as means of 3 independent experiments ± standard error of mean.Phytohormone extraction from seeds and detection by HPLC analysis
Sunflower seedling proteome analysis using two-dimensional electrophoresis
Statistical data analysis
References
Pietruszewski, S. & Martinez, E. Magnetic field as a method of improving the quality of sowing material: a review. Int. Agrophys. 29, 377–389 (2015).
Adamovich, I. et al. The 2017 Plasma roadmap: Low temperature plasma science and technology. J. Phys. D Appl. Phys. 50, 323001 (2017).
Maffei, M. E. Magnetic field effects on plant growth, development, and evolution. Front. Plant. Sci. 5, 445 (2014).
Sera, B. & Sery, M. Non-thermal plasma treatment as a new biotechnology in relation to seeds, dry fruits, and grains. Plasma Sci. Technol. 20, 044012 (2018).
Ohta, T. Plasma in agriculture in Cold plasma in food and agriculture: fundamentals and applications (eds Misra, N. N., Schlutter, O. & Culle, P. J.) 205–222 (Elsevier, 2016).
Henselova, M., Slovakova, L., Martinka, M. & Zahoranova, A. Growth, anatomy and enzyme activity changes in maize roots induced by treatment of seeds with low-temperature plasma. Biologia. 67, 490–497 (2012).
Abyanech, E. B. et al. Influence of the electromagnetic fields on some biological characteristics of Lepidium sativum. Advan. Environ. Biol. 8, 980–984 (2014).
Sujak, A., Dziwulska-Hunek, A. & Reszczynska, E. Effect of electromagnetic stimulation on selected Fabaceae plants. Pol. J. Environm. Stud. 22, 893–898 (2013).
Jiafeng, J. et al. Effect of cold plasma treatment on seed germination and growth of wheat. Plasma Sci. Technol. 16, 54 (2014).
Tong, J. et al. Effects of atmospheric pressure air plasma pretreatment on the seed germination and early growth of Andrographis paniculata. Plasma Sci. Technol. 16, 260 (2014).
Mildaziene, V. et al. Pre-sowing seed treatment with cold plasma and electromagnetic field increases secondary metabolite content in purple coneflower (Echinacea purpurea) leaves. Plasma Process Polym. 15, 1700059 (2017).
Pauzaite, G. et al. Changes in Norway spruce germination and growth induced by pre-sowing seed treatment with cold plasma and electromagnetic field: Short-term versus long-term effects. Plasma Process Polym. 15, 1700068 (2018).
Qureshi, M. I., Qadir, S. & Zolla, L. Proteomics-based dissection of stress-responsive pathways in plants. J Plant Physiol. 164, 1239–1260 (2007).
Kosova, K. et al. Plant abiotic stress proteomics: The major factors determining alterations in cellular proteome. Front. Plant Sci. 9, 122 (2018).
Aksoy, H., Unal, F. & Ozcan, S. Genotoxic effects of electromagnetic fields from high voltage power lines on some plants. Int. J. Environ. Res. 4, 595–606 (2010).
Bagheri Abyaneh, E. Low frequency electromagnetic field induced oxidative stress in Lepidium sativum L. Iran J. Science Technol. 42, 1419–1426 (2018).
Radhakrishnan, R., Leelapriya, T. & Kumari, B. D. R. Effects of pulsed magnetic field treatment of soybean seeds on calli growth, cell damage, and biochemical changes under salt stress. Bioelectromagnetics. 33, 670–681 (2012).
Tkalec, M. et al. Effects of radiofrequency electromagnetic fields on seed germination and root meristematic cells of Allium cepa L. Mutat Res. 672, 76–81 (2009).
Mildaziene, V. et al. Response of perennial woody plants to seed treatment by electromagnetic field and low-temperature plasma. Bioelectromagnetics. 37, 536–548 (2016).
Shu, K., Liu, X. D., **e, Q. & He, Z. H. Two faces of one seed: Hormonal regulation of dormancy and germination. Mol. Plant. 9, 34–45 (2016).
Finch-Savage, W. E. & Leubner-Metzger, G. Seed dormancy and the control of germination. New Phytol. 171, 501–523 (2006).
Graeber, K. et al. Molecular mechanisms of seed dormancy. Plant Cell Environ. 35, 1769–1786 (2012).
**a, Q. et al. One way to achieve germination: common molecular mechanism induced by ethylene and after-ripening in sunflower seeds. Int. J. Mol. Sci. 19, 2464 (2018).
Baskin, C. & Baskin, J. M. Seeds: Ecology, biogeography, and evolution of dormancy and germination. 150–162 (Academic Press, 2014).
Badouin, H. et al. The sunflower genome provides insights into oil metabolism, flowering and Asterid evolution. Nature 546, 148–152 (2017).
Schwartz, R., Ting, C. S. & King, J. Whole proteome pI values correlate with subcellular localizations of proteins for organisms within the three domains of life. Genome Res. 11, 703–709 (2001).
Wu, S. et al. Multi-modality of pI distribution in whole proteome. Proteomics. 6, 449–455 (2006).
Courteille, A. et al. Thioredoxin m4 controls photosynthetic alternative electron pathways in Arabidopsis. Plant Physiol. 161, 508–520 (2013).
Ishikawa, A. et al. Molecular characterization of the ZKT gene encoding a protein with PDZ, K-Box, and TPR motifs in Arabidopsis. Biosci. Biotechnol. Biochem. 69, 972–978 (2005).
Maiwald, D. et al. Knock-out of the genes coding for the Rieske protein and the ATP-synthase delta-subunit of Arabidopsis. Effects on photosynthesis, thylakoid protein composition, and nuclear chloroplast gene expression. Plant Physiol. 133, 191–202 (2003).
Levesque-Tremblay, G., Havaux, M. & Ouellet, F. The chloroplastic lipocalin AtCHL prevents lipid peroxidation and protects Arabidopsis against oxidative stress. Plant J. 60, 691–702 (2009).
Chang, I. F., Szick-Miranda, K., Pan, S. & Bailey-Serres, J. Proteomic characterization of evolutionarily conserved and variable proteins of Arabidopsis cytosolic ribosomes. Plant Physiol. 137, 848–862 (2005).
Rajamaki, M. L. et al. Differential requirement of the ribosomal protein S6 and ribosomal protein S6 kinase for plant-virus accumulation and interaction of S6 kinase with potyviral VPg. Mol. Plant Microbe Interact. 30, 374–384 (2017).
Holtgrefe, S. et al. Regulation of plant cytosolic glyceraldehyde 3-phosphate dehydrogenase isoforms by thiol modifications. Physiol. Plant. 133, 211–228 (2008).
Vescovi, M. et al. Nuclear accumulation of cytosolic glyceraldehyde-3-phosphate dehydrogenase in cadmium-stressed Arabidopsis roots. Plant Physiol. 162, 333–346 (2013).
Lu, W. et al. Identification and characterization of fructose 1,6-bisphosphate aldolase genes in Arabidopsis reveal a gene family with diverse responses to abiotic stresses. Gene. 503, 65–74 (2012).
Qian, W. et al. Molecular and functional analysis of phosphomannomutase (PMM) from higher plants and genetic evidence for the involvement of PMM in ascorbic acid biosynthesis in Arabidopsis and Nicotiana benthamiana. Plant J. 49, 399–413 (2007).
Carroll, A. J., Heazlewood, J. L., Ito, J. & Millar, A. H. Analysis of the Arabidopsis cytosolic ribosome proteome provides detailed insights into its components and their post-translational modification. Mol. Cell. Proteomics. 7, 347–369 (2008).
Laxa, M., Konig, J., Dietz, K. J. & Kandlbinder, A. Role of the cysteine residues in Arabidopsis thaliana cyclophilin CYP20-3 in peptidyl-prolyl cis-trans isomerase and redox-related functions. Biochem. J. 401, 287–297 (2007).
Primm, T. P., Walker, K. W. & Gilbert, H. F. Facilitated protein aggregation. Effects of calcium on the chaperone and anti-chaperone activity of protein disulfide-isomerase. J. Biol. Chem. 271, 33664–33669 (1996).
Lu, D. P. & Christopher, D. A. Endoplasmic reticulum stress activates the expression of a sub-group of protein disulfide isomerase genes and AtbZIP60 modulates the response in Arabidopsis thaliana. Mol. Genet. Genomics. 280, 199–210 (2008).
Ishikawa, K. Plasma diagnostics in Cold plasma in food and agriculture: fundamentals and applications (eds Misra, N. N., Schluter, O. & Culle, P. J.) 117–142 (Elsevier, 2016).
Vashisth, A. & Nagarajan, S. Effect on germination and early growth characteristics in sunflower (Helianthus annuus) seeds exposed to static magnetic field. J. Plant Physiol. 167, 149–156 (2010).
Diaz-Vivancos, P., Barba-Espin, G. & Hernandez, J. A. Elucidating hormonal/ROS networks during seed germination: insights and perspectives. Plant Cell Rep. 32, 1491–1502 (2013).
Forcella, F., Benech Arnold, R. L., Sanchez, R. & Ghersa, C. M. Modeling seedling emergence. Field Crop. Res. 67, 123–139 (2000).
Vian, A., Davies, E., Gendraud, M. & Bonnet, P. Plant responses to high frequency electromagnetic fields. Biomed. Res. Int. 2016, 1830262 (2016).
Roux, D. et al. High frequency (900 MHz) low amplitude (5 V m-1) electromagnetic field: a genuine environmental stimulus that affects transcription, translation, calcium and energy charge in tomato. Planta. 227, 883–891 (2008).
Chandel, S. et al. Exposure to 2100 MHz electromagnetic field radiations induces reactive oxygen species generation in Allium cepa roots. J. Microsc. Ultrastruct. 5, 225–229 (2017).
Vian, A. et al. Microwave irradiation affects gene expression in plants. Plant Signal Behav. 1, 67–70 (2006).
Tang, C. et al. Electromagnetic radiation disturbed the photosynthesis of Microcystis aeruginosa at the proteomics level. Sci. Rep. 8, 479 (2018).
Beaubois, E. et al. Intercellular communication in plants: evidence for two rapidly transmitted systemic signals generated in response to electromagnetic field stimulation in tomato. Plant Cell Environ. 30, 834–844 (2007).
Afroz, A., Ali, G. M., Mir, A. & Komatsu, S. Application of proteomics to investigate stress-induced proteins for improvement in crop protection. Plant Cell Reports. 30, 745–763 (2011).
Paparella, S. et al. Seed priming: state of the art and new perspectives. Plant Cell Rep. 34, 1–13 (2015).
Misas-Villamil, J. C., van der Hoorn, R. A. & Doehlemann, G. Papain-like cysteine proteases as hubs in plant immunity. New Phytol. 212, 902–907 (2016).
Hideg, E., Jansen, M. A. & Strid, A. UV-B exposure, ROS, and stress: inseparable companions or loosely linked associates? Trends Plant Sci. 18, 107–115 (2013).
Le, M. B. et al. Tobacco chloroplast transformants expressing genes encoding dehydroascorbate reductase, glutathione reductase, and glutathione-S-transferase, exhibit altered anti-oxidant metabolism and improved abiotic stress tolerance. Plant Biotechnol. J. 9, 661–673 (2011).
George, S., Venkataraman, G. & Parida, A. A chloroplast-localized and auxin-induced glutathione S-transferase from phreatophyte Prosopis juliflora confer drought tolerance on tobacco. J. Plant Physiol. 167, 311–318 (2010).
Boeckx, T. et al. Polyphenol oxidase-mediated protection against oxidative stress is not associated with enhanced photosynthetic efficiency. Ann. Bot. 116, 529–540 (2015).
Kohzuma, K. et al. The role of light-dark regulation of the chloroplast ATP synthase. Front. Plant Sci. 8, 1248 (2017).
Rius, S. P., Casati, P., Iglesias, A. A. & Gomez-Casati, D. F. Characterization of Arabidopsis lines deficient in GAPC-1, a cytosolic NAD-dependent glyceraldehyde-3-phosphate dehydrogenase. Plant Physiol. 148, 1655–1667 (2008).
Carmo-Silva, A. E. & Salvucci, M. E. The regulatory properties of Rubisco activase differ among species and affect photosynthetic induction during light transitions. Plant Physiol. 161, 1645–1655 (2013).
Berkowitz, O., Jost, R., Pollmann, S. & Masle, J. Characterization of TCTP, the translationally controlled tumor protein, from Arabidopsis thaliana. Plant Cell. 20, 3430–3447 (2008).
Ermolayev, V., Weschke, W. & Manteuffel, R. Comparison of Al-induced gene expression in sensitive and tolerant soybean cultivars. J. Exp. Bot. 54, 2745–2756 (2003).
Vincent, D. et al. Proteomic analysis reveals differences between Vitis vinifera L. cv. Chardonnay and cv. Cabernet Sauvignon and their responses to water deficit and salinity. J. Exp. Bot. 58, 1873–1892 (2007).
Zhang, J. et al. Role of the translationally controlled tumor protein in DNA damage sensing and repair. Proc. Natl. Acad. Sci. USA 109, E926–E933 (2012).
Richards, F. J. A flexible growth function for empirical use. J. Exp. Bot. 10, 290–300 (1959).
Hara, Y. Calculation of population parameters using Richards function and application of indices of growth and seed vigor to rice plants. Plant Prod. Sci. 2, 129–135 (1999).
Bendokas, V. et al. Predicting apple tree (Malus×domestica Borkh.) canopy architecture: phytohormone balance in juvenile hybrids. Zemdirbyste-Agriculture. 101, 327–332 (2014).
Isaacson, T. et al. Sample extraction techniques for enhanced proteomic analysis of plant tissues. Nat. Protoc. 1, 769–774 (2006).
Tamosiune, I. et al. Endophytic Bacillus and Pseudomonas spp. modulate apple shoot growth, cellular redox balance, and protein expression under in vitro conditions. Front. Plant Sci. 9, 889 (2018).
Shevchenko, A. et al. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 1, 2856–2860 (2006).
Conesa, A. et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 21, 3674–3676 (2005).
Pesquita, C. et al. Semantic similarity in biomedical ontologies. PLOS Comp. Biol. 5, e1000443 (2009).
Szklarczyk, D. et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 43, D447–D452 (2015).
Acknowledgements
This research was funded by the Lithuanian Research Council grant No. S-MIP-17/53.
Author information
Authors and Affiliations
Contributions
V.M., D.B. and I.F. conceptualized and designed the experiments; V.A., G.P., R.Z., G.P., Z.N., V.L., P.H., I.T. acquired, analysed and interpreted the data; D.B. and V.M. drafted the manuscript; V.A., G.P., R.Z., G.P., Z.N., V.L., P.H., I.T. contributed to critical revision of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mildažienė, V., Aleknavičiūtė, V., Žūkienė, R. et al. Treatment of Common Sunflower (Helianthus annus L.) Seeds with Radio-frequency Electromagnetic Field and Cold Plasma Induces Changes in Seed Phytohormone Balance, Seedling Development and Leaf Protein Expression. Sci Rep 9, 6437 (2019). https://doi.org/10.1038/s41598-019-42893-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-42893-5
- Springer Nature Limited
This article is cited by
-
Application of cold argon plasma on germination, root length, and decontamination of soybean cultivars
BMC Plant Biology (2024)
-
Cold atmospheric plasma enhances morphological and biochemical attributes of tomato seedlings
BMC Plant Biology (2024)
-
Non-thermal plasma enhances rice seed germination, seedling development, and root growth under low-temperature stress
Applied Biological Chemistry (2024)
-
Heavy fuel oil-contaminated soil remediation by individual and bioaugmentation-assisted phytoremediation with Medicago sativa and with cold plasma-treated M. sativa
Environmental Science and Pollution Research (2024)
-
Review on the impact of cell phone radiation effects on green plants
Environmental Monitoring and Assessment (2024)