

Abstract
Epstein–Barr virus (EBV) can infect both B cells and epithelial cells (ECs), causing diseases such as mononucleosis and cancer. It enters ECs via Ephrin receptor A2 (EphA2). The function of interferon-induced transmembrane protein-1 (IFITM1) in EBV infection of ECs remains elusive. Here we report that IFITM1 inhibits EphA2-mediated EBV entry into ECs. RNA-sequencing and clinical sample analysis show reduced IFITM1 in EBV-positive ECs and a negative correlation between IFITM1 level and EBV copy number. IFITM1 depletion increases EBV infection and vice versa. Exogenous soluble IFITM1 effectively prevents EBV infection in vitro and in vivo. Furthermore, three-dimensional structure prediction and site-directed mutagenesis demonstrate that IFITM1 interacts with EphA2 via its two specific residues, competitively blocking EphA2 binding to EBV glycoproteins. Finally, YTHDF3, an m6A reader, suppresses IFITM1 via degradation-related DEAD-box protein 5 (DDX5). Thus, this study underscores IFITM1’s crucial role in blocking EphA2-mediated EBV entry into ECs, indicating its potential in preventing EBV infection.
Similar content being viewed by others


Main
Epstein–Barr virus (EBV) is the first oncogenic herpesvirus that targets epithelial cells (ECs) and B lymphocytes1. It infects ~95% of the population worldwide2 and is associated with a spectrum of severe diseases, especially mononucleosis and various forms of cancer, including nasopharyngeal carcinoma (NPC), gastric cancer, colorectal cancer and B cell lymphoma3,4,5,6. Effective prevention of EBV infection is a crucial public health issue.
During EBV infection, viral glycoproteins collaborate with host envelope proteins to enable membrane fusion and EBV entry into target host cells7. Core to this process is the glycoprotein B (gB) homotrimer and the glycoprotein H/L (gH/gL) heterodimer, facilitating fusion in ECs and B lymphocytes8,9,10,11. Over the past several decades, extensive research has been conducted on B cell receptors involved in EBV entry11,12,13,14,15. As to ECs, integrins were initially identified to be the primary receptors for EBV entry16,17, but they were later confirmed to be accessory receptors18,19. More recently, following the discovery of Neuropilin 1 (NRP1) and Non-muscle myosin IIA (NMHC-IIA, also known as MYH9)20,47,Western blotting Cell lysates were collected in RIPA buffer (Thermo Fisher, 89900) or cell lysis buffer and supplemented with a commercial protease inhibitor cocktail (Thermo Fisher, 78442) before use. Total protein was obtained by centrifugation and the protein concentration was determined by a Pierce BCA Protein Assay kit (Thermo Fisher, 23225). An equal amount of total protein was fractionated by 8~12% SDS–PAGE and transferred onto a 0.22 μm PVDF membrane (Millipore, ISEQ 00010). Membranes were immunoblotted with the indicated primary antibodies and then incubated with HRP-conjugated secondary antibody. Primary antibodies were as follows: mouse anti-IFITM1 (60074-1, Proteintech, 5B5E2, KD/KO validated, 1/1,000), rabbit anti-EphA2 (6997, CST, 1/1,000), rabbit anti-EGFR (18986-1, Proteintech, KD/KO validated, 1/1,000), rabbit anti-DDX5 (ab126730, Abcam, EPR7239, KD/KO validated, 1/1,000), rabbit anti-DDX6 (ab174277, Abcam, EPR12146, KD/KO validated, 1/1,000), rabbit anti-DDX17 (ab180190, Abcam, EPR13807(B), KD/KO validated, 1/1,000), rabbit anti-Actin (YT0096, ImmunoWay, 1/5,000) and rabbit anti-GAPDH (ab9485, Abcam, 1/5,000). Secondary antibodies were HRP-conjugated goat anti-rabbit IgG (SA00001-2, Proteintech, 1/5,000) and HRP-conjugated goat anti-mouse IgG (SA00001-1, Proteintech, 1/5,000). Blots were incubated with ECL substrate (BioRad, 1705061) and imaged with the ECL detection system (ChemiDoc, BioRad). To determine EBV infection rates, 1 × 106 cells incubated with EBV were collected and washed using 1×PBS containing 0.2% bovine serum albumin (BSA). Cells were then resuspended in 300 μl of 1×PBS containing 0.2% BSA. Data were acquired using an LE-SA3800 Spectral Analyzer (Sony) and FlowJo software was used for analysis. To detect the co-localization of IFITM1 and EphA2 or IFITM1 and EGFR, cells were plated on glass-bottom cell culture plates (801006, NEST) for 24 h. After brief washing twice with 1×PBS, cells were fixed with 4% paraformaldehyde (BioSharp) and permeabilized with 0.2% Triton X-100 (Thermo Fisher, 89900). Plates were washed gently and blocked with 3% normal goat serum (C0265, Beyotime). Subsequently, the cells were incubated with the primary antibody pairs overnight, followed by incubation with respective fluorophore-conjugated secondary antibodies (Alexa Fluor 488 goat anti-mouse IgG H&L and Alexa Fluor 647 goat anti-rabbit IgG H&L; 150113, 150079, Abcam) for 1 h and counterstained with DAPI (C1005, Beyotime) for 10 min at room temperature. To avoid false-positive cross-reactivity, primary antibody pairs were chosen from different species (mouse anti-IFITM1 and rabbit anti-EphA2, mouse anti-IFITM1 and rabbit anti-EGFR). Fluorescence images were recorded using the High Content Analysis System (CQ1, Yokogawa). Immunoprecipitation was performed using an IP kit (abs955, Absin) following supplier instructions. Briefly, whole-cell proteins were isolated in lysis buffer and the lysate was centrifuged; the supernatant was subsequently collected. To lower the background, 500 µl of supernatant containing ~500–1,000 µg protein was incubated with 5 µl of protein A and 5 µl of protein G for 1 h at 4 °C. Total protein lysates that removed unspecific binding proteins were obtained after centrifugation. For protein binding, 1–5 µg of the corresponding antibodies (to IFITM1 or EphA2, as mentioned above) and homologous IgG (eBiosciences) was added to pre-cleaned protein lysates. Immediately afterwards, samples were incubated at 4 °C overnight under gentle rotation. To precipitate the target proteins, 5 µl of protein A and 5 µl of protein G were added to bind the antigen–antibody complexes; the reaction was maintained at 4 °C for 3 h. After gently washing three times with wash buffer, the unbound proteins were removed and the pellet (agarose–antibody–antigen complex) was resolved in 20–40 µl of SDS-loading buffer for further western blotting with the indicated antibodies. For exogenous co-IP of IFITM1, EphA2, gH/gL and gB, HEK293 cells were transfected with the corresponding combination of Myc-gH/gL, Myc-gB, pSMCV-IFITM1, pSMCV-EphA2 and empty vector (see figure legends for details). IFITM1, gB, gH/gL and EphA2 DNAs were constructed into pGEX6p-1-GST/His vector and recombinant fusion proteins were purified in the Rosetta strain of E. coli. Briefly, after the constructed plasmids were cloned into pET28a vector, E. coli Rosetta star pLysS cells were transformed with these plasmids. Colonies were inoculated into 25 ml of Luria-Bertani (LB) media containing ampicillin (100 μg ml−1) and grown for 16 h at 37 °C; then 25 ml cultures were transferred into 500 ml of fresh LB medium and grown at 37 °C for 4 h, adding isopropyl-β-d-thiogalactopyranoside (IPTG, 1 mM) to continue cultivation for 24 h at 16 °C. Bacterial cells were collected by centrifugation and 6×His- or GST-tagged target proteins were purified through His-tag or GST-tag affinity chromatography (Qiagen) and desalted using an Amersham column. First, 96-well microtitre plates were coated with 200 ng of GST-EphA2 overnight at 4 °C, followed by blocking with 5% BSA in 1×PBS for 2 h at room temperature. Then, gradient concentrations of 6×His-IFITM1 and 6×His-gB or 6×His-gH/gL proteins were added and incubated for 2 h at room temperature. After washing with 1×BST (0.05% Tween-20 in 1×PBS), the plate was incubated with a rabbit anti-GST antibody or mouse anti-His antibody (1:4,000 dilution) for 2 h at room temperature. The plate was then washed and incubated at 37 °C for 1 h with HRP-conjugated goat anti-rabbit antibody or HRP-conjugated goat anti-mouse antibody. After adding the substrate tetramethyl-benzidine, the absorbance was measured at 450 nm using the Nanodrop One spectrophotometer (Thermo Fisher). Two main methods, I-TASSER from the University of Michigan and SWISS-model from the European Center for Bioinformatics, were used to select the model consistent with the local structure published in ref. 41. After selecting a single predicted structure, the 3D structures of the complex formed by IFITM1, EphA2 and gH/gL were calculated. The top 100 models were selected from the 2,000 rigid docking models optimized by geometry and electrostatics. On the basis of the accurately predicted binding sites and side-chain flexibility of the binding interface amino acids, 200,000 models were generated, and the 10 best models were selected after clustering and scoring. After comparing with the model published in ref. 41, the first model was selected for subsequent calculations. IFITM1 mutant primers were designed by the homologous recombination method, and IFITM1 wild-type plasmids were amplified by PCR using mutant-specific primers and the KOD-Plus-Neo amplification kit (Toyobo). After mutation, the PCR products were recombined at 37 °C for 30 min. The top 10 competent cells were added to the recombinant products, and the transformation mixture was evenly spread onto an LB plate containing ampicillin and incubated overnight at 37 °C. Single clones were selected for Sanger sequencing. m6A RNA immunoprecipitation was performed according to the Magna MeRIP m6A kit (17-10499, Merck) instructions. Total RNA was extracted by the Trizol method and then interrupted to form fragments of ~100 nt. Beads with anti-m6A antibodies were added and incubated at 4 °C for 8 h to form m6A RNA–antibody–magnetic bead complexes, which were adsorbed using a magnetic rack and washed several times to remove impurities. The m6A RNA was eluted by competitive binding and submitted for sequence analysis by RT−qPCR. RNA immunoprecipitation (RIP) was performed according to the instructions of the Magna RIP RNA-Binding Protein Immunoprecipitation kit (Merck). Cells lysed by lysis buffer were incubated with anti-YTHDF3 antibody and magnetic beads at 4 °C for 8 h. The magnetic bead–antibody–target protein–RNA complex was adsorbed with a magnetic rack and cleaned 5 times with wash buffer to remove impurities. RNA was extracted by Trizol, and the purified RNA was analysed by RT−qPCR or submitted for sequence analysis. Wide-type full-length YTHDF3 and its double m6A binding site defective mutants, designated YTHDF3WT and YTHDF3DM, were constructed into a tandem affinity purification vector pLVpuro-TAP (SBP-3HA). Then, three stably expressing TAP-Vector, TAP-YTHDF3WT OE and TAP-YTHDF3DM OE HK1 cell lines were subjected to 2 µg ml−1 puromycin selection for a week. The TAP affinities of the exogenously expressed TAP-Vector, -YTHDF3WT and -YTHDF3DM cells were extracted from the three cell lysates by excess streptavidin resin; then the endogenously expressed YTHDF3 partners were immunoprecipitated by YTHDF3 antibodies coupled to protein A/G beads. Finally, the YTHDF3 exogenously expressed TAP affinities and endogenously expressed immunoprecipitates were separately identified by mass spectrometry. To assess IFITM1 RNA stability, cells were incubated with actinomycin (ActD) to terminate transcription. Briefly, HK1 cells were incubated with ActD (5 μg ml−1) for 0, 30 and 60 min, and collected. Total RNA was extracted and the IFITM1 RNA expression was determined by RT−qPCR. Data are presented as mean ± s.e.m. derived from at least three independent experiments. All statistical analyses, including Pearson’s correlation coefficients (r), t-tests and so on were two-tailed and executed using GraphPad Prism 8. Although the data conformed to the assumptions of the statistical tests utilized, normality of data distribution was presumed but was not rigorously tested. In addition, the collection and analysis of data were carried out in a blinded manner relative to experimental conditions and no animal or data point was excluded from the analysis for any reason. Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.Flow cytometry
Indirect immunofluorescence assay
Co-immunoprecipitation
Protein expression and purification
Competitive binding assays
3D structure and spatial binding site prediction
Site-specific mutation
MeRIP-seq and MeRIP−qPCR
RIP-seq and RIP−qPCR
Tandem affinity purification pull-down and mass spectrometry
RNA stability assay
Statistical analyses
Reporting summary
Data availability
The datasets that support the findings of this study are available within the paper. Transcriptomic datasets generated in this study can be found on the NCBI Sequence Read Archive (SRA) under BioProject PRJNA946546 and PRJNA976759. The prediction of the protein–protein interactions was performed using the STRING database (http://string-db.org/). The prediction of the three-dimensional structure of IFITM1, EphA2 and gH/gL was performed using I-TASSER (https://zhanglab.ccmb.med.umich.edu/I-TASSER/) and SWISS-model (https://swissmodel.expasy.org/). The raw sequencing data of the MeRIP-seq can be found under PRJNA997768. Mass spectrometry datasets can be accessed via Harvard Dataverse at https://doi.org/10.7910/DVN/QHCEZI (ref. 57). Source data are provided with this paper.
References
Djaoud, Z. et al. Two alternate strategies for innate immunity to Epstein-Barr virus: one using NK cells and the other NK cells and γδ T cells. J. Exp. Med. 214, 1827–1841 (2017).
Young, L. S., Yap, L. F. & Murray, P. G. Epstein–Barr virus: more than 50 years old and still providing surprises. Nat. Rev. Cancer 16, 789–802 (2016).
Dawson, C. W., Port, R. J. & Young, L. S. The role of the EBV-encoded latent membrane proteins LMP1 and LMP2 in the pathogenesis of nasopharyngeal carcinoma (NPC). Semin. Cancer Biol. 22, 144–153 (2012).
Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 513, 202–209 (2014).
Murray, P. G. & Young, L. S. An etiological role for the Epstein-Barr virus in the pathogenesis of classical Hodgkin lymphoma. Blood 134, 591–596 (2019).
Farrell, P. J. Epstein–Barr virus and cancer. Annu. Rev. Pathol. 14, 29–53 (2019).
Connolly, S. A., Jardetzky, T. S. & Longnecker, R. The structural basis of herpesvirus entry. Nat. Rev. Microbiol. 19, 110–121 (2021).
Chen, J. & Longnecker, R. Epithelial cell infection by Epstein–Barr virus. FEMS Microbiol. Rev. 43, 674–683 (2019).
Krummenacher, C., Carfi, A., Eisenberg, R. J. & Cohen, G. H. Entry of herpesviruses into cells: the enigma variations. Adv. Exp. Med. Biol. 790, 178–195 (2013).
Chen, J., Schaller, S., Jardetzky, T. S. & Longnecker, R. Epstein–Barr virus gH/gL and Kaposi’s sarcoma-associated herpesvirus gH/gL bind to different sites on EphA2 to trigger fusion. J. Virol. 94, e01454-20 (2020).
Chesnokova, L. S. & Hutt-Fletcher, L. M. Epstein-Barr virus infection mechanisms. Chin. J. Cancer 33, 545–548 (2014).
Valencia, S. M. & Hutt-Fletcher, L. M. Important but differential roles for actin in trafficking of Epstein-Barr virus in B cells and epithelial cells. J. Virol. 86, 2–10 (2012).
Zhu, Q. Y. et al. Association between antibody responses to Epstein-Barr virus glycoproteins, neutralization of infectivity, and the risk of nasopharyngeal carcinoma. mSphere 5, e00901–e00920 (2020).
Fingeroth, J. D. et al. Epstein-Barr virus receptor of human B lymphocytes is the C3d receptor CR2. Proc. Natl Acad. Sci. USA 81, 4510–4514 (1984).
Nemerow, G. R., Mold, C., Schwend, V. K., Tollefson, V. & Cooper, N. R. Identification of gp350 as the viral glycoprotein mediating attachment of Epstein-Barr virus (EBV) to the EBV/C3d receptor of B cells: sequence homology of gp350 and C3 complement fragment C3d. J. Virol. 61, 1416–1420 (1987).
Campadelli-Fiume, G., Collins-McMillen, D., Gianni, T. & Yurochko, A. D. Integrins as herpesvirus receptors and mediators of the host signalosome. Annu. Rev. Virol. 3, 215–236 (2016).
Hutt-Fletcher, L. M. & Chesnokova, L. S. Integrins as triggers of Epstein-Barr virus fusion and epithelial cell infection. Virulence 1, 395–398 (2010).
Chen, J. et al. Ephrin receptor A2 is a functional entry receptor for Epstein–Barr virus. Nat. Microbiol. 3, 172–180 (2018).
Zhang, H. et al. Ephrin receptor A2 is an epithelial cell receptor for Epstein–Barr virus entry. Nat. Microbiol. 3, 1–8 (2018).
Wang, H. B. et al. Neuropilin 1 is an entry factor that promotes EBV infection of nasopharyngeal epithelial cells. Nat. Commun. 6, 6240 (2015).
**ong, D. et al. Nonmuscle myosin heavy chain IIA mediates Epstein–Barr virus infection of nasopharyngeal epithelial cells. Proc. Natl Acad. Sci. USA 112, 11036–11041 (2015).
Ziegler, P. et al. A primary nasopharyngeal three-dimensional air-liquid interface cell culture model of the pseudostratified epithelium reveals differential donor- and cell type-specific susceptibility to Epstein-Barr virus infection. PLoS Pathog. 17, e1009041 (2021).
Bailey, C. C., Zhong, G., Huang, I. C. & Farzan, M. IFITM-family proteins: the cell’s first line of antiviral defense. Annu. Rev. Virol. 1, 261–283 (2014).
Perreira, J. M., Chin, C. R., Feeley, E. M. & Brass, A. L. IFITMs restrict the replication of multiple pathogenic viruses. J. Mol. Biol. 425, 4937–4955 (2013).
Ishikawa, H., Ma, Z. & Barber, G. N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461, 788–792 (2009).
Diamond, M. S. & Farzan, M. The broad-spectrum antiviral functions of IFIT and IFITM proteins. Nat. Rev. Immunol. 13, 46–57 (2013).
Smith, S., Weston, S., Kellam, P. & Marsh, M. IFITM proteins — cellular inhibitors of viral entry. Curr. Opin. Virol. 4, 71–77 (2014).
Wilkins, C. et al. IFITM1 is a tight junction protein that inhibits hepatitis C virus entry. Hepatology 57, 461–469 (2013).
Shi, G. et al. Opposing activities of IFITM proteins in SARS‐CoV‐2 infection. EMBO J. 40, e1065011 (2020).
Smith, S. E. et al. Interferon-induced transmembrane protein 1 restricts replication of viruses that enter cells via the plasma membrane. J. Virol. 93, e02003–e02018 (2019).
Hussein, H. A. M. & Akula, S. M. miRNA-36 inhibits KSHV, EBV, HSV-2 infection of cells via stifling expression of interferon induced transmembrane protein 1 (IFITM1). Sci. Rep. 7, 17972 (2017).
Hussein, H. A. M., Briestenska, K., Mistrikova, J. & Akula, S. M. IFITM1 expression is crucial to gammaherpesvirus infection, in vivo. Sci. Rep. 8, 14105 (2018).
Liu, Y. et al. N6-methyladenosine-mediated gene regulation and therapeutic implications. Trends Mol. Med. 29, 454–467 (2023).
Zhang, Y. et al. RNA-binding protein YTHDF3 suppresses interferon-dependent antiviral responses by promoting FOXO3 translation. Proc. Natl Acad. Sci. USA 116, 976–981 (2019).
Hu, L. et al. Comprehensive profiling of EBV gene expression in nasopharyngeal carcinoma through paired-end transcriptome sequencing. Front. Med. 10, 61–75 (2016).
Zhang, P. F. et al. Nasopharyngeal brushing: a convenient and feasible sampling method for nucleic acid-based nasopharyngeal carcinoma research. Cancer Commun. 38, 8 (2018).
Shao, J. Y. et al. Comparison of Epstein-Barr virus DNA level in plasma, peripheral blood cell and tumor tissue in nasopharyngeal carcinoma. Anticancer Res. 24, 4059–4066 (2004).
Dickens, P., Srivastava, G., Loke, S. L., Chan, C. W. & Liu, Y. T. Epstein-Barr virus DNA in nasopharyngeal carcinomas from Chinese patients in Hong Kong. J. Clin. Pathol. 45, 396–397 (1992).
Wrensch, F. et al. Interferon-induced transmembrane proteins mediate viral evasion in acute and chronic hepatitis C virus infection. Hepatology 70, 1506–1520 (2019).
Zhao, X., Li, J., Winkler, C. A., An, P. & Guo, J. T. IFITM genes, variants, and their roles in the control and pathogenesis of viral infections. Front. Microbiol. 9, 3228 (2018).
Su, C. et al. Molecular basis of EphA2 recognition by gHgL from gammaherpesviruses. Nat. Commun. 11, 5964 (2020).
Zaccara, S. & Jaffrey, S. R. A unified model for the function of YTHDF proteins in regulating m6A-modified mRNA. Cell 181, 1582–1595.e18 (2020).
Lupberger, J. et al. EGFR and EphA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat. Med. 17, 589–595 (2011).
Swidergall, M. et al. Activation of EphA2-EGFR signaling in oral epithelial cells by Candida albicans virulence factors. PLoS Pathog. 17, e1009221 (2021).
Winchester, S, John, S., Jabbar, K & John, I. Clinical efficacy of nitric oxide nasal spray (NONS) for the treatment of mild COVID-19 infection. J. Infect. 83, 237–279 (2021).
Beeraka, N. M. et al. Strategies for targeting SARS CoV-2: small molecule inhibitors—the current status. Front. Immunol. 11, 552925 (2020).
Zheng, Q., Hou, J., Zhou, Y., Li, Z. & Cao, X. The RNA helicase DDX46 inhibits innate immunity by entrap** m(6)A-demethylated antiviral transcripts in the nucleus. Nat. Immunol. 18, 1094–1103 (2017).
**a, T. L. et al. N(6)-methyladenosine-binding protein YTHDF1 suppresses EBV replication and promotes EBV RNA decay. EMBO Rep. 22, e50128 (2021).
Tsao, S. W., Tsang, C. M. & Lo, K. W. Epstein–Barr virus infection and nasopharyngeal carcinoma. Phil. Trans. R. Soc. B 372, 20160270 (2017).
Bu, G.-L., **e, C., Kang, Y.-F., Zeng, M.-S. & Sun, C. How EBV infects: the tropism and underlying molecular mechanism for viral infection. Viruses 14, 2372 (2022).
Cifuentes-Munoz, N., El Najjar, F. & Dutch, R. E. Virus assembly and exit pathways. in Advances in Virus Research Vol. 108. (eds Kielian, M. et al.) 85–125 (Academic Press, 2020).
Tsao, S. W. et al. Establishment of two immortalized nasopharyngeal epithelial cell lines using SV40 large T and HPV16E6/E7 viral oncogenes. Biochim. Biophys. Acta 1590, 150–158 (2002).
Li, H. M. et al. Molecular and cytogenetic changes involved in the immortalization of nasopharyngeal epithelial cells by telomerase. Int. J. Cancer 119, 1567–1576 (2006).
Tsang, C. M. et al. Epstein-Barr virus infection in immortalized nasopharyngeal epithelial cells: regulation of infection and phenotypic characterization. Int. J. Cancer 127, 1570–1583 (2010).
Bo, H. & Gong, Z. et al. Upregulated long non-coding RNA AFAP1-AS1 expression is associated with progression and poor prognosis of nasopharyngeal carcinoma. Oncotarget 6, 20404–20418 (2015).
Sari, I. N. et al. Interferon-induced transmembrane protein 1 (IFITM1) is required for the progression of colorectal cancer. Oncotarget 7, 86039–86050 (2016).
Ding, T. External and endogenous mass spectrometry of YTHDF3, V1. Harvard Dataverse https://doi.org/10.7910/DVN/QHCEZI (2024).
Acknowledgements
This work was supported by the National Nature Science Foundation of China (No. 81902788 to Y.Y. and 82273134 to X. Li), the Shenzhen Key Laboratory of Viral Oncology (No. ZDSYS201707311140430 to X. Li), the Natural Science Foundation of Guangdong Province (No. 2020A1515010902 to Y.Y. and 2020A1515010008 to C. Li), the Science and Technology Planning Project of Shenzhen, China (No. JCYJ20210324130801004 to Y.Y.; JCYJ20190814111213287 to C. Li; and JCYJ20210324134602008 and JCYJ20230807142-311024 to T.G.), and the Research Foundation of Shenzhen Hospital of Southern Medical University (Nos. UN-KJ-KY200024-YYPT, PT2020GZR07, 22H3ATF05 and UN-KC-BHKY202205 to Y.Y.). The funders had no role in the conceptualization, design, data collection, analysis, decision to publish or preparation of the manuscript. We thank M.-S. Zeng and H. Zhang (Sun Yat-sen University Cancer Center) for gifting the EphA2, gB and gH/gL overexpression vectors; and S.-W. Tsao’s group (The University of Hong Kong, Pokfulam, Hong Kong SAR, China) for providing the cell lines specified above.
Author information
Contributions
X. Li, Q. Zheng, X.G. and D.W. conceived the study. Y.Y., T.D., Y.C., X. Lou, T.G., C. Li, Y.Z. and J.Z. designed, performed and interpreted the experiments. C. Liu, Y.Y., Y.C., C.N. and X. Zheng supervised the molecular virology experiments. Y.Y., T.D., Y.C. and X. Lou designed and performed the molecular virology experiments. T.W., Z.J., S.Y. and Q. Zhou collected, processed and analysed the clinical samples. All authors interpreted the data. X. Li, Y.Y., T.D., Y.C. and M.Z. wrote the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Microbiology thanks Yasuko Mori, Zhijian Qian and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 IFITM1 negatively correlates with EBV infection in ECs.
(a) Heatmap of the mRNA expression levels (FPKM) of IFITMs and the reported epithelial cell receptors in B cells, EBV-negative (EBV.N) cells and EBV-positive (EBV.P) cells, analyzed by RNA sequencing. Data were plotted as the log10 of transformed cells for ease of comparison and visualization. (b) Volcano plot of the differentially expressed genes in the EBV-negative (EBVN) and EBV-positive (EBVP) groups, with upregulated genes shown in red, downregulated genes in green, and no differentially expressed genes in blue. The horizontal-axis represents −log2 (fold-change), and vertical-axis represents −log10 (p-value). n = 2 independent experiments; *p < 0.05, two-tailed t-test. (c) The Western blot analysis of tissue lysates from three NPC and three nasopharynx (NP) samples for IFITM1 protein expression using actin as a loading control, n = 3 pairs. (d) The mRNA level of IFITM1 in nine NPC and nine NP samples was assayed by RT-qPCR, and B2M was used as a reference gene, n = 9 pairs. (e) The EBV copy numbers in nine NPC and nine NP samples were also assayed by TaqMan-qPCR using an EBV detection kit, n = 9 pairs. Data are presented as mean values ± s.e.m., n = 9 pairs, *p < 0.05, **p < 0.01, ***p < 0.001 (d, e), two-tailed t-test. (f) A correlation analysis was performed between the relative IFITM1 mRNA expression and EBV copy numbers in nine NPC samples (horizontal axis: relative IFITM1 mRNA levels; vertical axis: relative EBV copy numbers). The black dot denotes scattered samples. Data are presented as mean values ± s.e.m., n = 9, r = −0.87, p = 0.0021 (f), two-tailed t-test.
Extended Data Fig. 2 Knockdown IFITM1 facilitates EBV infection in ECs.
(a) NP69, HK1, and HEK293 cell lines were collected, and immunoblotting assayed expression of IFITM1 at the protein level. (b-c) Knockdown of IFITM1 in NP69, HK1, and HEK293 cells by infection with lentiviruses encoding shIFITM1 (1-shIFITM1 and 2-shIFITM1). shLacZ-transfected cells were included as a control. (b) Knockdown efficiency of IFITM1 at the mRNA level. Bars show the relative IFITM1 mRNA level detected by RT-qPCR and data are presented relative to B2M (2˗ΔCt). (c) Knockdown efficiency of IFITM1 at the protein level. (d) HEK293 and HK-1 cell lines were incubated with cell-free EBV-GFP for 3 hours, and EBV copy numbers were then measured by TaqMan-qPCR. (e-h) After 72 hours, flow cytometric analyses were performed to show the percentage of EBV-GFP-positive cells. Representative images of cells infected with EBV-GFP were recorded for HEK293 cells (i). Data are presented as mean values ± s.e.m., n ≥ 3 independent experiments, *p < 0.05, **p < 0.01, two-tailed t-test (b, d-h).
Extended Data Fig. 3 Overexpression IFITM1 inhibits EBV infection in ECs.
(a) The efficiency of overexpression of IFITM1 at the mRNA level. Bars show the relative IFITM1 mRNA level detected by RT-qPCR and data are presented relative to B2M (2˗ΔCt) and are expressed as the mean ± s.e.m. (n = 3 independent experiments, ***p < 0.001), two-tailed t-test. (b) Efficiency of overexpression of IFITM1 at the protein level. GAPDH was used as a control to indicate equivalent amounts of lysates. (c) NP69 and HEK293 cell lines were incubated with cell-free EBV-GFP for 3 hours, and the remaining extracellular viruses were removed by washing with 1×PBS. EBV copy numbers were then measured by TaqMan-qPCR. (d-g) After 72 hours, flow cytometric analyses were performed to show the percentage of EBV-GFP-positive cells. Representative images of cells infected with EBV-GFP were recorded for HEK293 cells (h). The results are expressed as the mean ± s.e.m. from at least three biological replicates. *p < 0.05, **p < 0.01 (n ≥ 3, a, b, d, e), two-tailed t-test. OE, overexpression.
Extended Data Fig. 4 sIFITM1 couldn’t inhibit EBV infection in B cells.
(a) SDS–PAGE gel shows the purified 6×His-IFITM1 protein (indicated by the red arrow). (b) Diagram of FCM analysis process. In this study, all FCM analyses were carried out according to Extended Data Fig. 4b. (c) Nude mice were xenografted with HEK293 to form tumor-like cell clusters followed by injection of sIFITM1 and EBV-GFP at set intervals. A control was included in which ddH2O was injected at the same interval. After treatment, equal amounts of blood were obtained from Nude mice to detect IFNβ and IFNγ by ELISA. Five mice were investigated for each group (n = 5, ns: no significant difference). Data are presented as mean values ± s.e.m., *p < 0.05, **p < 0.01 (d: n = 8, e: n = 5).
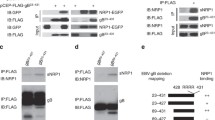
Extended Data Fig. 5 STRING interactome analysis of IFITM1 and proteins involved in EBV infection.
(a) EphA2, NRP1, MYH9, and ITGAV are EBV infection receptors in epithelial cells (dark blue). EGF and EGFR are also involved in EBV infection in epithelial cells (light blue). CR2, CD81, CD9, TLR2, and CIITA are B cell-related receptors (pink). IFITM1(green) and EphA2 are indicated by the red arrow. (b) Co-IP assays showed that endogenous IFITM1 co-immunoprecipitated with EphA2. NP69 and HK1 cell lysates were immunoprecipitated (IP) with IFITM1 antibody, followed by an immunoblotting (IB) assay with EphA2 antibody. IgG-IP samples were included as a control. (c) Recombinant His-IFITM1 protein expression detected by Coomassie Brilliant Blue staining. Lane 2: without IPTG induction, lane 3: IPTG induction, lane 1 purified fusion protein. (d) Recombinant GST-EphA2 protein expression detected by Coomassie Brilliant Blue staining. Lane 4: without IPTG induction, lane 3: IPTG induction, lane 2 purified fusion protein. (e) Recombinant His-gB, and His-gH/gL protein expression detected by Coomassie Brilliant Blue staining. Lane 1 and 7: without IPTG induction, lane 2 and 6: IPTG induction, lane 3 and 5 purified fusion protein. Representative of two independent experiments (b-e).
Extended Data Fig. 6 Overexpression of IFITM1 or/and EphA2 in NP69, HK1, and HEK293 cells.
(a–c) Lentiviruses encoding IFITM1 or EphA2 (referred to by ‘+’) and their corresponding unloaded lentiviruses (referred to by ‘−’) were transfected into NP69, HK1, and HEK293 cells. (a,b) Total RNA was obtained and RT-qPCR was performed to detect the relative mRNA levels of IFITM1 (a) and EphA2 (b). Data are presented relative to B2M (2˗ΔCt). All results are expressed as the mean ± s.e.m. from at least three biological replicates (n ≥ 3, a, b), two-tailed t-test. *p < 0.05, **p < 0.01, ***p < 0.001. (c) The corresponding IFITM1 protein levels in cells from (a) and (b). GAPDH was used as a control to indicate equivalent amounts of lysates. The results are from at least three biological replicates.
Extended Data Fig. 7 Site-directed mutation of 112Tyr and 104Leu on IFITM1.
(a) Schematic representation of the domain structure of EBV gH/gL; CT: C-terminal cytoplasmic tail domain, SP: signal peptide. (b) Cartoon representation of the structure of the EBV gH/gL-ligand-binding domain (LBD)41. EBV gH is indicated in green, gL is indicated in yellow, and the LBD of EphA2 is indicated in orange. (c) EphA2-overexpressing cells (NP69; HEK293) were transfected with plasmids corresponding to wild-type IFITM1 (IFITM1w), 112Tyr single-site mutation (IFITM1m1), 104Leu single-site mutation (IFITM1m2), or dual mutations (IFITM1m1+2). Experiments were implemented 72 hours after transfection. The cell lysates were harvested to test the IFITM1 level. GAPDH was used as a control to indicate equivalent amounts of lysates. The results are from at least three biological replicates.
Extended Data Fig. 8 IFITM1 expression is negatively regulated by YTHDF3.
(a) RT-qPCR shows the relative IFITM1 mRNA levels in siYTHDF1, siYTHDF2, and siYTHDF3 HEK293 cells. (b) Relative YTHDF3 mRNA levels in NP69, HK1, and HEK293 cells was checked by RT-qPCR after infecting with lentiviruses encoding shYTHDF3 or shLacZ. (c) Relative YTHDF3 mRNA levels in NP69, HK1, and HEK293 cells was checked by RT-qPCR after infecting with the control (Vector) or YTHDF3-overexpressing (YTHDF3 OE) lentiviruses. Data are presented relative to B2M (2˗ΔCt). Mean ± s.e.m., n ≥ 3 independent experiments, *p < 0.05, **p < 0.01, ***p < 0.001, two-tailed t-test (a-c). (d) RT-qPCR of YTHDF3 mRNA expression in nine nasopharyngeal carcinoma (NPC) and nine nasopharyngeal (NP) samples. Total RNA from each tissue was assayed by RT-qPCR and B2M was used as a reference gene. Data are presented as mean values ± s.e.m., n = 18, two-tailed t-test. (e) Correlation analysis of the relative YTHDF3 mRNA expression level and the EBV copy number (horizontal axis: YTHDF3 relative mRNA level; vertical axis: relative EBV copy number). Data are presented as mean values ± s.e.m., n = 8, r = –0.78, p = 0.021 (e).
Extended Data Fig. 9 IFITM1 expression is negatively regulated by YTHDF3 via RNA degradation.
(a) Fold enrichment of IFITM1 was analyzed by RIP-seq in shLacZ or shYTHDF3-HK1 cells. (b) Fold enrichment of IFITM1 was determined by MeRIP-qPCR in shLacZ or shYTHDF3-HEK293 cells and data was presented relative to B2M (2˗ΔCt). (c) KEGG pathway analysis of the 598 proteins from Fig. 6d. Proteins in the DDX family related to RNA degradation were detected. (d) Lentiviruses encoding shYTHDF3 or shDDX5 and their corresponding negative control lentiviruses (shLacZ + shLacZ) were transfected into HK1 cells, and immunoblotting assay were performed with anti-YTHDF3, DDX5, and IFITM1. (e) Cells from Extended Data Fig. 9d were treated with transcriptional inhibitor ActD, followed by IFITM1 detection using RT-qPCR. (f) Cells from Extended Data Fig. 9d were exposed to EBV-GFP for 3 hours and the remaining extracellular viruses were removed by washing with 1×PBS. EBV copy numbers were then measured by TaqMan-qPCR and results from control groups were taken as 100%. Data are presented as mean values ± s.e.m., n = 3 independent experiments, *p < 0.05, **p < 0.01, two-tailed t-test (a, b, e, f).
Extended Data Fig. 10 IFITM1 does not affect Epstein-Barr virus infection in B cells.
(a) Knockdown of IFITM1 in B cells by infection with lentiviruses encoding shIFITM1 (1-shIFITM1 and 2-shIFITM1), then cells were incubated with cell-free EBV-GFP for 3 hours and remaining extracellular viruses were removed by washing with 1×PBS. EBV copy numbers were then measured by TaqMan-qPCR. (b) B cells were infection with lentiviruses encoding wild-type IFITM1 (IFITM1w), or dual mutations (IFITM1m1+2), then cells were incubated with cell-free EBV-GFP for 3 hours and remaining extracellular viruses were removed by washing with 1×PBS. EBV copy numbers were then measured by TaqMan-qPCR. (c) Analysis of the anti-EBV effects of sIFITM1 on B cells, exposure of B cells to EBV-GFP for 3 hours, with sIFITM1 being added 2 hours in advance at different concentrations (0, 1, and 5 ng/µL), EBV copy numbers were then measured by TaqMan-qPCR. Data are presented as mean values ± s.e.m., n = 4 independent experiments, *p < 0.05, **p < 0.01 (a-c). (d) According to the method in Fig. 1k and l, anti-EBV effects of sIFITM1 was tested on B cells in vivo. (e) A schematic diagram showing two models of EBV infection: an insusceptibility model (left), in which IFITM1 inhibits EBV entry by competing with EBV gH/gL and gB for binding to EphA2; and a susceptibility model (right), in which YTHDF3 recognizes m6A modification sites on IFITM1 and interacts with RNA degradation-related proteins DDX5, then leads to the degradation of IFITM1. The loss of IFITM1 would result in exposure of EphA2, which may aid EBV entry.
Supplementary information
Source data
Source Data Fig. 1
Statistical source data.
Source Data Fig. 2
Unprocessed western blots and statistical source data.
Source Data Fig. 3
Statistical source data.
Source Data Fig. 4
Unprocessed western blots and statistical source data.
Source Data Fig. 5
Statistical source data.
Source Data Fig. 6
Unprocessed western blots and statistical source data.
Source Data Extended Data Fig. 1
Unprocessed western blots and statistical source data.
Source Data Extended Data Fig. 2
Unprocessed western blots and statistical source data.
Source Data Extended Data Fig. 3
Unprocessed western blots and statistical source data.
Source Data Extended Data Fig. 4
Statistical source data.
Source Data Extended Data Fig. 5
Unprocessed western blots.
Source Data Extended Data Fig. 6
Unprocessed western blots and statistical source data.
Source Data Extended Data Fig. 7
Unprocessed western blots.
Source Data Extended Data Fig. 8
Statistical source data.
Source Data Extended Data Fig. 9
Unprocessed western blots and statistical source data.
Source Data Extended Data Fig. 10
Statistical source data.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yang, Y., Ding, T., Cong, Y. et al. Interferon-induced transmembrane protein-1 competitively blocks Ephrin receptor A2-mediated Epstein–Barr virus entry into epithelial cells. Nat Microbiol 9, 1256–1270 (2024). https://doi.org/10.1038/s41564-024-01659-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41564-024-01659-0
- Springer Nature Limited