

Abstract
Cranial neural crest cells are an evolutionary innovation of vertebrates for craniofacial development and function, yet the mechanisms that govern the cell fate decisions of postmigratory cranial neural crest cells remain largely unknown. Using the mouse molar as a model, we perform single-cell transcriptome profiling to interrogate the cell fate diversification of postmigratory cranial neural crest cells. We reveal the landscape of transcriptional heterogeneity and define the specific cellular domains during the progression of cranial neural crest cell-derived dental lineage diversification, and find that each domain makes a specific contribution to distinct molar mesenchymal tissues. Furthermore, IGF signaling-mediated cell-cell interaction between the cellular domains highlights the pivotal role of autonomous regulation of the dental mesenchyme. Importantly, we reveal cell-type-specific gene regulatory networks in the dental mesenchyme and show that Foxp4 is indispensable for the differentiation of periodontal ligament. Our single-cell atlas provides comprehensive mechanistic insight into the cell fate diversification process of the cranial neural crest cell-derived odontogenic populations.
Similar content being viewed by others

Introduction
Craniofacial structures are highly complex tissues comprised of various cell types. The establishment of cranial neural crest cells (CNCCs) was a critical step in the evolution of the vertebrate head1. CNCCs are a uniquely multipotent population that give rise to numerous cell types in the vertebrate craniofacial complex, including bones and cartilage, teeth, cranial ganglia, glial cells and pigment cells2. Unlike trunk neural crest cells, CNCCs migrate throughout the head, differentiate into different cell types upon reaching their final destinations. Defects in the migration or differentiation of CNCCs lead to a variety of developmental abnormalities in craniofacial structures, such as cleft palate and tooth malformation3.
Although extensive knowledge about CNCCs has accumulated over last several decades, the mechanisms that control late-stage CNCC fate decisions during craniofacial morphogenesis remain to be elucidated4. Recently, a single-cell study of pre-migratory and migratory neural crest cells (NCCs) revealed that early NCCs undergo a sequential series of binary cell fate decisions, and that, compared to the trunk neural crest, CNCCs have a lineage bias towards mesenchymal fate when they delaminate from the neural tube5. Interestingly, Zalc et al. have found that CNCC precursors undergo in vivo programming by reactivating pluripotency factors to acquire the ectomesenchymal fate6. Our recent study has shown that postmigratory CNCCs in the first pharyngeal arch undergo dynamic intrapharyngeal arch movement and contribute to multiple lineages as they undergo a sequence of bifurcating fate decisions7. Nonetheless, how these postmigratory CNCCs further diversify after acquiring mesenchymal fate and eventually give rise to committed cell types that form functional adult tissues remains unclear.
Gene regulatory networks (GRNs) are the information-processing system that cells utilize to govern complex spatiotemporal developmental programs8. Williams et al. have recently portrayed the GRN of early CNCCs by combining epigenomic and transcriptional profiling approaches9. They found that multiple regulatory layers including cis regulatory elements and transcription factors govern the neural crest ontogeny. However, despite advances in our knowledge of the mechanisms that control early CNCC fate decisions, limited information is available about how postmigratory CNCCs acquire cell-fate-specific programs10. Furthermore, the GRNs of postmigratory CNCCs at the single-cell level remain largely unknown11.
The tooth is a vertebrate-specific organ and, similar to other ectodermal organs, including the hair and mammary gland, its development involves sequential, reciprocal epithelial–mesenchymal interactions12. Therefore, studies of the tooth model provide broadly applicable insights into organogenesis. Each tooth can be anatomically separated into two components: the crown which development begins during embryonic stage and the root which starts to develop postnatally13. Tooth development initiates with dental lamina which is derived from thickened oral epithelium and will invaginate into the underlying dental mesenchyme to form the tooth bud. The dental mesenchyme then condenses around the epithelial tooth bud and instructs tooth morphogenesis through the bud, cap, bell and crown-root transition stages14. The dental mesenchyme is derived from CNCCs after their migration into the oral region of first pharyngeal arch15; subsequently, it segregates into dental papilla and follicle lineages. Dental papilla cells give rise to dental pulp and odontoblasts, whereas dental follicle cells differentiate into periodontal tissues including the periodontal ligament (PDL), cementum, and alveolar bone16. The multipotency of dental mesenchyme therefore enables tooth to be an excellent model to investigate the lineage diversification of postmigratory CNCC. Moreover, the anatomy and developmental stages of tooth are well established, further underlining the competence of tooth to be an ideal model to study the late-stage CNCC. Progenitor cells in the dental mesenchyme are highly heterogeneous in vivo17,18,19 and the cellular heterogeneity of mouse incisors and adult human teeth has been recently evaluated20,21; however, the landscapes of transcriptional heterogeneity and GRNs across mouse molar development at single-cell resolution have not yet been revealed. Furthermore, the mechanisms that control cellular domain establishment and lineage development during mouse molar formation remain unclear.
In this study, we used the mouse molar as a model to interrogate the cellular heterogeneity and GRNs that govern postmigratory CNCC fate decisions. We transcriptionally profiled single cells from mouse molars at several critical developmental stages and identified the landscape of cellular heterogeneity within the dental mesenchyme across mouse molar development from embryonic to postnatal stages. We defined the cellular domains through newly identified signature genes in the dental mesenchyme as the molar develops and found that each one makes a specific contribution to mouse molar formation. Furthermore, we investigated the cell-cell interaction in the dental mesenchyme and found that disruption of IGF signaling-mediated cell–cell interaction disturbed PDL development, highlighting the importance of cell-autonomous regulation for postmigratory CNCCs. Importantly, we identified cell population-specific regulons in the dental mesenchyme and found that loss of a critical regulon, Foxp4, can lead to developmental defects in PDL lineage. Thus, this single-cell atlas reveals previously unidentified heterogeneity within the mesenchyme of the develo** molar and provides comprehensive understanding of the mechanisms governing CNCC-derived cell lineage diversification during molar morphogenesis.
Results
Postmigratory CNCC segregation and lineage commitment during early tooth development
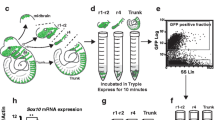
Postmigratory CNCCs commit to the dental mesenchymal lineage after they arrive in the oral region of the first pharyngeal arch. The dental tissues then further differentiate through a series of developmental steps until the tooth erupts. To comprehensively reveal the cellular heterogeneity and function of postmigratory CNCCs during mouse molar development, we sequenced the individual cell transcriptomes in the tooth and surrounding tissue from E13.5 to P7.5 (Fig. 1a). We first analyzed the cell populations at E13.5, the onset of tooth development. Leveraging unsupervised clustering and marker analysis, we identified a large group of CNCC-derived cells which include chondrogenic cells, osteogenic cells, dermal fibroblasts and dental mesenchyme (Supplementary Fig. 1a–c). Through further subclustering analysis with a known early dental mesenchyme marker, Tfap2b22, one cell population (Tfap2b+/Lhx6+) was identified as the dental mesenchyme at E13.5 (Fig. 1b), suggesting that dental mesenchymal cells are relatively homogenous right after postmigratory CNCCs commit to the dental fate at E13.5. Consistent with marker analysis, RNAscope in situ staining of these marker genes showed they are expressed in the dental mesenchyme surrounding the dental epithelium (Fig. 1c–f). The cellular domain of the dental mesenchyme in the mouse molar at E13.5 is illustrated in Fig. 1g. To confirm that this cell population we identified at E13.5 is the progenitor population that contributes to the tooth formation, we performed lineage tracing experiments with Lhx6-CreERT2;tdT mice. Tamoxifen was injected at E12.5 to enable the activation of CreERT2 and we verified that Lhx6+ cells are present in the dental mesenchyme at E14.0 (Fig. 1h). We found that Lhx6+ cells can give rise to all the lineages in the dental mesenchyme by P21.5 (Fig. 1i–m), suggesting that the Lhx6+ cell population at E13.5 is indeed the dental mesenchymal progenitor population that contributes to tooth formation.

a Schematic drawings illustrating the experimental design in this study. b UMAP plot of the cell cluster of the dental mesenchyme in the mouse molar at E13.5. c–f Feature plot and RNAscope staining of Tfap2b and Lhx6 in the mouse molar at E13.5 (n = 3). g Schematic drawing of the cellular domain in the dental mesenchyme of the mouse molar at E13.5. h–m Lineage tracing experiment with Lhx6-CreERT2;tdT mouse molars (n = 3). Injection protocol is illustrated underneath the figures. White dotted lines in d, f, h, i and j outline the dental epithelial cells and in l and m outline the PDL in the mouse molar. White arrows point to the positive signals. Scale bars, 100 μm.
In order to interrogate the lineage development of dental mesenchymal cells, we further analyzed the cell populations in the mouse molar at E14.5. Utilizing known dental mesenchyme markers at the cap stage, Tfap2b and Pax923, we were able to pinpoint the dental mesenchymal cell populations more specifically at this stage (Fig. 2a, b and Supplementary Fig. 2a–c). Consistent with previous studies, we found that Pax9 is expressed in both the dental follicle and papilla at E14.5. To confirm that Pax9+ cells in the mouse molar at E14.5 are dental mesenchymal progenitor cells, we performed lineage tracing experiments with Pax9-CreERT2;tdT mice induced with tamoxifen at E13.5. The results clearly showed that Pax9+ cells are located in the dental mesenchyme at E14.5 and they are indeed progenitor cells, because they can give rise to all the mesenchymal lineages in the mouse molar at P21.5 (Fig. 2c–f). Through subclustering analysis of Pax9+ cells, we found two well-separated clusters corresponding to dental follicle and dental papilla cell populations, suggesting these two lineages segregate at E14.5 (Fig. 2g and Supplementary Fig. 3a–f). We identified the top enriched genes in these two cell populations and found previously unknown unique markers for these two lineages with unbiased marker analysis (Fig. 2h, i). Through RNAscope in situ staining, we found dental papilla marker genes (Crym+/Egr3+/Fgf3+) that are specifically expressed in the dental papilla and absent from the dental follicle (Fig. 2j–l and Supplementary Fig. 3a–c). Conversely, we found dental follicle marker genes (Epha3+/Fxyd7+/Foxf1+) that are expressed in the dental follicle cell domain surrounding the dental epithelium and dental papilla (Fig. 2m–o and Supplementary Fig. 3d–f). These two distinct cellular domains of the dental papilla and follicle in the mouse molar at E14.5 are illustrated in Fig. 2p.

a, b Feature plot and RNAscope staining of Pax9 in the mouse molar at E14.5 (n = 3). c–f Lineage tracing analysis of Pax9+ cells in the dental mesenchyme of the mouse molar from E14.5 to P21.5 (n = 3). g UMAP plot of the dental mesenchyme in the mouse molar at E14.5. h, i Heatmap and Dotplot of signature genes in the dental papilla and follicle of the mouse molar at E14.5. j–l Feature plot and RNAscope staining of dental papilla markers in the mouse molar at E14.5 (n = 3). m–o Feature plot and RNAscope staining of dental follicle markers of the mouse molar at E14.5 (n = 3). p Schematic drawing of the cellular domain in the dental mesenchyme of the mouse molar at E14.5. White dotted lines in b, c, d, k, l, n and o outline the epithelial cells and in f outline the PDL in the mouse molar. White arrows point to the positive signals. Scale bars, 100 μm.
Evolution and spatial separation of cellular domains in the mouse molar
At E16.5, the mouse molar reaches the bell stage of development, at which the crown patterning is determined. Through subclustering and unsupervised marker analysis, we were able to identify four cell populations in the dental mesenchyme at E16.5 (Fig. 3a, Supplementary Fig. 4a–c and Supplementary Fig. 5a–l). Varied signature genes were revealed among these cell clusters (Fig. 3b, c). At this stage, the dental papilla cell population has formed separate coronal (Lmo1+/Fgf3+/Smpd3+) and apical domains (Lhx6+/Fst+/Gldn+) (Fig. 3d–i and Supplementary Fig. 5a–f), a reflection of further differentiation of the dental papilla lineage compared to E14.5. Likewise, the dental follicle cells have also evolved into two cellular domains which are located in the lateral (Lepr+/Foxf1+/Bmp3+) and apical regions (Aldh1a2+/Rasl11a+/Sgk1+) of the dental follicle, respectively (Fig. 3j–o and Supplementary Fig. 5g–l), implying they contribute to the development of spatially distinct regions within the tooth. In order to validate the contributions of the dental follicle cell populations, we generated Lepr-Cre;tdT mice to investigate the contribution of Lepr+ lateral follicle cells during mouse molar development. Lepr+ cells have been reported to be bone marrow mesenchymal stem cells in the long bone24. We found Lepr+ cells in the dental follicle can give rise to the periodontal tissue including PDL and alveolar bone, but these cells were absent from the dental papilla-derived dental pulp and odontoblasts in the root region of the mouse molar, indicating Lepr+ cells are indeed dental follicle progenitor cells that contribute to development of the periodontal tissue in the mouse molar (Fig. 3p–s). The cellular domains of the dental papilla and follicle in the mouse molar at E16.5 are illustrated in Fig. 3t.

a UMAP plot of cell clusters in the dental mesenchyme of the mouse molar at E16.5. b, c Heatmap and Dotplot of signature genes in the dental papilla and follicle of the mouse molar at E16.5. d–o Feature plot and RNAscope staining of dental papilla and dental follicle markers in the mouse molar at E16.5 (n = 3). p–s Lineage tracing experiment performed with Lepr-Cre;tdT mice (n = 3). t Schematic drawing of the cellular domain in the dental mesenchyme of the mouse molar at E16.5. White dotted lines outline the dental epithelial cells and PDL in the mouse molar. White arrows point to the positive signals. Yellow and green arrows in s point to the positive signals in PDL and alveolar bone in the root furcation region, respectively. Scale bars, 100 μm.
To better understand the cellular heterogeneity and role of dental mesenchyme during mouse molar root development, we isolated cells from the molar at P3.5, at which time root development is about to initiate. Six cell populations were assigned to the dental mesenchyme according to the unsupervised clustering and newly identified signature gene analysis at P3.5 (Fig. 4a–c and Supplementary Fig. 6). A lateral follicle domain (Bmp3+/Tnmd+) and an apical follicle domain (Smoc2+/Slc1a3+) were defined based on the expression patterns of these newly identified signature genes at P3.5 (Fig. 4d–k). The follicle pattern appeared similar to that observed at E16.5, suggesting that the cellular domains in the dental follicle are already established at E16.5. In order to examine the contribution of dental follicle cells to tooth root development at P3.5, we first analyzed the expression of Pthlh (the gene encoding Pthrp), a known marker of the dental follicle in the mouse molar17, and confirmed it is expressed in both the lateral and apical follicle domains (Supplementary Fig. 7a, b). To validate the contribution of Pthrp+ cells, we performed a lineage tracing experiment with Pthrp-CreERT2;tdT mice. Tamoxifen was injected at P3.5 to enable the activation of CreERT2 and, consistent with its expression pattern, we found Pthrp+ cells in both the lateral and apical domains of the dental follicle two days after the tamoxifen induction (Supplementary Fig. 7c). We also revealed that Pthrp+ cells can give rise to periodontal tissue in both the furcation and root-forming regions of the mouse molar (Supplementary Fig. 7d, e). To further understand the contributions of the apical domains in the dental follicle, we generated Slc1a3-CreERT2;tdT mice to target the apical follicle domain and performed lineage tracing. Slc1a3 is a known marker for astrocytes in the nervous system25. The cell lineage tracing data showed that Slc1a3+ cells are located in the apical region of the dental follicle two days after tamoxifen induction and that they largely contribute to the PDL and alveolar bone formation in the root furcation region where the two roots of mouse molar are separated (Fig. 4m–o), suggesting dental follicle cells in the apical domain govern the cell lineages for root furcation development. The cellular domains of the dental mesenchyme at P3.5 were illustrated in Fig. 4q based on the analysis of the expression of signature genes and lineage tracing data (Fig. 4d–p and Supplementary Fig. 8).

a UMAP plot of cellular domains in the mouse molar at P3.5. b, c Heatmap and Dotplot of signature genes of dental mesenchymal cell populations in the mouse molar. d–f Feature plot and RNAscope staining of lateral follicle markers in the mouse molar at P3.5 (n = 3). g Schematic drawing of the lateral follicle domain in the mouse molar at P3.5. h–j Feature plot and RNAscope staining of apical follicle markers in the mouse molar at P3.5 (n = 3). k Schematic drawing of the apical follicle domain in the mouse molar at P3.5. l–o Feature plot of Slc1a3 and lineage tracing analysis of Slc1a3+ cells in the mouse molar from P3.5 to P21.5 (n = 3). p UMAP plot of cellular domains in the mouse molar at P3.5 with annotation. q Schematic drawing of the cellular domains in the mouse molar at P3.5. Injection protocol is illustrated underneath the figures. Black dotted lines outline the cellular domains in the Feature plots. The white box in n is magnified in o. White dotted lines in o outline the PDL in the mouse molar. White arrows point to the positive signals. Yellow and green arrows in o point to the positive signals in PDL and alveolar bone in the root furcation region, respectively. Scale bars, 100 μm.
Postnatal apical papilla domain retains bipotent progenitor cells that give rise to dental pulp cells and odontoblasts during mouse molar development
In analyzing the cellular heterogeneity within the CNCC-derived dental papilla, we identified four cellular domains with newly identified marker genes in the dental papilla of the mouse molar at P3.5 through unsupervised marker analysis. Odontoblasts (Phex+/Ifitm5+) are terminally differentiated dentin-forming cells that are located underneath the dentin in the mouse molar (Supplementary Fig. 8a-d). Cell populations within the coronal dental papilla (Enpp6+/Fabp7+), middle dental papilla (Nnat+/Rab3b+) and apical dental papilla (Aox3+/Tac1+) are dental papilla cells with different differentiation status (Supplementary Fig. 8e–p). In order to understand the differentiation trajectory of these cell populations in the dental papilla, we took advantage of Velocity analysis, which infers the differentiation status of the cells based on the ratio of spliced and unspliced mRNA26. The results indicated that apical dental papilla cells are progenitor cells that can give rise to odontoblasts and the dental pulp lineage (Fig. 5a). Because highly proliferative cells are located in the apical region of the dental papilla in the mouse molar (Fig. 5b, c), in order to confirm the differentiation trajectory in vivo, we first examined whether EdU+ cycling cells can contribute to the dental pulp and odontoblast formation through an EdU tracing experiment. The results suggested that EdU+ cells within the apical dental papilla domain indeed give rise to odontoblast and pulp lineages nine days after tracing (Fig. 5d, e). In addition, we found that Fgf3, encoding a ligand for FGF signaling39,40,41 and the data clearly depict the organization of the cellular domains in these cranial crest derived structures, highlighting the progressive establishment of cellular domains by postmigratory CNCCs. CNCCs migrate into the first pharyngeal arch and establish four patterning domains in the mandible along the proximal-distal and oral-aboral axes42. Our previous study has identified the cellular heterogeneity within the patterning domains of the mandible and has demonstrated that the cells residing in the proximal domain are progenitors that give rise to major lineages in the mandible, highlighting a spatiotemporal sequence of fate decisions during postmigratory CNCC lineage development7.
Postmigratory CNCCs undergo cell fate restrictions and ultimately commit to the dental fate after they arrive in the oral domain of the first pharyngeal arch. In this study, we have also uncovered the gradual establishment of the cellular domains in the dental mesenchyme. Our data suggest that the cell populations at E13.5 are relatively homogenous and they maintain multipotency necessary to establish different lineages at later stages. The segregation of dental mesenchymal cells into dental papilla and dental follicle lineages started at E14.5, a first sign of lineage diversification in the dental mesenchyme during molar development. We show numerous different transcription factors enriched in the dental papilla and follicle, and the key determinant for the cell fate choice between these two lineages still needs to be further investigated in the future. Interestingly, the dental follicle cells establish two distinct cellular domains from E16.5, a reflection of different functional deployment in these two cell populations. Indeed, the lineage tracing data from Slc1a3+ cells indicates that the apical follicle domain largely contributes to the periodontal tissue formation in the furcation region of the mouse molar, suggesting that the apical follicle domain governs the lineage development in the root furcation region. Nonetheless, the exact functional difference between these two follicle cellular domains remains to be elucidated in the future.
The cell populations in the dental mesenchyme keep evolving until P3.5, by which time the growth of the tooth root initiates, suggesting that forming the tooth root is more demanding in terms of functional requirements from complex cell populations than forming the crown. However, the mechanisms that maintain the boundaries between different cellular domains in the dental mesenchyme still need to be further explored in the future. Consistent with our findings, the progenitor cells in the dental mesenchyme that contribute to tooth root formation have been shown to be highly heterogeneous43,44. Interestingly, we have discovered that apical papilla cells are a uniquely bipotent population that gives rise to both dental pulp and odontoblast lineages at P3.5, providing more comprehensive understanding about the dental papilla lineages beyond what has been previously reported19. Collectively, our results reveal that the lineages in the dental mesenchyme are progressively diversified and determined after the commitment of postmigratory CNCCs to the odontogenic fate. The dental mesenchyme in the mouse molar thus provides an excellent model in which to study the lineage development of postmigratory CNCCs.
The development and differentiation of CNCCs are controlled via complex GRNs, comprising transcription factors, cis regulatory elements and epigenetic modifiers45. Numerous studies have identified the GRNs in premigratory and migratory CNCCs, expanding our knowledge of the mechanisms that control CNCC development at early stages46. Simoes-Costa et al. revealed a cranial neural crest-specific transcriptional circuit which includes Sox8, Tfap2b and Ets1, and found that this circuit not only determines the chondrogenic fate of CNCCs but can also reprogram trunk neural crest cells into craniofacial cartilage, highlighting the power of this GRN to alter cell identity in the neural crest47. Early CNCCs maintain patterning plasticity and also respond to local cues to induce positionally specific transcriptional programs. These distinct transcriptional programs are selectively activated in specific CNCCs but silenced in others. A recent study has shown that positional genes of CNCCs depend on bivalent poised chromatin organization and have accessible H3K27me3/H3K4me2 bivalent distal regulatory elements48. The differentiation of CNCCs into distinct derivatives is mediated by gene regulatory programs that compartmentalize these cells into different territories, each with a specific and unique regulatory state. Attanosio et al. have identified the active enhancers in the craniofacial skeleton and their different domains of activity, and showed that craniofacial malformations can arise from deletion of combined enhancer activity49.
GRNs in the enamel knot of the dental epithelium have been shown to be important for tooth development and patterning. For instance, disruption of Eda signaling leads to a reduced number of cusps and disorganized tooth patterning50. In this study, we have identified GRNs in the dental mesenchyme across mouse molar development for the first time. Numerous regulons identified in this study, such as DLX family members Dlx1, Dlx3 and Dlx5, have been shown to play pivotal roles in tooth development. GRNs have also been proposed to be key coordinators during tooth evolution51. FOX family genes are also broadly distributed in the identified regulon dataset. FOX transcription factors are evolutionarily conserved across species52 and they are important for regulating a wide variety of biological functions both during development and in adult organisms. Mutations of FOX genes can have profound effects, seen in human diseases such as Rieger syndrome and lymphedema–distichiasis syndrome53. The Foxp family, which is composed of Foxp1, Foxp2, Fxop3 and Foxp4, is also involved in diverse biological processes including development, immune disorders and cancer progression54. For example, loss of Foxp1/Foxp2/Foxp4 can lead to disturbed bone and cartilage formation55. In this study, we found that Foxp4 is expressed in the dental mesenchyme and is required for the PDL differentiation largely in the root furcation region, highlighting the lineage-specific requirement for root development.
The interaction between different NCC lineages is known to be important for NCC development and is implicated in related diseases56. Tissue-tissue interactions between dental mesenchyme and epithelium play a key role in guiding the entire tooth developmental process. However, the cell-cell interaction within the dental mesenchymal linages is poorly understood. The cell-cell interaction network revealed in this study suggests that cell-autonomous regulation within the dental mesenchyme is also important for tooth development. The single-cell atlas established in this study provides a valuable resource for understanding the development of CNCC-derived dental mesenchymal lineages during tooth development. The cellular heterogeneity and domains identified in the dental mesenchyme enable the mouse molar to serve as an excellent model to investigate the lineage development and cell fate decisions of postmigratory CNCCs. The mechanisms uncovered in this study may also have important implications for other CNCC-derived tissues, organs and related diseases.
Methods
Animal studies
Igf1flox/flox, Igf1rflox/flox, Lepr-Cre, Lhx6–CreERT2, Rosa2 <fstdTomat>, and Slc1a3-CreERT2 mouse strains were purchased from Jackson Laboratory. Pthrp-CreERT2 mouse line was generously shared by Dr. Wanida Ono17. Foxp4flox/flox mouse strain was shared by Dr. Edward Morrisey, University of Pennsylvania57. Osr2-Cre mouse line was shared by Dr. Rulang Jiang, Cincinnati Children’s Hospital Medical Center58. Fgf3-CreERT2 mouse line was generously shared by Dr. Hu Zhao, Chinese Institute for Brain Research. A Pax9-CreERT2 mouse line was generated for this study. Pax9 sgRNA (TCTGATGGGAGCGTCCACTCCGG) was designed to target the 3’ end of Pax9, 34 bp downstream of the target site for the previously generated Pax9-CreER mouse23. This Pax9-CreERT2 mouse line was generated and genotyped as previously described23, except that 20 ng/μl Cas9 Nuclease v3 and Pax9 sgRNA (0.61pmol/μl crRNA+ 0.61pmol/μl trancrRNA) were used in the injection mix. One male founder mouse was generated for breeding. The tdT reporter mouse line was mated with the Pax9-CreERT2 line to validate whether the CreERT2 activity is comparable to endogenous Pax9 expression. The reagents used for the generation of this Pax9-CreERT2 line were as below: Alt-RTM S.p. HiFi Cas9 Nuclease v3 (1081060, Integrated DNA Technologies IDT), Alt-R® CRISPR-Cas9 tracrRNA (0000531479, Integrated DNA Technologies IDT) and crRNA (283557335, Integrated DNA Technologies IDT). Pax9-CreERT2 mouse line will be shared upon request through the corresponding author. All mouse procedures were approved by the Department of Animal Resources and Institutional Animal Care and Use Committee of the University of Southern California (protocol 20299).
All mice were housed under specific-pathogen-free (SPF) conditions in a 12 h light/12 h dark cycle, 18–23 °C and 40–60% humidity. Individuals were identified via ear tags and analyzed in a mixed background. Genoty** was performed as follows: tail biopsies were lysed by incubating at 55 °C overnight in DirectPCR tail solution (Viagen 102T) and heat-inactivated for 30 min at 85 °C for PCR-based genoty** (GoTaq Green MasterMix, Promega, and C1000 Touch Cycler, Bio-rad). Mice were euthanized by CO2 overdose followed by cervical dislocation. All mice were used for analysis regardless of sex. To activate CreERT2, tamoxifen (Sigma-Aldrich) dissolved in corn oil (Sigma-Aldrich, 20 mg/ml) was injected intraperitoneally (200 mg/kg).
MicroCT analysis
Imaging was performed on a SCANCO μCT50 device at the University of Southern California Molecular Imaging Center with the X-ray source at 70 kVp and 114 μA. The resolution of images was 10 μm. Images were reconstructed in 3D using AVIZO 7.1 (Visualization Sciences Group).
Immunostaining and RNAscope in situ hybridization
Mouse mandibles were dissected and fixed in 4% PFA (VWR) overnight. After decalcification in 10% EDTA (Sigma-Aldrich) for 4 weeks, samples were incubated in 15% sucrose (VWR) for 2 h followed by 30% sucrose overnight, then embedded in OCT (VWR). Frozen tissue blocks were sectioned at 10 μm on a cryostat (Leica) and mounted on SuperFrost Plus slides (Thermo Fisher Scientific). The tissue sections were blocked for 1 h at room temperature in blocking solution (Vector Laboratories). Sections were then incubated with Rabbit anti-Foxp4 (1:200, HPA007176, MilliporeSigma), Rabbit anti-Periostin (1:100, ab14041, Abcam) and Rabbit anti-Ki67 (1:100, ab15580, Abcam) antibodies diluted in blocking solution at 4 °C overnight. Sections were washed three times with PBS (Thermo Fisher Scientific) prior to incubation with Goat anti-Rabbit secondary antibodies (1:200, A-11011, Thermo Fisher Scientific) in blocking solution at room temperature for 1 h. DAPI (Thermo Fisher Scientific) stained the nuclei. Images were acquired using a Keyence microscope (Carl Zeiss). Non-immune immunoglobulins of the same isotype as the primary antibodies were used as negative controls.
ISH was performed using an RNAscope Multiplex Fluorescent Assay (Advanced Cell Diagnostics). Briefly, tissues were fixed in 4% PFA overnight at room temperature before cryosectioning to 10 μm thickness. Target retrieval was performed on the sections for 5 min at 95–100 °C, followed by protease (Advanced Cell Diagnostics) treatment for 7 min at 40 °C. Probes (Advanced Cell Diagnostics) were then hybridized for 2 h at 40 °C followed by RNAscope amplification reagents provided in RNAscope Multiplex Fluorescent Reagent Kit v2 (Advanced Cell Diagnostics). The signal was detected by TSA Plus Cyanine 3 (PerkinElmer).
EdU incorporation and staining
Mouse pups at P3.5 were injected intraperitoneally with EdU (Thermo Fisher Scientific, 10 μg/g body weight) before being euthanized. Mandibles were then fixed and decalcified as described above. Click-iT plus EdU cell proliferation kit (Thermo Fisher Scientific, C10637) was used to detect EdU according to the manufacturer’s instructions.
ImageJ analysis
ImageJ was used to determine the tooth root length and PDL area. Images of the controls and mutants were first converted to 8-bit binary. The root length and PDL area were then calculated by the Analyze function.
Statistics and reproducibility
GraphPad Prism 9 was used to perform all statistical analyses and the data are presented as mean ± standard deviation unless otherwise stated. Two-tailed Student’s t test or one-way ANOVA were used for comparisons, with P < 0.05 considered statistically significant. All immunostaining, RNAscope staining and lineage tracing experiments were performed with at least three experimental replicates.
Single-cell RNA sequencing
Isolation of cells and sequencing
Single-cell transcriptomes were obtained from the digestion of the tooth and its surrounding tissue at different stages (E13.5, E14.5, E16.5, P3.5, and P7.5). In brief, the tooth and its surrounding tissue were placed in 4 mg/ml Dispase (Roche) and 2 mg/ml Collagenase I (Gibco) on a thermomixer (Eppendorf) at 37 °C for 15 to 30 min depending on the stage of the sample to release cells from the tissue. All our samples had more than 70% viable cells. For each sample, we targeted 20,000 cells for scRNA sequencing. The numbers of actually sequenced cells were 21,416 cells for E13.5, 26,461 cells for E14.5, 29,766 cells for E16.5, 19,134 cells for P3.5, and 15,462 cells for P7.5. On average, we had a sequencing depth of 80,000 read pairs per cell. At each stage, we used two biological replicates for sequencing analysis. Quality control, map**, and count table assembly of the library were performed using the CellRanger pipeline version 3.1.0.
Identifying variable genes and dimensionality reduction
Raw read counts from the cells at each stage were analyzed using the Seurat 4.0R package59. Low gene expression cells were filtered out following standard Seurat object generation. Poor-quality cells with less than 200 genes and mitochondrial gene percentages >50 were removed. Sctransform was applied for normalization and cell cycle regression. RunPCA and RunUMAP were performed for dimensionality reduction and final visualization of the clustering.
Subcluster analysis
Subcluster analysis was performed to investigate the heterogeneity within the dental mesenchymal populations. Published markers for the dental mesenchyme identified the dental mesenchymal cell populations in the mouse molar.
Integrative analysis of samples at different stages
Seurat 4 was used to combine the single-cell data from five stages and perform integration analysis. The PrepSCTIntegration function was performed before identifying anchors with the function FindIntegrationAnchors. Seurat objects were then returned by passing these anchors to the IntegrateData function. RunPCA and RunUMAP visualization were used for downstream analysis and visualization.
Monocle trajectory analysis
We used Monocle 3 to generate the pseudotime trajectory across the dental mesenchymal cells. The cells were input to Monocle to infer cluster and lineage relationships within a given cell type. UMAP embeddings and cell subclusters generated from Seurat were converted to a cell_data_set object using SeuratWrappers (v.0.1.0) and then trajectory graph learning and pseudotime measurement were performed through reversed graph embedding. The estimation of the root node of the trajectory was based on the early dental mesenchyme marker Tfap2b.
GeneSwitches analysis
Following Monocle trajectory analysis, pseudo-times were subsetted using choose_graph_segments() and expression plots were generated using plot_cells(). Analysis of the genes switching on and off in pseudo-time was performed using the GeneSwitches package.
Velocity analysis
Python-based Velocyto command-line tool and Velocyto R package were used as instructed for RNA velocity analysis. Velocyto calculated the single-cell trajectory/directionality using spliced and unspliced reads. From loom files generated by the command-line tool, we extracted the dental papilla cells in R to perform the Velocity analysis using the SeuratWrappers package. RNA velocity was estimated using a gene-relative model with kNN cell pooling (k = 25). RNA velocity was visualized on the UMAP embedding with the parameter n = 200. scVelo was used to perform Velocity analysis of integrative datasets combining different stages.
Gene regulatory network inference
To better understand the transcription factors activating gene expression in the dental mesenchymal cells, we utilized the R package SCENIC, which is a computational method to detect gene regulatory networks. The network activity was analyzed in the dental mesenchymal cells to identify recurrent cellular states. Transcription factors were identified using GENIE3 and compiled into regulons, then subjected to cis-regulatory motif analysis. Regulon activity was then scored using AUCell.
Ligand–receptor interaction analysis
CellChat was performed to identify the potential ligand-receptor interactions in the dental mesenchymal cells. To infer potential cell–cell communication networks, we followed the standard workflow, imported the Seurat object as the input into CellChat and used preprocessing functions identifyOverExpressedGenes, identifyOverExpressedInteractions and projectData with standard parameters set. We then ran the core functions computeCommunProb, computeCommunProbPathway and aggregateNet with standard parameters. Finally, the functions netVisual_circle, netVisual_bubble, netAnalysis_signalingRole_heatmap and netAnalysis_signalingRole_network were applied to the network to determine the senders and receivers.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All single-cell RNA-seq datasets are available through the GEO database under accession code “GSE189381” and FaceBase data repository (Record ID A-QVNG, https://doi.org/10.25550/A-QVNG). All other relevant data supporting the key findings of this study are available within the article and its Supplementary Information files or from the corresponding author upon reasonable request. Source data are provided with this paper.
References
Square, T., Jandzik, D., Romasek, M., Cerny, R. & Medeiros, D. M. The origin and diversification of the developmental mechanisms that pattern the vertebrate head skeleton. Dev. Biol. 427, 219–229 (2017).
Chai, Y. et al. Fate of the mammalian cranial neural crest during tooth and mandibular morphogenesis. Development 127, 1671–1679 (2000).
Siismets, E. M. & Hatch, N. E. Cranial neural crest cells and their role in the pathogenesis of craniofacial anomalies and coronal craniosynostosis. J. Dev. Biol. 8, 18 (2020).
Parker, H. J., Pushel, I. & Krumlauf, R. Coupling the roles of Hox genes to regulatory networks patterning cranial neural crest. Dev. Biol. 444, S67–S78 (2018).
Soldatov, R. et al. Spatiotemporal structure of cell fate decisions in murine neural crest. Science 364, eaas9536 (2019).
Zalc, A. et al. Reactivation of the pluripotency program precedes formation of the cranial neural crest. Science 371, eabb4776 (2021).
Yuan, Y. et al. Spatiotemporal cellular movement and fate decisions during first pharyngeal arch morphogenesis. Sci. Adv. 6, eabb0119 (2020).
Thompson, D., Regev, A. & Roy, S. Comparative analysis of gene regulatory networks: from network reconstruction to evolution. Annu. Rev. Cell Dev. Biol. 31, 399–428 (2015).
Williams, R. M. et al. Reconstruction of the global neural crest gene regulatory network in vivo. Dev. Cell 51, 255–276.e7 (2019).
Betancur, P., Bronner-Fraser, M. & Sauka-Spengler, T. Assembling neural crest regulatory circuits into a gene regulatory network. Annu. Rev. Cell Dev. Biol. 26, 581–603 (2010).
Artinger, K. B. & Monsoro-Burq, A. H. Neural crest multipotency and specification: power and limits of single cell transcriptomic approaches. Fac. Rev. 10, 38 (2021).
Yu, T. & Klein, O. D. Molecular and cellular mechanisms of tooth development, homeostasis and repair. Development 147, dev.184754 (2020).
Li, J., Parada, C. & Chai, Y. Cellular and molecular mechanisms of tooth root development. Development 144, 374–384 (2017).
Jussila, M. & Thesleff, I. Signaling networks regulating tooth organogenesis and regeneration, and the specification of dental mesenchymal and epithelial cell lineages. Cold Spring Harb. Perspect. Biol. 4, a008425 (2012).
Cobourne, M. T. & Sharpe, P. T. Tooth and jaw: molecular mechanisms of patterning in the first branchial arch. Arch. Oral. Biol. 48, 1–14 (2003).
Krivanek, J., Adameyko, I. & Fried, K. Heterogeneity and developmental connections between cell types inhabiting teeth. Front. Physiol. 8, 376 (2017).
Takahashi, A. et al. Autocrine regulation of mesenchymal progenitor cell fates orchestrates tooth eruption. Proc. Natl Acad. Sci. U.S.A. 116, 575–580 (2019).
Feng, J. et al. BMP signaling orchestrates a transcriptional network to control the fate of mesenchymal stem cells in mice. Development 144, 2560–2569 (2017).
Sharpe, P. T. Dental mesenchymal stem cells. Development 143, 2273–2280 (2016).
Krivanek, J. et al. Dental cell type atlas reveals stem and differentiated cell types in mouse and human teeth. Nat. Commun. 11, 4816 (2020).
Pagella, P., Roditi, L. D., Stadlinger, B., Moor, A. E. & Mitsiadis, T. A. A single-cell atlas of human teeth. Iscience 24, 102405 (2021).
Woodruff, E. D., Gutierrez, G. C., Van Otterloo, E., Williams, T. & Cohn, M. J. Anomalous incisor morphology indicates tissue-specific roles for Tfap2a and Tfap2b in tooth development. Dev. Biol. 472, 67–74 (2021).
Feng, J. et al. Generation and characterization of tamoxifen-inducible Pax9-CreER knock-in mice using CrispR/Cas9. Genesis 54, 490–496 (2016).
Zhou, B. O., Yue, R., Murphy, M. M., Peyer, J. G. & Morrison, S. J. Leptin-receptor-expressing mesenchymal stromal cells represent the main source of bone formed by adult bone marrow. Cell Stem Cell 15, 154–168 (2014).
Batiuk, M. Y. et al. Identification of region-specific astrocyte subtypes at single cell resolution. Nat. Commun. 11, 1220 (2020).
Bergen, V., Lange, M., Peidli, S., Wolf, F. A. & Theis, F. J. Generalizing RNA velocity to transient cell states through dynamical modeling. Nat. Biotechnol. 38, 1408–1414 (2020).
**e, Y. et al. FGF/FGFR signaling in health and disease. Signal Transduct. Target. Ther. 5, 181 (2020).
Neves, V. C. M. & Sharpe, P. T. Regulation of reactionary dentine formation. J. Dent. Res. 97, 416–422 (2018).
Zaugg, L. K. et al. Translation approach for centine regeneration using GSK-3 antagonists. J. Dent. Res. 99, 544–551 (2020).
Butler, A., Hoffman, P., Smibert, P., Papalexi, E. & Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420 (2018).
Cao, E. Y., Ouyang, J. F. & Rackham, O. J. L. GeneSwitches: ordering gene expression and functional events in single-cell experiments. Bioinformatics 36, 3273–3275 (2020).
**, S. Q. et al. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 12, 1088 (2021).
Tamura, M. & Nemoto, E. Role of the Wnt signaling molecules in the tooth. Jpn. Dent. Sci. Rev. 52, 75–83 (2016).
Alberio, R. Regulation of cell fate decisions in early mammalian embryos. Annu. Rev. Anim. Biosci. 8, 377–393 (2020).
Aibar, S. et al. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods 14, 1083–1086 (2017).
Zhao, Z. Y., Stock, D. W., Buchanan, A. V. & Weiss, K. M. Expression of Dlx genes during the development of the murine dentition. Dev. Genes Evol. 210, 270–275 (2000).
Tao, H. et al. Klf4 promotes dentinogenesis and odontoblastic differentiation via modulation of TGF-beta signaling pathway and interaction with histone acetylation. J. Bone Miner. Res. 34, 1502–1516 (2019).
Lam, E. W., Brosens, J. J., Gomes, A. R. & Koo, C. Y. Forkhead box proteins: tuning forks for transcriptional harmony. Nat. Rev. Cancer 13, 482–495 (2013).
DeSisto, J. et al. Single-cell transcriptomic analyses of the develo** meninges reveal meningeal fibroblast diversity and function. Dev. Cell 54, 43–59 (2020).
Holmes, G. et al. Integrated transcriptome and network analysis reveals spatiotemporal dynamics of calvarial suturogenesis. Cell Rep. 32, 107871 (2020).
Farmer, D. T. et al. The develo** mouse coronal suture at single-cell resolution. Nat. Commun. 12, 4797 (2021).
Xu, J. Y. et al. Hedgehog signaling patterns the oral-aboral axis of the mandibular arch. Elife 8, e40315 (2019).
He, J. et al. Lhx6 regulates canonical Wnt signaling to control the fate of mesenchymal progenitor cells during mouse molar root patterning. PLoS Genet. 17, e1009320 (2021).
Ono, W., Sakagami, N., Nishimori, S., Ono, N. & Kronenberg, H. M. Parathyroid hormone receptor signalling in osterix-expressing mesenchymal progenitors is essential for tooth root formation. Nat. Commun. 7, 11277 (2016).
Murillo-Rincon, A. P. & Kaucka, M. Insights into the complexity of craniofacial development from a cellular perspective. Front. Cell Dev. Biol. 8, 620735 (2020).
Simoes-Costa, M. & Bronner, M. E. Establishing neural crest identity: a gene regulatory recipe. Development 142, 242–257 (2015).
Simoes-Costa, M. & Bronner, M. E. Reprogramming of avian neural crest axial identity and cell fate. Science 352, 1570–1573 (2016).
Minoux, M. et al. Gene bivalency at Polycomb domains regulates cranial neural crest positional identity. Science 355, eaal2913 (2017).
Attanasio, C. et al. Fine tuning of craniofacial morphology by distant-acting enhancers. Science 342, 1241006 (2013).
Tucker, A. S. et al. Edar/Eda interactions regulate enamel knot formation in tooth morphogenesis. Development 127, 4691–4700 (2000).
Sadier, A., Santana, S. E. & Sears, K. E. The role of core and variable Gene Regulatory Network modules in tooth development and evolution. Integr. Comp. Biol. 6, icaa116 (2020).
Golson, M. L. & Kaestner, K. H. Fox transcription factors: from development to disease. Development 143, 4558–4570 (2016).
Hannenhalli, S. & Kaestner, K. H. The evolution of Fox genes and their role in development and disease. Nat. Rev. Genet. 10, 233–240 (2009).
Kim, J. H. et al. Molecular networks of FOXP family: dual biologic functions, interplay with other molecules and clinical implications in cancer progression. Mol. Cancer 18, 180 (2019).
Zhao, H. X. et al. Foxp1/2/4 regulate endochondral ossification as a suppresser complex. Dev. Biol. 398, 242–254 (2015).
Takahashi, Y., Sipp, D. & Enomoto, H. Tissue interactions in neural crest cell development and disease. Science 341, 860–863 (2013).
Li, S. et al. Foxp1/4 control epithelial cell fate during lung development and regeneration through regulation of anterior gradient 2. Development 139, 2500–2509 (2012).
**g, J. et al. Antagonistic interaction between Ezh2 and Arid1a coordinates root patterning and development via Cdkn2a in mouse molars. Elife 8, 46426 (2019).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e29 (2021).
Acknowledgements
We thank Bridget Samuels for critical reading of the manuscript. We thank Dr. Hu Zhao and Yuling Wang from the Chinese Institute for Brain Research for collecting samples from Fgf3-CreERT2;tdT mice. We acknowledge USC Libraries Bioinformatics Service for assisting with data analysis. The bioinformatics software and computing resources used in the analysis are funded by the USC Office of Research and the Norris Medical Library. This study was supported by grants from the National Institute of Dental and Craniofacial Research, National Institutes of Health (R01 DE022503 to Y.C. and R01 DE012711 to Y.C.).
Author information
Authors and Affiliations
Contributions
J.J. and Y.C. designed the study. J.J. carried out most of the experiments and analyzed the data. J.F., Y.Y., T.G., J.L., and F.P. participated in the sample collection. J.F. participated in the generation of Pax9-CreERT2 mouse line. T-V.H. participated in the microCT analysis. J.J. and Y.C. co-wrote the paper. Y.C. supervised the research.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer review reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
**g, J., Feng, J., Yuan, Y. et al. Spatiotemporal single-cell regulatory atlas reveals neural crest lineage diversification and cellular function during tooth morphogenesis. Nat Commun 13, 4803 (2022). https://doi.org/10.1038/s41467-022-32490-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-022-32490-y
- Springer Nature Limited
This article is cited by
-
FGF signaling modulates mechanotransduction/WNT signaling in progenitors during tooth root development
Bone Research (2024)
-
PRX1-positive mesenchymal stem cells drive molar morphogenesis
International Journal of Oral Science (2024)
-
Notum regulates the cusp and root patterns in mouse molar
Scientific Reports (2024)
-
The role of WWP1 and WWP2 in bone/cartilage development and diseases
Molecular and Cellular Biochemistry (2024)
-
Identification of a novel de novo mutation in SOX4 for syndromic tooth agenesis
Clinical Oral Investigations (2024)