

Abstract
Efficient search for DNA damage embedded in vast expanses of the DNA genome presents one of the greatest challenges to DNA repair enzymes. We report here crystal structures of human 8-oxoguanine (oxoG) DNA glycosylase, hOGG1, that interact with the DNA containing the damaged base oxoG and the normal base G while they are nested in the DNA helical stack. The structures reveal that hOGG1 engages the DNA using different protein-DNA contacts from those observed in the previously determined lesion recognition complex and other hOGG1-DNA complexes. By applying molecular dynamics simulations, we have determined the pathways taken by the lesion and normal bases when extruded from the DNA helix and their associated free energy profiles. These results reveal how the human oxoG DNA glycosylase hOGG1 locates the lesions inside the DNA helix and facilitates their extrusion for repair.
Similar content being viewed by others


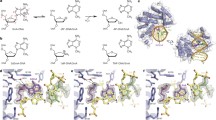
Introduction
The chemical instability of the genome toward attack by a variety of reactive species with which it comes into routine contact has given rise to dedicated surveillance and maintenance systems that seek out DNA damage and repair it. Most forms of damage to the heterocyclic nucleobases of DNA are repaired by mechanistically related pathways collectively referred to as the base-excision repair (BER) mechanism. Both the recognition of damaged sites and initiation of repair by BER are performed by DNA glycosylases, lesion-specific enzymes that recognize specific nucleobase damages in the genome and catalyze their excision through cleavage of the glycosidic bond1,2,3,4.
DNA glycosylases carry out the formidable task of locating, on average, one aberrant base embedded among ~106–107 normal bases5. Many of these lesions differ from their undamaged counterparts by at most a few atoms and cause no frank distortion or little energetic destabilization of the DNA duplex6,7,8,9. Among them, 8-oxoguanine (oxoG, OG) presents the greatest challenge to the DNA glycosylases responsible for its repair, due to its unique combination of structural innocuousness, extrordinary mutagenicity, and chronic low-level production. Arising through the attack of the reactive by-products of aerobic respiration on guanine (G), oxoG differs from G by only two “atoms”, =O versus –H at C8, and lone-pair versus –H at N7 (Fig. 1a). Also, oxoG is highly mutagenic and mis-pairs with adenine at a greater than 90% frequency during processive DNA replication10. For these reasons, oxoG is believed to be responsible for the majority of G:C to T:A transversion mutations, which are the second most common type of spontaneous genetic change in humans11. The G:C to T:A transversion mutation has also been found in codon 12 of the highly oncogenic protein K-ras, which resulted in the formation of lung tumors in mice deficient in the oxidative DNA repair genes, myh and ogg41, the entire process is accelerated by DNA bending and the extent of protein–DNA contacts on the minor groove face of DNA. For example, hOGG1 uses a non-specific breakage of the target base-pair, assisted by N149. In addition, K249 contacts the 3′-side phosphate of oxoG/G at the beginning of the process (Figs. 5b, 6), thereby establishing a pivot for base extrusion. H270 is the first residue that specifically interacts with extruded oxoG, followed by K249 with C8=O of oxoG. This suggests that H270 and K249 function as a “cherry-picking” residue in hOGG1, with a similar role of R112 in MutM via significantly different mechanisms22. By contrast, in the case of G, its extrusion cannot be stabilized by the two residues and competes with the translocation of the enzyme along the DNA strand.
Figure 5 also suggests that hOGG1 extrudes the oxoG through the major groove, in accordance with the previously determined hOGG1/DNA complex structure32 with a barrier of 8.1 kcal/mol. The free energy profile for the minor-groove oxoG extrusion is also presented in Fig. 5a, and the free energy profiles along the entire base extrusion process are shown in Supplementary Fig. 9. The barrier for the minor groove extrusion is 17.9 kcal/mol. This result can be compared with the different results reported for MutM between the major30,42 and minor groove base extrusions22,29.
In summary, we present X-ray crystallographic structures of human DNA glycosylase hOGG1 interrogating DNA lesions in their intrahelical position, achieved by covalent trap** of an ordinarily transient state in DNA recognition. They reveal how hOGG1 discriminates oxoG from G while both are embedded in the DNA duplex. Specifically, the enzyme utilizes unique protein/DNA contacts to induce DNA bending at the target site. This bending brings the repulsive functional group of oxoG to the immediate vicinity of the DNA backbone, resulting in an oxoG specific distortion of the DNA backbone in its intrahelical orientation. In silico molecular dynamics simulations and free energy calculations corroborate the structural results and help to elucidate the role of the human enzyme in discriminating oxoG from G prior to a complete extrusion from the DNA stack. The results presented here broaden our understanding of one of the earliest events that occur as this extraordinary enzyme patrols genome in its surveillance of DNA damage.
Methods
Cross-linked complex formation and crystallization
A fragment of hOGG1 (amino acids 12–327, UniProtKB-015527) bearing the Y207C, Y207C/C253W, and Y207C/K249Q mutation was expressed in Escherichia coli BL21(DE3)pLysS cells. The cells were lysed by sonication in solution of 50 mM sodium phosphate pH 8.0, 10 mM imidazole, 500 mM NaCl, 5 mM BME, and 10% glycerol. The protein was immobilized by Ni-NTA resin (Qiagen) and eluted with 50 mM sodium phosphate pH 8.0, 250 mM imidazole, 500 mM NaCl, 5 mM BME, and 10% glycerol. Protein was concentrated, centrifuged, and diluted with 10 mM Tris pH 7.4 to 50 mM NaCl, loaded to Hi-Trap SP column (GE Healthcare) and eluted with increasing NaCl concentration. The N-terminal histidine-tag was cleaved by enterokinase digestion (New England Biolabs) using a 1:1 solution of 1 M CaCl2 for 36 h at 4 °C. Protein was further purified by Superdex-200 gel filtration chromatography (GE Healthcare) equilibrated with 10 mM Tris 7.4, 100 mM NaCl, 1 mM EDTA, and 10% glycerol43. Each mutant was prepared using QuickChange mutagenesis kit (Stratagene) and confirmed by sequencing (see Supplementary Table 2 for primer sequences used in mutagenesis).
Phosphoramidite derivatives of 8-oxoG and 2-F-dI were purchased from Glen research. DNA oligomers 5′-AGCGTCCAXG*TCTACC-3′, where X denotes 8-oxoG or G and G* refers the site of modification with the thiol-bearing tether, were synthesized using ABI Expedite 8909 DNA synthesizer and functionalized with X8 (NH2CH2CH2OCH2CH2OCH2CH2S–)2 using post synthetic modification44. DNA oligonucleotides were deprotected with ammonium hydroxide and purified in 20% denaturing urea polyacrylamide gel electrophoresis (PAGE). For DNA containing tether and oxoG on the same strand, 50 μM β-me was added to prevent oxidative degradation of 8-oxoG. DNA was purified by 20% urea-PAGE and dissolved in 10 mM Tris, pH 8.0, 1 mM EDTA, and annealed with complimentary strand 5′-TGGTAGACCTGGACGC-3′.
Cross-linked complexes were formed by mixing duplex DNA with 2-fold molar excess protein and incubating at 4 °C for several days. Unreacted DNA and protein were removed by Mono Q chromatography (GE Healthcare). The purified complexes were buffer-exchanged to 10 mM Tris 7.4 and 100 mM NaCl, concentrated and crystallized by hanging droplet vapor diffusion at 20 °C. For each complex, crystals were allowed to grow for several days, transferred to a cryoprotectant solution containing mother liquor supplemented with 25% glycerol, and frozen in liquid nitrogen for data collection. For LRC (Y207C/K249Q), diffraction quality crystals appeared with 16.4 mg/mL complex concentration (protein concentration measured using Bradford assay) within a few days in well solution containing 100 mM sodium cacodylate, pH 6.5, 200 mM MgCl2 and 18% polyethylene glycol 8000. For IC (Y207C), diffraction quality crystals appeared with 12 mg/mL complex concentration within a few days in well solution containing 200 mM NH4NO3, and 20% polyethylene glycol 3350. For EC (Y207C/C253W), diffraction quality crystals appeared with ~17 mg/mL complex concentration within a few days in well solution containing 100 mM sodium cacodylate, pH 6.1, 200 mM MgOAc, and 17% polyethylene glycol 8000.
Structure determination
Diffraction datasets were collected at −170 °C at the 24-ID-C and 24-ID-E beamlines (NE-CAT) of the Advanced Photon Source and processed using the HKL program suites45. Initial molecular replacement solutions were obtained by PHASER in the CCP4 suite46,47, using the coordinates of previously determined hOGG1 structure (PDB ID: 1EBM16) but omitting DNA as search models. Each hOGG1–DNA model was built through iterative cycles of manual model building in COOT48 and structure refinement using REFMAC549,50 and PHENIX51. The Ramachandran plots, calculated by MolProbity (http://molprobity.biochem.duke.edu), confirmed no residues in disallowed regions for all structures. Full details on the data collection and structure refinement are provided in Supplementary Table 1. PyMol (The PyMOL Molecular Graphics System, Version 2.0, Schrödinger, LLC.) was used to prepare all structure model figures presented in the paper.
System preparation for molecular dynamics (MD) simulations
Two systems were prepared based on the intrahelical IC (for G) and EC (for oxoG) structures, respectively. In the preparation of the IC system, we further refined the IC X-ray structure to build more base pairs. Although this resulted in slightly lower quality of DNA structure, the core DNA base pairs were essentially unchanged. We used this refined structure in the IC system building. In each system, protonation states of all ionizable residues were determined based on their hydrogen bonding interactions deduced from the X-ray structures as well as on their pKa values in water. All crystal waters were included. For DNA, the central 14 base pairs of the sequence presented in Fig. 1d were used in the simulations, in which any missing nucleotide coordinates from the crystal structures were model built as the standard B-form DNA. Then, the HBUILD facility of the CHARMM program52,53 was used to assign atomic coordinates of hydrogen atoms. The resulting systems were solvated with a rhombic dodecahedron (RHDO) box of 11,712 TIP3P water molecules54 and any water molecules42 within 2.5 Å from any heavy atom of protein, DNA, and crystal water were removed, leaving, for example, 9554 TIP3P waters for the IC system. Finally, each system was neutralized by adding 50 Na+ and 26 Cl− ions at random positions, making its ionic concentration equal to 150 mM. In addition, two additional systems were prepared based on the LRC X-ray structure (PDB-ID: 1YQR)23 to be used as the reference state of the targeted molecular dynamics (TMD) simulations55 (see the Supplementary Methods for details).
MD simulations
Each system was first energy minimized for 5000 steps and equilibrated for 500 ps at 300 K. The energy minimization and equilibration procedures were very similar to those employed in our previous studies of MutM/DNA complexes22,29. Then, the production MD was carried out for 500 ns for the IC and EC systems, during which the atomic coordinates of the entire system were saved at every 2 ps for later analysis. The all-atom CHARMM2256 and 27 force fields57 were used to represent the protein, DNA, and ions, the CMAP correction58 for protein backbone dihedrals, and the TIP3P water model54 for water molecules, respectively. For oxoG, we used the force field parameters developed in our previous study22. The RHDO periodic boundary conditions were imposed with the lattice length parameter of 78.5 Å. Electrostatic interactions were evaluated using the smooth particle mesh Ewald (PME) sum method59 and van der Waals interactions were evaluated using a switching function between 9.0 Å and 11.0 Å. All MD simulations were performed with a 2 fs integration time step and SHAKE60 applied to all bonds involving hydrogen atoms. The Langevin thermostat was used to maintain the system temperature at 300 K. In all simulations, we also applied harmonic restraints to the terminal base pairs to avoid them fraying away from each other.
System preparations, trajectory analysis, the TMD simulations, and the string method simulations36 (see below) were carried out using the CHARMM program (version c37a1)52,53 and the 500 ns production MD simulations were performed using the NAMD program60.
String method (SM) simulations
The base extrusion pathways for G and oxoG were determined by applying the string method in collective variables (SMCV)36. In SMCV, a path connecting two end state conformations (i.e., the intrahelical IC/EC and LRC conformations) is represented by N discretized images (called MD replicas), which are evenly distributed along the path. In the present work, we used N = 64 discretized images to represent the entire base extrusion pathway described by a total of 45 CVs defined in Supplementary Fig. 10a. Starting from the initial path generated by the TMD simulation (Supplementary Methods), each path was optimized for 25 ns in an iterative manner. A total of 1.28 μs path optimization MD was performed. Then, the Markovian milestoning simulations with Voronoi tessellations36,61 were performed for 10 ns for each MD replica (thus, 0.64 μs MD simulations collectively) to determine the free energy change along the optimized base extrusion paths. The details of the SMCV path optimization and Voronoi tessellations simulations are provided in the Supplementary Methods.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
References
Schärer, O. D. & Jiricny, J. Recent progress in the biology, chemistry and structural biology of DNA glycosylases. Bioessays 23, 270–281 (2001).
Krokan, H. E., Nilsen, H., Skorpen, F., Otterlei, M. & Slupphaug, G. Base excision repair of DNA in mammalian cells. Febs Lett. 476, 73–77 (2000).
Fromme, J. C. & Verdine, G. L. Base excision repair. Adv. Protein Chem. 69, 1–41 (2004).
Fromme, J. C., Banerjee, A. & Verdine, G. L. DNA glycosylase recognition and catalysis. Curr. Opin. Struc Biol. 14, 43–49 (2004).
Lindahl, T. Instability and decay of the primary structure of DNA. Nature 362, 709–715 (1993).
Bowman, B. R., Lee, S., Wang, S. & Verdine, G. L. Structure of the E. coli DNA glycosylase AlkA bound to the ends of duplex DNA: a system for the structure determination of lesion-containing DNA. Structure 16, 1166–1174 (2008).
Ezaz-Nikpay, K. & Verdine, G. L. The effects of N7-methylguanine on duplex DNA structure. Chem. Biol. 1, 235–240 (1994).
Lipscomb, L. A. et al. X-ray structure of a DNA decamer containing 7,8-dihydro-8-oxoguanine. Proc. Natl Acad. Sci. USA 92, 719–723 (1995).
Plum, G. E., Grollman, A. P., Johnson, F. & Breslauer, K. J. Influence of the oxidatively damaged adduct 8-oxodeoxyguanosine on the conformation, energetics, and thermodynamic stability of a DNA duplex. Biochem.-us 34, 16148–16160 (1995).
Shibutani, S., Takeshita, M. & Grollman, A. P. Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG. Nature 349, 431–434 (1991).
Hollstein, M. et al. Somatic point mutations in the p53 gene of human tumors and cell lines: updated compilation. Nucleic Acids Res. 24, 141–146 (1996).
**e, Y. et al. Deficiencies in mouse Myh and Ogg1 result in tumor predisposition and G to T mutations in codon 12 of the K-Ras oncogene in lung tumors. Cancer Res. 64, 3096–3102 (2004).
Ohno, M. et al. A genome-wide distribution of 8-oxoguanine correlates with the preferred regions for recombination and single nucleotide polymorphism in the human genome. Genome Res. 16, 567–575 (2006).
Ohno, M. et al. 8-oxoguanine causes spontaneous de novo germline mutations in mice. Sci. Rep. 4, 4689 (2014).
Fromme, J. C., Banerjee, A., Huang, S. J. & Verdine, G. L. Structural basis for removal of adenine mispaired with 8-oxoguanine by MutY adenine DNA glycosylase. Nature 427, 652–656 (2004).
Bruner, S. D., Norman, D. P. G. & Verdine, G. L. Structural basis for recognition and repair of the endogenous mutagen 8-oxoguanine in DNA. Nature 403, 859–866 (2000).
Wang, L., Chakravarthy, S. & Verdine, G. L. Structural basis for the lesion-scanning mechanism of the MutY DNA glycosylase. J. Biol. Chem. 292, 5007–5017 (2017).
Parikh, S. S. et al. Uracil-DNA glycosylase–DNA substrate and product structures: Conformational strain promotes catalytic efficiency by coupled stereoelectronic effects. Proc. Natl Acad. Sci. USA 97, 5083–5088 (2000).
Blainey, P. C., Oijen, A. M., van, Banerjee, A., Verdine, G. L. & **e, X. S. A base-excision DNA-repair protein finds intrahelical lesion bases by fast sliding in contact with DNA. Proc. Natl Acad. Sci. USA 103, 5752–5757 (2006).
Bowman, B. R., Lee, S., Wang, S. & Verdine, G. L. Structure of Escherichia coli AlkA in complex with undamaged DNA. J. Biol. Chem. 285, 35783–35791 (2010).
Huang, H., Chopra, R., Verdine, G. L. & Harrison, S. C. Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase: implications for drug resistance. Science 282, 1669–1675 (1998).
Qi, Y. et al. Encounter and extrusion of an intrahelical lesion by a DNA repair enzyme. Nature 462, 762–766 (2009).
Banerjee, A., Yang, W., Karplus, M. & Verdine, G. L. Structure of a repair enzyme interrogating undamaged DNA elucidates recognition of damaged DNA. Nature 434, 612–618 (2005).
Banerjee, A., Santos, W. L. & Verdine, G. L. Structure of a DNA glycosylase searching for lesions. Science 311, 1153–1157 (2006).
Yi, C. et al. Iron-catalysed oxidation intermediates captured in a DNA repair dioxygenase. Nature 468, 330–333 (2010).
Yang, C.-G. et al. Crystal structures of DNA/RNA repair enzymes AlkB and ABH2 bound to dsDNA. Nature 452, 961–965 (2008).
Lee, S.-J., Sung, R.-J. & Verdine, G. L. Mechanism of DNA lesion homing and recognition by the Uvr nucleotide excision repair system. Res. 2019, 5641746 (2019).
Kuznetsov, N. A. et al. Active destabilization of base pairs by a DNA glycosylase wedge initiates damage recognition. Nucleic Acids Res. 43, 272–281 (2015).
Nam, K., Verdine, G. L. & Karplus, M. Analysis of an anomalous mutant of MutM DNA glycosylase leads to new insights into the catalytic mechanism. J. Am. Chem. Soc. 131, 18208–18209 (2009).
Song, K. et al. Molecular simulations reveal a common binding mode for glycosylase binding of oxidatively damaged DNA lesions. J. Am. Chem. Soc. 129, 14536–14537 (2007).
Radom, C. T., Banerjee, A. & Verdine, G. L. Structural characterization of human 8-oxoguanine DNA glycosylase variants bearing active site mutations. J. Biol. Chem. 282, 9182–9194 (2007).
Banerjee, A. & Verdine, G. L. A nucleobase lesion remodels the interaction of its normal neighbor in a DNA glycosylase complex. Proc. Natl Acad. Sci. USA 103, 15020–15025 (2006).
Crenshaw, C. M. et al. Enforced presentation of an extrahelical guanine to the lesion recognition pocket of human 8-oxoguanine glycosylase, hOGG1. J. Biol. Chem. 287, 24916–24928 (2012).
Dalhus, B. et al. Separation-of-function mutants unravel the dual-reaction mode of human 8-oxoguanine DNA glycosylase. Structure 19, 117–127 (2011).
Robey-Bond, S. M., Barrantes-Reynolds, R., Bond, J. P., Wallace, S. S. & Bandaru, V. Clostridium acetobutylicum 8-oxoguanine DNA glycosylase (Ogg) differs from eukaryotic Oggs with respect to opposite base discrimination. Biochem. 47, 7626–7636 (2008).
Ovchinnikov, V., Karplus, M. & Vanden-Eijnden, E. Free energy of conformational transition paths in biomolecules: the string method and its application to myosin VI. J. Chem. Phys. 134, 085103 (2011).
Kemp, P. A., van der, Charbonnier, J., Audebert, M. & Boiteux, S. Catalytic and DNA‐binding properties of the human Ogg1 DNA N‐glycosylase/AP lyase: biochemical exploration of H270, Q315 and F319, three amino acids of the 8‐oxoguanine‐binding pocket. Nucleic Acids Res. 32, 570–578 (2004).
Fedorova, O. S. et al. Stopped-flow kinetic studies of the interaction between Escherichia coli Fpg protein and DNA substrates. Biochem. 41, 1520–1528 (2002).
Koval, V. V. et al. Pre-steady-state kinetics shows differences in processing of various DNA lesions by Escherichia coli formamidopyrimidine-DNA glycosylase. Nucleic Acids Res. 32, 926–935 (2004).
Priyakumar, U. D. & MacKerell, A. D. NMR imino proton exchange experiments on duplex DNA primarily monitor the opening of purine bases. J. Am. Chem. Soc. 128, 678–679 (2006).
Lemkul, J. A., Savelyev, A. & MacKerell, A. D. Induced polarization influences the fundamental forces in DNA base flip**. J. Phys. Chem. Lett. 5, 2077–2083 (2014).
Song, K. et al. Computational analysis of the mode of binding of 8-oxoguanine to formamidopyrimidine-DNA glycosylase. Biochem. 45, 10886–10894 (2006).
Norman, D. P. G., Chung, S. J. & Verdine, G. L. Structural and biochemical exploration of a critical amino acid in human 8-oxoguanine glycosylase. Biochem. 42, 1564–1572 (2003).
Erlanson, D. A., Glover, J. N. M. & Verdine, G. L. Disulfide cross-linking as a mechanistic probe for the B ↔ Z transition in DNA. J. Am. Chem. Soc. 119, 6927–6928 (1997).
Otwinowski, Z. & Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 (1997).
McCoy, A. J. et al. Phaser crystallographic software. J. Appl Crystallogr 40, 658–674 (2007).
Winn, M. D. et al. Overview of the CCP4 suite and current developments. Acta Crystallogr Sect. D. Biol. Crystallogr 67, 235–242 (2011).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D. Biol. Crystallogr. 66, 486–501 (2010).
Vagin, A. A. et al. REFMAC 5 dictionary: organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr. Sect. D. Biol. Crystallogr. 60, 2184–2195 (2004).
Murshudov, G. N., Vagin, A. A. & Dodson, E. J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. Sect. D. Biol. Crystallogr. 53, 240–255 (1997).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D. Biol. Crystallogr. 66, 213–221 (2010).
Brooks, B. R. et al. CHARMM: the biomolecular simulation program. J. Comput Chem. 30, 1545–1614 (2009).
Brooks, B. R. et al. CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J. Comput Chem. 4, 187–217 (1983).
Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W. & Klein, M. L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 (1983).
Schlitter, J., Engels, M., Krüger, P., Jacoby, E. & Wollmer, A. Targeted molecular dynamics simulation of conformational change-application to the T ↔ R Transition in Insulin. Mol. Simula. 10, 291–308 (1993).
MacKerell, A. D. et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 102, 3586–3616 (1998).
Foloppe, N. & Mackerell, AlexanderD. Jr All-atom empirical force field for nucleic acids: I. Parameter optimization based on small molecule and condensed phase macromolecular target data. J. Comput Chem. 21, 86–104 (2000).
Mackerell, A. D., Feig, M. & Brooks, C. L. Extending the treatment of backbone energetics in protein force fields: Limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput Chem. 25, 1400–1415 (2004).
Essmann, U. et al. A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8593 (1995).
Phillips, J. C. et al. Scalable molecular dynamics with NAMD. J. Comput Chem. 26, 1781–1802 (2005).
Vanden-Eijnden, E. & Venturoli, M. Markovian milestoning with Voronoi tessellations. J. Chem. Phys. 130, 194101 (2009).
Acknowledgements
This work was supported by National Institutes of Health (CA100742 to G.L.V., and R01GM132481 to K.N.), the University of Texas at Arlington (K.N.), AACR Anna D. Barker Postdoctoral fellowship to U.K.S., and the CHARMM Development Project at Harvard (M.K.). We thank the staff at the NE-CAT at the advanced Photon Source for Assistance during data collection. Computer resources were provided by the Swedish National Infrastructure for Computing (SNIC) at the High Performance Computing Center North (HPC2N), by the National Energy Research Scientific Computing Center (NERSC), which is supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231, and by the Oak Ridge Leadership Computing Facility (OLCF) at Oak Ridge National Laboratory (ORNL), which is supported by the Office of Science of the Department of Energy under Contract DE-AC05-00OR22725.
Author information
Authors and Affiliations
Contributions
U.K.S. and G.L.V. conceived and designed the research. U.K.S. and J.A.S. prepared samples and performed all laboratory experiments. S.J.L. helped with X-ray diffraction data collection and structure determination. V.O. M.K. and K.N. conceived and designed computational experiments. V.O. and K.N. performed and analyzed molecular dynamics simulations. U.K.S. K.N. and G.L.V. wrote the paper. All authors provided critical feedback, revised, approved, and helped shape the research and the analysis embodied in the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks Kenichi Miyazono and the other, anonymous reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shigdel, U.K., Ovchinnikov, V., Lee, SJ. et al. The trajectory of intrahelical lesion recognition and extrusion by the human 8-oxoguanine DNA glycosylase. Nat Commun 11, 4437 (2020). https://doi.org/10.1038/s41467-020-18290-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-020-18290-2
- Springer Nature Limited
This article is cited by
-
DNA damage and repair in the nucleosome: insights from computational methods
Biophysical Reviews (2024)
-
Unexpected benzene oxidation in collisions with superoxide anions
Scientific Reports (2021)