

Abstract
DUSP22 is a dual-specificity phosphatase that inhibits T cell activation by inactivating the kinase Lck. Here we show that the E3 ubiquitin ligase UBR2 is a positive upstream regulator of Lck during T-cell activation. DUSP22 dephosphorylates UBR2 at specific Serine residues, leading to ubiquitin-mediated UBR2 degradation. UBR2 is also modified by the SCF E3 ubiquitin ligase complex via Lys48-linked ubiquitination at multiple Lysine residues. Single-cell RNA sequencing analysis and UBR2 loss of function experiments showed that UBR2 is a positive regulator of proinflammatory cytokine expression. Mechanistically, UBR2 induces Lys63-linked ubiquitination of Lck at Lys99 and Lys276 residues, followed by Lck Tyr394 phosphorylation and activation as part of TCR signalling. Inflammatory phenotypes induced by TCR-triggered Lck activation or knocking out DUSP22, are attenuated by genomic deletion of UBR2. UBR2-Lck interaction and Lck Lys63-linked ubiquitination are induced in the peripheral blood T cells of human SLE patients, which demonstrate the relevance of the UBR2-mediated regulation of inflammation to human pathology. In summary, we show here an important regulatory mechanism of T cell activation, which finetunes the balance between T cell response and aggravated inflammation.
Similar content being viewed by others


Introduction
Lymphocyte cell-specific protein-tyrosine kinase (Lck), a Src-family tyrosine kinase, is phosphorylated and activated in the propagation of T-cell receptor (TCR) signaling. Lck kinase activity is dynamically regulated through conformational changes by phosphorylation and dephosphorylation of its tyrosine residues1,2. Phosphorylation of Lck Tyr505 by C-terminal Src kinase (CSK) results in a closed and inactive conformation of Lck in resting T cells1,2. Dephosphorylation of Lck Tyr505 by CD45 results in an open conformation of Lck, leading to trans-autophosphorylation of Tyr394 and subsequent activation of Lck1,2. Lys48-linked ubiquitination of Lck by the E3 ubiquitin ligase Cbl leads to proteasomal degradation of Lck3. It was unclear whether Lck kinase activity is regulated by ubiquitination (such as Lys63-linked ubiquitination).
JNK pathway-associated phosphatase (JKAP, also named DUSP22) belongs to the dual-specificity phosphatase (DUSP) family4,5. DUSPs dephosphorylate threonine/serine and/or tyrosine residues of their substrates6,7. Dysregulation of DUSP22 in T cells contributes to the development of several human diseases7, including peripheral T-cell lymphoma8, inflammatory bowel disease (IBD)9, systemic lupus erythematosus (SLE)10, and ankylosing spondylitis (AS)11. DUSP22 dephosphorylates and inactivates focal adhesion kinase, resulting in the suppression of cancer cell motility and liver cancer progression12,13. DUSP22 dephosphorylates the tyrosine kinase Lck at Tyr394 in the turn-off stage of TCR signaling, leading to the suppression of T cell-mediated immune responses14. Conversely, DUSP22-deficient T cells display induction of the Th1 cytokines IFN-γ and the Th17 cytokine IL-1710,11,14,15. Thus, DUSP22 is an important inhibitor of T-cell activation by dephosphorylating and inhibiting Lck in the attenuation phase of TCR signaling.
In the ubiquitin-proteasome pathway, E3 ubiquitin ligases ubiquitinate the substrate with Lys48-linked ubiquitin chains, targeting the substrate for proteasomal degradation. SKP1–CUL1–F-box proteins (SCF) E3 ubiquitin ligase complexes catalyze the ubiquitination of proteins targeted for 26 S proteasomal degradation16. In SCF complexes, the F-box proteins are the substrate-recognition subunits. SKP1 is an adaptor protein essential for the binding of F-box proteins. CUL1 is the major structural scaffold of the SCF complexes. RBX1 (RING-box protein 1) or RBX2 (RING-box protein 2), containing a RING domain, interacts with an E2 ubiquitin-conjugating enzyme and transfers ubiquitin from E2 to a lysine residue on target proteins15.
Ubiquitin Protein Ligase E3 Component N-Recognin 2 (UBR2), a RING domain E3 ubiquitin ligase, is a component of the N-end rule pathway of targeted proteolysis17. UBR2 restricts mobilization of LINE-1 (L1) retrotransposons by inducing the protein ubiquitination and proteasomal degradation of L1-ORF1p18. UBR2 also induces the protein degradation of the cleaved N-terminal NLRP1B fragment, leading to inflammasome activation19. In addition, UBR2 is associated with cancer progression18,20,21, cancer cachexia autoimmune pancreatitis22, amyotrophic lateral sclerosis23, and T-cell chronic lymphocytic leukemia24. The predominant expression of UBR2 in immune cells25 suggests a role of UBR2 in regulating immune cell functions; however, the roles and functions of UBR2 in lymphocytes have not been investigated.
Here, we report that DUSP22 dephosphorylates UBR2 and blocks UBR2-mediated Lck K63-ubiquitination and activation, leading to the reduction of T-cell activation and cytokine production. Our findings on the regulation of Lck activation by UBR2 may provide therapeutic approaches to treat autoimmune diseases.
Results
DUSP22 induces UBR2 proteasomal degradation
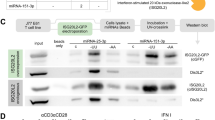
To search for regulators that control DUSP22 protein levels, anti-Myc- DUSP22 immunocomplexes from transfected HEK293T cells were precipitated and then subjected to proteomics. The E3 ubiquitin-protein ligase UBR2 was identified as a DUSP22-interacting protein (Fig. 1a). We first examined whether UBR2 induces DUSP22 protein degradation. Myc-DUSP22 and Flag-UBR2 plasmids were co-transfected into HEK293T cells, and cell extracts were subjected to immunoblotting analysis. UBR2 overexpression did not affect DUSP22 protein levels (Fig. 1b). Surprisingly, DUSP22 overexpression resulted in a dose-dependent decrease of UBR2 protein levels (Fig. 1c). To test whether DUSP22 phosphatase activity is required for the induction of UBR2 degradation, the DUSP22 phosphatase-dead mutant Myc-DUSP22 (C88S)14 and Flag-UBR2 plasmids were co-transfected into HEK293T cells. The result showed that overexpression of DUSP22 mutant (C88S) did not inhibit, but increased, UBR2 protein levels in a dose-dependent manner (Fig. 1d), suggesting that DUSP22 phosphatase activity is required for the induction of UBR2 degradation. To examine whether UBR2 protein levels are increased by DUSP22 knockout, we generated DUSP22 knockout mice by TALEN-mediated gene targeting (Fig. 1e). DUSP22 knockout was confirmed by PCR and immunoblotting analyses (Fig. 1f). The deletion of 4 bp (GATC) in DUSP22 exon 1 resulted in a 24-amino acid frame-shift/truncated mutant (Fig. 1e). The mutant proteins were not detected by an anti-DUSP22 antibody that recognized the C-terminus of murine DUSP22 proteins (Fig. 1g). Although UBR2 is predominately expressed in immune cells25, UBR2 proteins were detected in T cells and multiple non-lymphoid tissues of wild-type mice (Fig. 1g). The protein levels of UBR2 in multiple tissues of DUSP22 knockout mice were increased compared to those of wild-type mice (Fig. 1g). The purified T cells of DUSP22 knockout mice also showed increased UBR2 protein levels compared to those of wild-type mice (Fig. 1g). These results support that DUSP22 induces UBR2 degradation. In addition to immune cells, UBR2 protein may also play a role in other tissues or cell types.

a The identified peptide sequences of endogenous UBR2 protein by mass spectrometry-based analyses using Myc-tagged immunocomplexes from the lysate of Myc-DUSP22-transfected HEK293T cells. b-d Immunoblotting of Flag-tagged UBR2, Myc-tagged DUSP22, and tubulin proteins in HEK293T cells co-transfected with different amounts of Flag-UBR2 plus either Myc-DUSP22 (b and c) or Myc-DUSP22 (C88S) (d) plasmids. Tubulin immunoblotting was performed by reprobing the Flag (UBR2) immunoblot membrane (b and d). Arrow, intact UBR2 protein; asterisk, degraded UBR2 protein. e Schematic diagram of the murine DUSP22 wild-type (WT) alleles and the targeted DUSP22 mutant alleles. P1 and P2, the primers for PCR. f Characterization of DUSP22-knockout mice. PCR analyses of wild-type and DUSP22 mutant alleles in the genomic DNA from mouse tails. The PCR product of the upper band (300 bp) denotes the wild-type allele, and the lower band (145 bp) denotes the DUSP22 mutant allele. g Immunoblotting analysis of UBR2 protein levels in multiple tissues of wild-type and DUSP22 knockout mice. h, i DUSP22-induced UBR2 proteasomal degradation. Flag-UBR2 and Myc-DUSP22 plasmids were co-transfected into HEK293T cells. The transfected cells were treated with MG132 (25 µM), Z-VAD-FMK (50 µM), or chloroquine (50 µM) for the indicated time points and then subjected to immunoblotting analysis. Anti-tubulin immunoblotting was performed by reprobing the anti-Myc (DUSP22) immunoblot membrane (h). Vinculin immunoblotting was performed by reprobing the anti-Flag (UBR2) immunoblot membrane (i). j, k UBR2 interacted with DUSP22 or DUSP22 (C88S) mutant proteins. Coimmunoprecipitation and immunoblotting analyses of UBR2, DUSP22, and DUSP22 (C88S) mutant proteins in HEK293T cells co-transfected with Flag-UBR2 plasmid plus either Myc-DUSP22 or Myc-DUSP22 (C88S) mutant plasmid. l Proximity ligation assays (PLA) showed in vivo UBR2-DUSP22 interaction in Jurkat T cells. Red fluorescence represents interactions ( < 40 nm) of Flag-UBR2 and Myc-DUSP22 proteins. Images were captured by confocal microscope (Leica TCS SP5 II). Original magnification, 400X. Cell nucleus was stained with DAPI. Scale bar, 10 μm. m In vitro binding assays of purified Flag-tagged UBR2 and recombinant GST-tagged DUSP22 proteins.
To study whether DUSP22 induces UBR2 degradation by a caspase-, lysosome-, or proteasome-mediated pathway, cells were treated with the caspase inhibitor Z-VAD-FMK, the lysosomal enzyme inhibitor chloroquine, or the proteasome inhibitors MG132 and carfilzomib. The data showed that DUSP22-induced UBR2 proteasomal degradation was reversed by treatment of MG132 (Fig. 1h) or carfilzomib (Supplementary Fig. 1a); however, the degradation was not affected by Z-VAD-FMK or chloroquine treatment (Fig. 1i). The data suggest that DUSP22 induces UBR2 proteasomal degradation. We next confirmed the interaction between DUSP22 and UBR2 in cells using reciprocal coimmunoprecipitation assays. Flag-UBR2 plasmid was co-transfected with either Myc-DUSP22 or Myc-DUSP22 (C88S) plasmid into HEK293T cells. The transfected cell lysates were subjected to immunoprecipitation with either anti-Flag antibody (Fig. 1j) or anti-Myc antibody (Fig. 1k), followed by immunoblotting. These assays confirmed the interaction between DUSP22 and UBR2 proteins, as well as between DUSP22 (C88S) mutant and UBR2 proteins (Fig. 1j, k). As expected, the levels of coimmunoprecipitated UBR2 proteins by DUSP22 substrate-trap** (C88S) mutant26 were higher than that of wild-type DUSP22 (Fig. 1j, k). This result also ruled out the possibility that the DUSP22 (C88S) mutant26 reversed UBR2 degradation due to its inability in binding to UBR2. To further demonstrate the interaction between DUSP22 and UBR2 proteins in vivo, we performed in situ proximity ligation assay (PLA), which detects two molecules in close proximity ( < 40 nm) in cells with paired antibody-conjugated probes27. The data showed strong PLA signals of DUSP22-UBR2 interaction in MG132-treated Jurkat T cell but not in other negative control T cells (Fig. 1l). We further examined whether DUSP22 (C88S) mutant competes with wild-type DUSP22 for UBR2 binding. The PLA data showed that the interaction between DUSP22 and UBR2 proteins in HEK293T cells was decreased by DUSP22 (C88S) overexpression (Supplementary Fig. 1b). To confirm the direct interaction of DUSP22 with UBR2, in vitro binding assays were performed using purified proteins. Purified Flag-tagged UBR2 proteins and recombinant GST-tagged DUSP22 proteins were subjected to in vitro binding assays. The results showed that GST-tagged DUSP22 proteins strongly interacted with Flag-tagged UBR2 proteins (Fig. 1m). Collectively, these results showed that DUSP22 directly interacted with UBR2.
Lys94, Lys779, and Lys1599 residues of UBR2 are responsible for DUSP22-induced UBR2 degradation
Next, to study whether DUSP22 induces Lys48-linked ubiquitination of UBR2, we immunoprecipitated Flag-tagged UBR2 proteins from HEK293T cells co-transfected with Flag-UBR2 and either Myc-DUSP22 or vector control plasmids. The data showed that DUSP22 overexpression enhanced UBR2 Lys48-linked ubiquitination (Fig. 2a). Heterotypic Lys11-linked ubiquitination also serves as a targeting signal for proteasome28, thus we also tested whether DUSP22 enhances UBR2 Lys11-linked ubiquitination. Flag-tagged UBR2 proteins were immunoprecipitated from HEK293T cells co-transfected with Lys11-only ubiquitin (K11-Ub) mutant, Flag-UBR2, and either Myc-DUSP22 or vector control plasmids. The data showed that DUSP22 overexpression did not enhance UBR2 Lys11-linked ubiquitination (Supplementary Fig. 2a). To confirm that these DUSP22-induced ubiquitinated protein bands were UBR2 itself but not UBR2-interacting proteins, we performed two rounds of immunoprecipitations. The anti-Flag-UBR2 immunoprecipitates were denatured using 1% SDS and boiling to dissociate UBR2-interacting proteins, the proteins were renatured through serial dilution, and the renatured Flag-UBR2 proteins were re-immunoprecipitated. Ubiquitinated UBR2 bands were still detected in the second-round anti-Flag-UBR2 immunoprecipitates (Fig. 2b), indicating that UBR2 itself is ubiquitinated. To study whether DUSP22 is responsible for inducing UBR2 Lys48-linked ubiquitination, we depleted DUSP22 using DUSP22 shRNAs and then assessed the levels of UBR2 ubiquitination. DUSP22 shRNA knockdown decreased Lys48-linked ubiquitination of UBR2 (Fig. 2c). Furthermore, DUSP22 (C88S) overexpression reduced Lys48-linked ubiquitination of UBR2 (Supplementary Fig. 2b), suggesting that DUSP22 phosphatase activity is required for the induction of UBR2 Lys48-linked ubiquitination. To identify the DUSP22-induced Lys48-linked ubiquitination sites responsible for UBR2 degradation, we immunoprecipitated Flag-tagged UBR2 proteins from HEK293T cells co-transfected with Flag-UBR2, Lys48-only ubiquitin (K48-Ub) mutant, and Myc-DUSP22 plasmids. The immunoprecipitated Flag-tagged UBR2 proteins were then subjected to mass spectrometry analysis. The result showed that UBR2 was ubiquitinated at Lys94 only when co-transfected with Lys48-only ubiquitin (K48-Ub) plus DUSP22 under MG132 treatment (Fig. 2d and Supplementary Table 1). The Lys255 residue was ubiquitinated when co-transfected with Lys48-only ubiquitin (K48-Ub) in the absence of DUSP22, but undetectable in the presence of DUSP22, suggesting that Lys255 residue was not responsible for DUSP22-induced UBR2 ubiquitination (Supplementary Fig. 3a and Supplementary Table 1). We also analyzed the mass spectrometry data from the experiment using HEK293T cells transfected with Flag-UBR2 and Myc-DUSP22 plasmids without Lys48-only ubiquitin (K48-Ub) mutant plasmid. The result showed that UBR2 was ubiquitinated at 9 lysine residues, including Lys158, Lys165, Lys248, Lys470, Lys488, Lys568, Lys789, Lys1496, and Lys1689 (Supplementary Fig. 3b; Supplementary Table 2). Besides mass spectrometry data, Lys731, Lys798, Lys958, and Lys1142 residues were predicted as UBR2 ubiquitination sites using web-based softwares29 (UbiSite, http://csb.cse.yzu.edu.tw/UbiSite/). PhosphoSitePlus (https://www.phosphosite.org/homeAction) website shows additional 14 UBR2 ubiquitination sites; however, the functional consequences of these ubiquitination sites have not been studied. To study whether ubiquitination of any of the 24 mapped and 4 web-predicted residues induces DUSP22-mediated UBR2 degradation, these lysine residues were individually mutated to arginine residues. Individual UBR2 (K94R), UBR2 (K779R), and UBR2 (K1599R) mutations significantly blocked DUSP22-mediated UBR2 degradation (Fig. 2e). In the absence of DUSP22 overexpression, the expression of each UBR2 mutant alone was comparable to that of wild-type UBR2 (Supplementary Fig. 3c). Furthermore, UBR2 triple mutations (K94/779/1599R) blocked DUSP22-induced UBR2 degradation in Jurkat T cells (Fig. 2f). The Lys48-linked ubiquitination of UBR2 (K94/779/1599R) mutant was abolished compared to that of wild-type UBR2 in HEK293T cells (Fig. 2g). The data suggest that Lys48-linked ubiquitination at Lys94, Lys779, and Lys1599 residues of UBR2 are responsible for DUSP22-mediated UBR2 degradation.

a DUSP22 induced UBR2 Lys48-linked ubiquitination. UBR2 was immunoprecipitated with anti-Flag antibody and then immunoblotted with anti-ubiquitin (Lys48) antibody or anti-Flag antibody. b UBR2 was ubiquitinated. UBR2 was first immunoprecipitated with anti-Flag antibody (1st IP); half of anti-Flag immunoprecipitates were denatured, renatured, followed by a second immunoprecipitation (2nd IP) with anti-Flag antibody. Anti-Flag immunoprecipitates were immunoblotted with anti-ubiquitin antibody or anti-Flag antibody. c DUSP22 knockdown reduced UBR2 Lys48-linked ubiquitination. Flag-UBR2 and individual DUSP22 shRNAs were co-transfected into HEK293T cells. The cells were treated with 25 µM MG132. UBR2 was immunoprecipitated with anti-Flag antibody and then immunoblotted with anti-ubiquitin (Lys48) antibody or anti-Flag antibody. Vinculin immunoblotting was performed by reprobing the Flag (UBR2) immunoblot membrane with anti-vinculin antibody. d Lys94 residue was identified as a K48-linked ubiquitination site of UBR2. HEK293T cells were transfected with Flag-UBR2 plus Lys48-only ubiquitin mutant (K48-Ub) plasmids and with or without DUSP22 plasmid, followed by treatment with or without 25 µM MG132 for 4 h. Flag-tagged UBR2 proteins were immunoprecipitated with anti-Flag antibody and then subjected to mass spectrometry analysis. The MS/MS fragmentation spectra displayed UBR2 tryptic peptides containing the ubiquitinated residue Lys94. K(ub) denotes the ubiquitinated lysine residue. e K94R, K779R, or K1599R mutation of UBR2 reversed DUSP22-mediated degradation of UBR2. Myc-DUSP22 and Flag-UBR2 mutant plasmids were co-transfected into HEK293T cells. The levels of Flag-tagged UBR2 proteins were examined by immunoblotting analysis. f Triple mutations (K94/779/1599R) of UBR2 blocked DUSP22-mediated degradation of UBR2. Myc-DUSP22 plus either Flag-UBR2 or Flag-UBR2 (K94/779/1599R) mutant plasmids were co-transfected into Jurkat T cells. The levels of UBR2 proteins were determined by immunoblotting analysis. g Triple mutations (K94/779/1599R) of UBR2 reduced UBR2 Lys48-linked ubiquitination. Myc-DUSP22 plus either Flag-UBR2 or Flag-UBR2 mutant (K94/779/1599R) plasmids were co-transfected into HEK293T cells. Flag-tagged UBR2 wild-type (WT) or (K94/779/1599R) mutant proteins were immunoprecipitated with anti-Flag antibody and then immunoblotted with anti-ubiquitin (Lys48) antibody or anti-Flag antibody. Arrow, intact UBR2 protein; asterisk, degraded UBR2 protein.
CUL1-βTrCP is identified as an E3 ubiquitin ligase complex of UBR2
To identify the E3 ubiquitin ligase of UBR2, we searched for UBR2-interacting proteins using HEK293T cells transfected with Flag-UBR2. UBR2-interacting proteins were coimmunoprecipitated with anti-Flag antibody and followed by mass spectrometry analysis. We found that CUL1, a component of SKP1-CUL1-F-box protein (SCF) E3 ubiquitin ligase complex43,44. The peptide data were analyzed by MASCOT MS/MS Ions Search (Matrix Science) under the following conditions: peptide mass tolerance, 20 ppm; fragment MS/MS tolerance, 0.6 Da; allow up to one missed cleavage; peptide charge, 2+, 3+, and 4+.
In situ proximity ligation assay (PLA)
The HEK293T cells coexpressing the molecules (Flag-tagged UBR2 plus Myc-tagged DUSP22; Flag-tagged UBR2 plus either Myc-tagged CUL1 or F-box proteins; Flag-tagged UBR2 plus Myc-tagged βTrCP and either GFP-tagged DUSP22 or GFP-tagged DUSP22 (C88S); Flag-tagged UBR2, Flag-tagged UBR2 (S1694D/Y1697D), or Flag-tagged UBR2 (S1694A/Y1697F) plus Myc-tagged βTrCP and GFP-tagged DUSP22; Flag-tagged UBR2 plus Myc-tagged DUSP22 and GFP-tagged DUSP22 (C88S)) were fixed with 4 % formaldehyde for 1 h. The fixed cells were permeabilized by 0.1% Triton X-100 at room temperature for 2 h, and then washed twice by PBS, followed by blocking with PLA blocking buffer. The cells were hybridized with anti-Flag and anti-Myc antibodies, followed by rabbit- and mouse-specific secondary antibodies conjugated with oligonucleotides (PLA probes; Sigma-Aldrich). For human primary T cells, T cells were fixed with cold methanol for 2 min. The fixed cells were permeabilized by 0.1% triton X-100 at room temperature for 2 h, and then washed twice by PBS, followed by blocking with PLA blocking buffer. For murine primary T cells and human Jurkat T cells, the transcription factor buffer set (#562574, BD Pharmingen™) was used for fixation and permeabilization, followed by blocking with PLA blocking buffer. The K63-linked ubiquitination of the endogenous Lck proteins were recognized by anti-Lck and anti-Lys63-specific ubiquitin antibodies, followed by rabbit- and mouse-specific secondary antibodies conjugated with oligonucleotides (PLA probes; Sigma-Aldrich). The endogenous Lck Tyr394 phosphorylation was recognized by anti-Lck and anti-phospho-Lck (Y394) antibodies, followed by rabbit- and mouse-specific secondary antibodies conjugated with oligonucleotides (PLA probes; Sigma-Aldrich). After the reaction of ligation and amplification, the PLA signals from the pair of PLA probes in close proximity less than 40 nm were visualized27. For fluorescent signal in cells, images were detected by fluorescence microscope (DM2500; Leica Microsystems, Buffalo Grove, IL, USA), confocal microscope Leica TCS SP5, and Leica TCS SP5 II. Images were analyzed by LAS X software (v3.7.4). The intensity value was obtained from area multiplying by mean.
IHC
Kidneys, livers, and lungs were harvested from mice and fixed in 10% neutral-buffered formalin for 3–7 days at room temperature, and then transferred to 75% alcohol for further processing. Tissue processing, embedding, and H&E staining were performed by the NHRI Pathology Core.
In vitro binding assay
Flag-UBR2 plasmid was transfected into HEK293T cells. Transfected cells were lysed in RIPA buffer (1% sodium deoxycholate, 1 mM EDTA, 150 mM NaCl, 1% Triton X-100, 0.1% SDS, 50 mM Tris-HCl) supplemented with 0.5% protease inhibitor cocktail, 1% phosphatase inhibitor cocktail, and 1 mM Na3VO4 at 4 °C for 30 min. Flag-tagged UBR2 proteins were immunoprecipitated with anti-Flag agarose beads in 1 ml of lysis buffer at 4 °C for 2 h. Flag-tagged UBR2 proteins were eluted by 3X Flag peptide (F4799, Sigma-Aldrich). GST-DUSP22 plasmid was transferred into Escherichia coli strain BL21 by transformation. GST-tagged DUSP22 proteins were precipitated by glutathione-conjugated sepharose beads (#17-0756-01, GE Healthcare), and then purified with glutathione (#G4251, Sigma) in GST elution buffer. Purified Flag-tagged UBR2 proteins were incubated with the GST-tagged DUSP22 proteins. The reaction mixtures were immunoprecipitated with anti-Flag agarose beads or anti-GST glutathione-sepharose at 4 °C for 2 h. The reaction mixtures were then subjected to immunoblotting analyses.
In vitro ubiquitination assay
Purified Flag-tagged UBR2 proteins, SKP1-CUL1-βTrCP-RBX1 complex, E1 (0.1 μM UBE1), E2 (0.35 μM UBE2D3 or 1 μM CDC34), and His-ubiquitin (20 μM) were incubated at 37 °C for 1 h in 35 μl buffer containing 50 mM Tris (pH 7.5), 5 mM MgCl2, 2 mM ATP, and 2 mM DTT. The reactant products were subjected to immunoblotting analyses. UBE1, UBE2D3, CDC34, and His-ubiquitin proteins were purchased from R&D Systems.
In vitro ubiquitin E3 ligase assay in combination with in vitro kinase assay
Purified Myc-tagged UBR2 proteins, Flag-tagged Lck proteins, E1 (0.1 μM UBE1), E2 (0.98 μM UBE2N), and His-ubiquitin (20 μM) were incubated at 37 °C for 1 h in buffer containing 50 mM Tris (pH 8), 10 mM MgCl2, 1 mM ATP, 1 mM DTT, and 50 mM NaCl. After in vitro E3 ligase assay, the reactants were incubated in kinase buffer containing 25 mM Tris-HCl (pH 7.5), 20 mM MgCl2, and 5 mM beta-glycerophosphate at 37 °C for 0.5 h. The reaction mixtures were subjected to immunoblotting analyses. UBE1, UBE2N, and His-ubiquitin proteins were purchased from R&D Systems.
In vitro phosphatase assay
Flag-UBR2, Myc-DUSP22, and Myc-DUSP22 (C88S) plasmids were individually transfected into HEK293T cells. Transfected cells were lysed in RIPA buffer. Flag-tagged UBR2 proteins were immunoprecipitated with anti-Flag agarose beads, and Myc-tagged DUSP22 proteins were immunoprecipitated with anti-Myc agarose beads in 1 ml of lysis buffer at 4 °C for 2 h. Flag-tagged UBR2 immunocomplexes were then incubated with Myc-tagged DUSP22 wild-type or mutant (C88S) proteins at 37 °C for 30 min in buffer containing imidazole (50 mM, pH 7.5) and DTT (10 mM). The reactant products were subjected to immunoblotting analysis.
In vitro kinase assay
For kinase preparation, Myc-UBR2 plasmid plus either Flag-Lck, Flag-Lck (K99/276R), or Flag-Lck (Y394F) plasmid were co-transfected into Jurkat T cells. Transfected cells were lysed in RIPA buffer (1% sodium deoxycholate, 1 mM EDTA, 150 mM NaCl, 1% Triton X-100, 0.1% SDS, 50 mM Tris-HCl) supplemented with 0.5% protease inhibitor cocktail (#539134, EMD Millipore), 1% phosphatase inhibitor cocktail (#524629, EMD Millipore), and 1 mM Na3VO4 at 4 °C for 30 min. For coimmunoprecipitation, 1 mg protein lysates were incubated with anti-Flag agarose beads (clone M2, Sigma-Aldrich) in 1 ml of lysis buffer at 4 °C for 1-2 h. The immunocomplexes were washed three times with lysis buffer. Flag-tagged wild-type, K99/276R, or Y394F Lck immunocomplexes from the lysates of UBR2-overexpressing Jurkat T cells were subjected to peptide elution by Flag peptides. For substrate preparation, Flag-Lck plasmid was transfected into HEK293T cells. Transfected cell lysates were extracted by RIPA buffer described above. Additional Flag-tagged wild-type Lck immunocomplex was isolated from the lysates of HEK293T cells using anti-Flag agarose beads. Purified Flag-tagged Lck proteins were incubated with the substrate Flag-tagged Lck for 40 min at 37 °C with 1 μM ATP in 60 μl kinase buffer. The reactant products were subjected to immunoblotting analyses.
Purification of T cells
Peripheral blood was collected from mice or human individuals, and then red blood cells were lysed by ACK (ammonium-chloride-potassium) lysis buffer. To purify murine T cells, T cells were isolated by negative selection using a cocktail of biotin-conjugated antibodies against B220, CD49b, CD11b, CD11c, and TER-119 on a magnetic cell separation column (Miltenyi Biotec). To purify human T cells45,46,47, peripheral blood T cells were negatively selected from 10 ml of whole blood from participants using a cocktail of biotin-conjugated antibodies against CD14, CD11b, CD11c, CD19, and CD235a on a magnetic cell separation column (Miltenyi Biotec).
Enzyme-linked immunosorbent assays (ELISAs)
Serum levels of IFN-γ, and TNF-α were analyzed by individual ELISA kits purchased from eBioscience. The IL-17A levels were determined using an ELISA kit from BioLegend.
Single-cell RNA sequencing analyses
Single-cell RNA sequencing was performed using peripheral blood T cells (total bulk T cells including CD4+ and CD8+ T cells) of 12- to 14- week-old UBR2 knockout and wild-type mice. The purified T cells were labeled by BD AbSeq Ab-Oligos reagents according to the manufacturer’s protocol (BD Biosciences, #633793). Next, single-cell capture and cDNA synthesis were performed using the BD Rhapsody Single-Cell Analysis System (210966 v1.0). cDNA libraries were constructed by combining Sample Tag and BD AbSeq libraries (BD Biosciences, Cat 23-21752-00). cDNA libraries of individual cells were constructed using BD Rhapsody WTA Amplification Kit (Cat 633801). Paired-end sequencing was performed on a HiSeq X Ten sequencer (Illumina). Data analysis and quality control were performed following the BD Biosciences Rhapsody pipeline. Uniform Manifold Approximation and Projection (UMAP) generation were conducted with the R package Seurat (v3.0).
T-cell development
Thymocytes, splenocytes, lymph node cells, and bone-marrow-derived cells were harvested from 5-week-old mice. For surface staining, cells were washed by PBS and were stained with antibodies at room temperature for 1 h (anti-CD3, anti-CD4, anti-CD8, anti-B220, and anti-IgM antibodies). After incubation, cells were washed twice with PBS buffer to remove unbound antibodies. For intracellular Foxp3 staining, cells were permeabilized using Foxp3 Fix/Perm solution (BioLegend) and then incubated with diluted anti-Foxp3 antibodies in Foxp3 Perm buffer (BioLegend) at 4 °C for overnight. After incubation, cells were washed twice with Foxp3 Perm wash buffer (BioLegend) to remove unbound antibodies. For flow cytometry, data were acquired with a FACS CantoII (BD Biosciences) and analyzed with FlowJo (v10.8.1) analytical software.
Crystal structure of human Lck protein
The Lck monomer structure model (63 to 509 a.a.) was generated by online homology modeling server, SWISS-MODEL48,49. The structure template was referred to hematopoietic cell kinase (Hck, PDB ID 1QCF, https://www.rcsb.org/structure/1QCF)50 from RCSB Protein Data Bank (RSCB PDB, https://www.rcsb.org/). Lck protein sequence (UniProt ID P06239) was aligned to chicken Src (UniProt ID P00523) by using the online server Clustal Omega Multiple Sequence Alignment51. The interaction sites between SH3 domain, linker and catalytic domain according to the chicken Src structure (PDB ID 2PTK, https://www.rcsb.org/structure/2PTK)36,37 were mapped on the aligned Lck sequence. The ubiquitin structure (PDB ID 3HMH, https://www.rcsb.org/structure/3HMH) was downloaded from RSCB PDB52. All the structures were operated by UCSF Chimera molecular structure interactive visualization and analysis program53, followed by Visual Molecular Dynamics (VMD) molecular visualization program (http://www.ks.uiuc.edu/).
T-cell activation assays
Human Jurkat T cells were stimulated with 5 μg/ml of anti-CD3 (clone OKT3) for the appropriate time at 37 °C. For supernatant collection, purified murine T cells were costimulated with 20 μg/ml of plate-bound anti-CD3 (clone 145-2C11)/anti-CD28 antibodies (clone 37.51). For PLA assay, purified T cells were stimulated with 3 μg/ml of biotin-conjugated anti-CD3 (clone 500A2, Cat 553239, Lot 0314175) plus 3 μg/ml of streptavidin (Sigma, Cat 85878-5MG, Lot 049k8616v).
T-cell differentiation assays
In vitro differentiation assays of Th1, Th17, and Treg cells were performed using the methods described previously47.
EAE induction and MOG restimulation
Mice used in each experiment were 4- to 12-month-old female littermates. Experimental autoimmune encephalomyelitis (EAE) was induced by subcutaneous injection of mice (four per group) with 200 μg myelin oligodendrocyte glycoprotein (MOG) peptides (amino acids 35–55: MEVGWYRSPFSRVVHLYRNGK; Genemed Synthesis)54 emulsified in CFA (Chondrex). Mice were also intraperitoneally injected with 200 ng pertussis toxin (List Biological Laboratories) on days 0, 1, and 2. Antigen-specific T cells were isolated from lymph nodes of MOG-immunized mice at day 14. Antigen-specific T cells were cultured in the presence of 0, 20, 50 μg/ml MOG for 72 h. Culture supernatants from MOG-restimulated T cells were analyzed by ELISA.
Statistical analysis and reproducibility
Statistical analyses were performed by using Excel (v16.7) or BD SEQGEQ (v1.8.0). The statistical significance between two unpaired groups was analyzed using one- or two-tailed Student’s t test. P values of less than 0.05 were considered statistically significant. Symbols of p values by one-tailed Student’s t test represent §p < 0.05 (Fig. 7d, middle). Symbols of p values by two-tailed Student’s t test represent *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 (Figs. 5g, 5h, 7b, 7c, and 7i). Wilcoxon’s rank-sum test was used to analyze violin plots. Symbols of p values by Wilcoxon’s rank-sum test represent *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 (Fig. 5g). For reproducibility, experiments were performed 3 independent times with n = 6 (IFN-γ and IL-17A) or n = 7 (TNF-ɑ) (Fig. 5h); 6 independent times with n = 10 (Fig. 7b-c); 3 independent times with n = 6 (wild-type), n = 4 (DUSP22 KO), or n = 2 (DUSP22/UBR2 double KO) (Fig. 7d); 3 independent times with n = 6 (Fig. 7i). The data shown are representatives of 2 independent experiments (Fig. 2c and e; Fig. 6d, i, and j; Fig. 7e) and 3 independent experiments (Fig. 1b–d, f, g–m; Fig. 2a, b, f, and g; Fig. 3b–i; Fig. 4a, c–f; Fig. 5b, c; Fig. 6a–c, f–h; Fig. 7a, f–h). Data are presented as mean values ± SEM (Figs. 5h, 7b, 7c, 7d, 7i).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data are available in the main text or the supplementary materials. The raw single cell RNA sequencing data generated in this study have been deposited in the Sequence Read Archive database under accession code of SRR25120110. The raw proteomic mass data generated in this study have been deposited in the ProteomeXchange Consortium via the PRIDE proteomics IDEntifications database under accession codes of PXD043490, PXD043463, PXD043457, and PXD043454. The protein structure was modeled by SWISS-MODEL web server and visualized by Visual Molecular Dynamics (VMD) molecular visualization program (http://www.ks.uiuc.edu/). The chicken Src structure (PDB ID 2PTK) was download from [https://www.rcsb.org/structure/2PTK]. The ubiquitin structure (PDB ID 3HMH) was downloaded from RSCB PDB [https://www.rcsb.org/structure/3HMH]. Source data are provided with this paper.
References
Gaud, G., Lesourne, R. & Love, P. E. Regulatory mechanisms in T cell receptor signalling. Nat. Rev. Immunol. 18, 485–497 (2018).
Courtney, A. H., Lo, W.-L. & Weiss, A. TCR signaling: mechanisms of initiation and propagation. Trends Biochem. Sci. 43, 108–123 (2018).
Rao, N. et al. Negative regulation of Lck by Cbl ubiquitin ligase. Proc. Natl. Acad. Sci. USA 99, 3794–3799 (2002).
Chen, A. J. et al. The dual specificity JKAP specifically activates the c-Jun N-terminal kinase pathway. J. Biol. Chem. 277, 36592–36601 (2002).
Chuang, H. C. & Tan, T. H. MAP4K family kinases and DUSP family phosphatases in T-cell signaling and systemic lupus erythematosus. Cells 8, 1433 (2019).
Huang, C. Y. & Tan, T. H. DUSPs, to MAP kinases and beyond. Cell Biosci. 2, 24 (2012).
Chen, H.-F., Chuang, H.-C. & Tan, T.-H. Regulation of dual-specificity phosphatase (DUSP) ubiquitination and protein stability. Int. J. Mol. Sci. 20, 2668 (2019).
Melard, P. et al. Molecular alterations and tumor suppressive function of the DUSP22 (dual specificity phosphatase 22) gene in peripheral T-cell lymphoma subtypes. Oncotarget 7, 68734–68748 (2016).
Zhou, R. et al. JNK pathway-associated phosphatase/DUSP22 suppresses CD4+ T-cell activation and Th1/Th17-cell differentiation and negatively correlates with clinical activity in inflammatory bowel disease. Front. Immunol. 8, 781 (2017).
Chuang, H.-C. et al. Downregulation of the phosphatase JKAP/DUSP22 in T cells as a potential new biomarker of systemic lupus erythematosus nephritis. Oncotarget 7, 57593 (2016).
Chen, M.-H., Chuang, H.-C., Yeh, Y.-C., Chou, C.-T. & Tan, T.-H. Dual-specificity phosphatases 22-deficient T cells contribute to the pathogenesis of ankylosing spondylitis. BMC Med. 21, 1–15 (2023).
Li, J.-P., Fu, Y.-N., Chen, Y.-R. & Tan, T.-H. JNK pathway-associated phosphatase dephosphorylates focal adhesion kinase and suppresses cell migration. J. Biol. Chem. 285, 5472–5478 (2010).
Ge, C. et al. Hepatocyte phosphatase DUSP22 mitigates NASH-HCC progression by targeting FAK. Nat. Commun. 13, 1–21 (2022).
Li, J.-P. et al. The phosphatase JKAP/DUSP22 inhibits T-cell receptor signalling and autoimmunity by inactivating Lck. Nat. Commun. 5, 1–13 (2014).
Zhou, W., Wei, W. & Sun, Y. Genetically engineered mouse models for functional studies of SKP1-CUL1-F-box-protein (SCF) E3 ubiquitin ligases. Cell Res. 23, 599–619 (2013).
Skaar, J. R., Pagan, J. K. & Pagano, M. Mechanisms and function of substrate recruitment by F-box proteins. Nat. Rev. Mol. Cell Biol. 14, 369–381 (2013).
Tasaki, T. et al. A family of mammalian E3 ubiquitin ligases that contain the UBR box motif and recognize N-degrons. Mol. Cell. Biol. 25, 7120–7136 (2005).
MacLennan, M. et al. Mobilization of LINE-1 retrotransposons is restricted by Tex19. 1 in mouse embryonic stem cells. eLife 6, e26152 (2017).
Xu, H. et al. The N-end rule ubiquitin ligase UBR2 mediates NLRP1B inflammasome activation by anthrax lethal toxin. EMBO J. 38, e101996 (2019).
Kwak, K. S. et al. Regulation of protein catabolism by muscle-specific and cytokine-inducible ubiquitin ligase E3α-II during cancer cachexia. Cancer Res. 64, 8193–8198 (2004).
Mao, J. et al. UBR2 enriched in p53 deficient mouse bone marrow mesenchymal stem cell-exosome promoted gastric cancer progression via Wnt/β-catenin pathway. Stem Cells 35, 2267–2279 (2017).
Bulajic, M., Panic, N. & Lohr, J. M. Helicobacter pylori and pancreatic diseases. World J. Gastrointest. Pathophysiol. 5, 380–383 (2014).
Mougeot, J.-L. C., Li, Z., Price, A. E., Wright, F. A. & Brooks, B. R. Microarray analysis of peripheral blood lymphocytes from ALS patients and the SAFE detection of the KEGG ALS pathway. BMC Med. Genomics 4, 1–19 (2011).
Santos, A. et al. Comprehensive comparison of large-scale tissue expression datasets. PeerJ 3, e1054 (2015).
Su, A. I. et al. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc. Natl. Acad. Sci. USA 101, 6062–6067 (2004).
Lountos, G. T., Cherry, S., Tropea, J. E. & Waugh, D. S. Structural analysis of human dual-specificity phosphatase 22 complexed with a phosphotyrosine-like substrate. Acta Crystallogr. F Struct. Biol. Commun. 71, 199–205 (2015).
Soderberg, O. et al. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat. Methods 3, 995–1000 (2006).
Kwon, Y. T. & Ciechanover, A. The ubiquitin code in the ubiquitin-proteasome system and autophagy. Trends Biochem. Sci. 42, 873–886 (2017).
Radivojac, P. et al. Identification, analysis, and prediction of protein ubiquitination sites. Proteins 78, 365–380 (2010).
**e, C. M., Wei, W. & Sun, Y. Role of SKP1-CUL1-F-box-protein (SCF) E3 ubiquitin ligases in skin cancer. J. Genet. Genomics 40, 97–106 (2013).
Skaar, J. R., D’Angiolella, V., Pagan, J. K. & Pagano, M. SnapShot: F box proteins II. Cell 137, 1358–1358.e1351 (2009).
Kwon, Y. T. et al. Female lethality and apoptosis of spermatocytes in mice lacking the UBR2 ubiquitin ligase of the N-end rule pathway. Mol. Cell. Biol. 23, 8255–8271 (2003).
Shah, K., Al-Haidari, A., Sun, J. & Kazi, J. U. T cell receptor (TCR) signaling in health and disease. Signal. Transduc. Target. Ther. 6, 412 (2021).
Madiraju, C., Novack, J. P., Reed, J. C. & Matsuzawa, S. I. K63 ubiquitination in immune signaling. Trends Immunol 43, 148–162 (2022).
Miller, S. D., Karpus, W. J. & Davidson, T. S. Experimental autoimmune encephalomyelitis in the mouse. Curr. Protoc. Immunol. 88, 15.1 (2010).
Gonfloni, S. et al. The role of the linker between the SH2 domain and catalytic domain in the regulation and function of Src. EMBO J. 16, 7261–7271 (1997).
Williams, J. C. et al. The 2.35 Å crystal structure of the inactivated form of chicken Src: a dynamic molecule with multiple regulatory interactions. J. Mol. Biol. 274, 757–775 (1997).
Nika, K. et al. Constitutively active Lck kinase in T cells drives antigen receptor signal transduction. Immunity 32, 766–777 (2010).
Philipsen, L. et al. De novo phosphorylation and conformational opening of the tyrosine kinase Lck act in concert to initiate T cell receptor signaling. Sci. Signal. 10, eaaf4736 (2017).
Li, J. P., Fu, Y. N., Chen, Y. R. & Tan, T. H. JNK pathway-associated phosphatase dephosphorylates focal adhesion kinase and suppresses cell migration. J. Biol. Chem. 285, 5472–5478 (2010).
Abraham, R. T. & Weiss, A. Jurkat T cells and development of the T-cell receptor signalling paradigm. Nat. Rev. Immunol. 4, 301–308 (2004).
Marhaba, R. et al. The protein tyrosine kinase p56lck regulates the serine-base exchange activity in Jurkat T cells. FEBS Lett. 405, 163–166 (1997).
Chuang, H.-C. et al. MAP4K3/GLK promotes lung cancer metastasis by phosphorylating and activating IQGAP1. Cancer Res. 79, 4978–4993 (2019).
Chuang, H. C. et al. SARS‐CoV‐2 spike protein enhances MAP4K3/GLK‐induced ACE2 stability in COVID‐19. EMBO Mol. Med. 14, e15904 (2022).
Chuang, H. C. et al. Induction of interferon‐γ and tissue inflammation by overexpression of eosinophil cationic protein in T cells and exosomes. Arthritis Rheum. 74, 92–104 (2022).
Chuang, H.-C. et al. BPI overexpression suppresses Treg differentiation and induces exosome-mediated inflammation in systemic lupus erythematosus. Theranostics 11, 9953 (2021).
Chuang, H.-C. et al. DUSP8 induces TGFβ-stimulated IL-9 transcription and Th9-mediated allergic inflammation by promoting nuclear export of Pur-α. J. Clin. Invest. 133, e166269 (2023).
Bienert, S. et al. The SWISS-MODEL Repository—new features and functionality. Nucleic Acids Res. 45, D313–D319 (2017).
Studer, G. et al. QMEANDisCo—distance constraints applied on model quality estimation. Bioinformatics 36, 1765–1771 (2020).
Schindler, T. et al. Crystal structure of Hck in complex with a Src family–selective tyrosine kinase inhibitor. Mol. Cell 3, 639–648 (1999).
Madeira, F. et al. Search and sequence analysis tools services from EMBL-EBI in 2022. Nucleic Acids Res. 50, W276–W279 (2022).
Datta, A. B., Hura, G. L. & Wolberger, C. The structure and conformation of Lys63-linked tetraubiquitin. J. Mol. Biol. 392, 1117–1124 (2009).
Pettersen, E. F. et al. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Shui, J.-W. et al. Hematopoietic progenitor kinase 1 negatively regulates T cell receptor signaling and T cell–mediated immune responses. Nat. Immunol. 8, 84–91 (2007).
Acknowledgements
The authors thank the Transgenic Mouse Core (NHRI, Taiwan) for generation of DUSP22 knockout mice and UBR2 knockout mice. The authors thank the NHRI Laboratory Animal Center for mouse housing. The authors thank the Institute of Biological Chemistry of Academia Sinica for mass spectrometry analyses. The authors thank the core facilities of NHRI for confocal microscopy and fluorescence microscope. The authors thank members of Tan Lab for technical assistance, including Ms. Jhih-Yu Yang and Chia-Hsin Hsueh for in vitro binding assays, Mr. Pu-Ming Hsu for immunoprecipitation assays, Yu-Zhi **ao for human patients PLA assays, and Ms. Ching-Yi Tsai for ELISAs. The authors also thank Dr. Hsien-Yi Chiu and Dr. Ming-Han Chen for providing clinical samples. This work was supported by grants from NHRI (IM-112-SP-01 to T.-H.T.) and the National Science and Technology Council, Taiwan (MOST-107-2314-B-400-008, and MOST-110-2320-B-400-018, NSTC-111-2320-B-400-003, and NSTC-112-2320-B-400-023) to T.-H.T.; MOST-108-2628-B-400-001 and NSTC-112-2320-B-400-024 to H.-C.C.). T.-H.T. is a Taiwan Bio-Development Foundation (TBF) Endowed Chair Professor in Biotechnology.
Author information
Authors and Affiliations
Contributions
H.-C.C., Y.-C.S, and H.-F.C. designed and performed experiments, analyzed and interpreted data, and wrote the manuscript; C.-Y.W. and Y.-R.C. performed experiments; C.-W.W. analyzed the crystal structure of human Lck protein; T.-H.T. conceived of the study, supervised experiments, and wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Hyunjoo Cha-Molstad, José González-Navajas and Pierre Lutz for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shih, YC., Chen, HF., Wu, CY. et al. The phosphatase DUSP22 inhibits UBR2-mediated K63-ubiquitination and activation of Lck downstream of TCR signalling. Nat Commun 15, 532 (2024). https://doi.org/10.1038/s41467-024-44843-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-44843-w
- Springer Nature Limited