
Abstract
LKB1 and KRAS are the third most frequent co-mutations detected in non-small cell lung cancer (NSCLC) and cause aggressive tumor growth. Unfortunately, treatment with RAS-RAF-MEK-ERK pathway inhibitors has minimal therapeutic efficacy in LKB1-mutant KRAS-driven NSCLC. Autophagy, an intracellular nutrient scavenging pathway, compensates for Lkb1 loss to support Kras-driven lung tumor growth. Here we preclinically evaluate the possibility of autophagy inhibition together with MEK inhibition as a treatment for Kras-driven lung tumors. We found that the combination of the autophagy inhibitor hydroxychloroquine (HCQ) and the MEK inhibitor Trametinib displays synergistic anti-proliferative activity in KrasG12D/+;Lkb1-/- (KL) lung cancer cells, but not in KrasG12D/+;p53-/- (KP) lung cancer cells. In vivo studies using tumor allografts, genetically engineered mouse models (GEMMs) and patient-derived xenografts (PDXs) showed anti-tumor activity of the combination of HCQ and Trametinib on KL but not KP tumors. We further found that the combination treatment significantly reduced mitochondrial membrane potential, basal respiration, and ATP production, while also increasing lipid peroxidation, indicative of ferroptosis, in KL tumor-derived cell lines (TDCLs) and KL tumors compared to treatment with single agents. Moreover, the reduced tumor growth by the combination treatment was rescued by ferroptosis inhibitor. Taken together, we demonstrate that autophagy upregulation in KL tumors causes resistance to Trametinib by inhibiting ferroptosis. Therefore, a combination of autophagy and MEK inhibition could be a novel therapeutic strategy to specifically treat NSCLC bearing co-mutations of LKB1 and KRAS.
Similar content being viewed by others
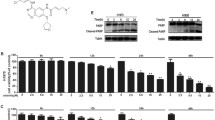

Introduction
Lung cancer is a leading contributor to cancer-attributed deaths in the United States, with an overall survival rate of 17% at any stage and 4% for more advanced stages [1]. About 85% of lung cancers are characterized as non-small cell lung carcinoma (NSCLC) [2]. KRAS mutations in NSCLC usually signify a poor prognosis and are linked with resistance to multiple cancer treatments [3]. Functional, predictive biomarkers play an essential role in predicting response to different treatments to enhance prognosis, thereby enabling the formulation of individualized therapies. Mutations in the epidermal growth factor receptor (EGFR), KRAS, Fibroblast growth factor receptor 1 (FGFR1), Anaplastic lymphoma kinase (ALK), MET, PIK3CABRAF, ROS1, NTRK, RET, HER2, LKB1, and TP53 [4,5,6,7] have been used as biomarkers to guide targeted cancer treatment. In particular, tumors with LKB1 mutations, which are detected in 15–30% of NSCLC, including tumors with co-occurring KRAS mutations detected in 5–10% of NSCLC [8, 9], unfortunately lack effective targeted therapies and are associated with the resistance to immunotherapy [6].
TP53 and LKB1 mutations in KRAS-mutant NSCLC show distinct genetic profiles and responses to therapies [6]. While co-mutations in KRAS and LKB1 are found in tumors of squamous cell carcinoma and adenocarcinoma, KRAS and TP53 co-mutations are found only in adenocarcinoma [10, 11]. Whereas Kras-p53 mutant tumors respond to combination therapy with docetaxel and the MEK inhibitor Selumetinib, Kras-Lkb1 mutant tumors do not [12]. Kras-p53 mouse lung tumors are also more responsive to radiotherapy compared to tumors harboring Kras-Lkb1 mutations [13, 14]. Therefore, there is an urgent need for precision therapy for different subgroups of KRAS-driven NSCLC.
Except for KRASG12C [15, 16], targeting RAS directly remains a challenge for NSCLC therapy [17]. Moreover, inhibiting downstream effectors of RAS signaling such as the MAP kinase pathway has not produced durable responses [18,19,20]. The contemporary strategies for lung cancer therapy are centered on the rationale of combining several different treatment modalities that produce the best patient outcomes [21]. Treatment with MEK inhibitors often forms the basis for lung cancer combination therapies [22, 23]. Trametinib, a MEK1/2 inhibitor, was the first agent approved by the FDA in 2013 for the treatment of BRAF V600E-mutant metastatic melanoma, followed by Cobimetinib [23, 24]. A combination of Dabrafenib and Trametinib was approved by the FDA in 2017 for the treatment of metastatic NSCLC harboring BRAF V600E-mutations [24]. The use of Trametinib as a single agent has been shown to induce resistance via overactivation of BRAF or PI3K/Akt pathways [25] and activation of the IGF1R–MEK5–Erk5 pathway [26]. In addition, the adverse side effects of MEK inhibitors [27] prompt the urgent need to develop other novel combination strategies to reduce drug-induced toxicities and suppress conventional therapy-induced resistance.
Autophagy -a lysosomal-mediated cannibalization process- captures, degrades, and recycles subcellular macromolecules and organelles [28]. Using genetically engineered mouse models (GEMMs) to conditionally delete essential autophagy genes, our group and others have demonstrated that autophagy is upregulated and promotes tumorigenesis in different types of cancer by multiple tumor cell-autonomous mechanisms [29,30,31,32,Full size image
Combination of HCQ and Trametinib impaired KL TDCL mitochondrial metabolism
Autophagy is essential to maintain mitochondrial metabolism for cancer cell survival during metabolic stress [5, 30, 32, 45]. To understand the mechanism of synergistic anti-proliferation by the combination treatment in KL TDCLs, KL and KP TDCLs were treated with the single agents or their combination for 6 h and the mitochondrial function was examined using the Seahorse XFe24 Analyzer (Fig. 1F). Combination treatment significantly inhibited basal respiration and ATP production compared with the vehicle control or Trametinib alone in KL TDCLs (Fig. 1G), but not in KP TDCLs (Fig. 1H). Further, metabolomics analysis (Fig. 1I) of TDCLs observed that treatment with Trametinib significantly increased the levels of several glycolytic and TCA cycle intermediates in KL TDCLs, which were reduced upon co-treatment with HCQ (Fig. 1J). In connection with these changes, Trametinib treatment suppressed AMP levels and increased ATP levels in KL cells, which was corrected by co-treatment with HCQ (Fig. 1J). The mitochondrial membrane potential generated by proton pumps is an essential component in the process of energy storage during oxidative phosphorylation. Together with the proton gradient, mitochondrial membrane potential forms the transmembrane potential of hydrogen ions which is harnessed to make ATP [46]. Hence, we measured the mitochondrial membrane potential using MitoTracker Red CMXRos and observed that Trametinib treatment alone significantly increased mitochondrial membrane potential compared to vehicle control in KL TDCLs, which was significantly dampened by the combination of HCQ and Trametinib (Fig. 1K), but not in KP TDCLs (Fig. 1L). Such reduced mitochondrial membrane potential by the combination treatment in KL TDCLs, not in KP TDCLs, was further validated using a standard less oxidative stress-sensitive dye TMRM (Fig. 1M, N). The combination treatment also reduced the levels of several amino acids that were upregulated upon treatment with Trametinib (Fig. 1J). Taken together, we found that the combination of HCQ and Trametinib in KL cells impairs functional mitochondria, leading to energy crises that could prevent cell proliferation and induce cell death.
Combination of HCQ and Trametinib synergistically inhibited KL allograft tumor growth, but not KP allograft tumor growth
To further validate our hypothesis that combining autophagy and MEK inhibitors could be a novel therapeutic strategy specifically in the treatment of KL NSCLC, we first assessed the anti-tumor effect of the combination therapy in established KL and KP TDCL-induced allograft tumors in immunodeficient NCr nude mice. When the allograft tumors were measurable, the mice were treated with HCQ or Trametinib alone, or with a combination of both (Fig. 2A). Although the single agents had no effect in reducing tumor growth, the combination treatment significantly reduced KL allograft tumor growth (Fig. 2B–D), which was accompanied by reduced cell proliferation (Ki67) and pErk in these tumors compared to the tumors collected from the mice treated with the single agents or vehicle control (Fig. 2E). Moreover, p62 accumulation, indicative of autophagy blockade, was observed in the tumors collected from the mice treated with HCQ or the combination (Fig. 2E). Compared to KP lung tumors, KL lung tumors have increased metastasis [11]. We observed that the combination treatment significantly inhibited spontaneous lung metastasis migrated from KL allograft tumors in NCr nude mice assessed by the quantification of tumor number and tumor burden of scanned lung sections (Fig. 2F–H). However, combination treatment did not cause any tumor growth inhibition of KP allograft tumors in NCr nude mice (Fig. 2I–L).

A Scheme of an allograft mouse model in immunodeficient Ncr nude mice treated with vehicle control, HCQ (50 mg/kg, daily, I.P.), Trametinib (1 mg/kg, 5 days/week, oral gavage), or the combination. B Graph of KL allograft tumor growth in Ncr nude mice treated with vehicle control, HCQ, Trametinib, or the combination. C Gross pathology of KL allograft tumors in Ncr nude mice treated with vehicle control, HCQ, Trametinib or the combination. D Tumor weight of KL allograft tumors in Ncr nude mice treated with vehicle control, HCQ, Trametinib or the combination. E IHC for p62, pERK and Ki67 of KL allograft tumor from (C) (left panel) and quantification of pERK and Ki67 (right panel). F Representative H&E staining of lung tissues shows spontaneous lung metastasis from KL allograft tumors in Ncr nude mice. G&H. Quantification of tumor number (G) and tumor burden (H) from (F). I Graph of KP allograft tumor growth in Ncr nude mice treated with vehicle control, HCQ (50 mg/kg, daily, I.P.), Trametinib (1 mg/kg, 5 days/week, oral gavage), or the combination. J Gross pathology of KP allograft tumors in Ncr nude mice treated with vehicle control, HCQ, Trametinib or the combination. K Tumor weight of KP allograft tumors in Ncr nude mice treated with vehicle control HCQ, Trametinib or the combination. L IHC for p62, pERK and Ki67 of KP allograft tumor from (J) (top panel) and quantification of pERK and Ki67 (bottom panel). Data are mean ± s.e.m. *P < 0.05; **P < 0.01; ***P < 0.001.
Combination of HCQ and Trametinib synergistically inhibited established KL lung tumor growth, but not KP lung tumor growth
To better determine whether the combination of HCQ and Trametinib is a novel specific approach for treating KL lung tumors, we induced KL lung tumors in KrasLSL-G12D/+;Lkb1Flox/Flox GEMMs via a single Lentiviral Cre-recombinase intranasal infection. At 12 weeks post tumor induction, when lung tumors were developed [5], mice were treated with vehicle, Trametinib, HCQ or the combination therapy for four weeks (Fig. 3A) and the tumor growth was assessed by gross lung pathology, wet lung weight (Fig. 3B), and quantification of tumor burden and tumor number of scanned lung sections with H&E staining (Fig. 3C–E). As expected, Trametinib did not inhibit the growth of KL lung tumors; however, HCQ showed a tendency to inhibit the growth of KL lung tumors. Most excitingly, the combination therapy significantly suppressed the growth of KL lung tumors compared with the vehicle control or the single agents (Fig. 3B–E). In addition, there was no significant difference in mouse body weight between the four treatment groups, indicating lack of drug toxicity (Fig. 3F). The treatment with Trametinib significantly reduced the phosphorylation of ERK, whereas HCQ treatment caused the accumulation of p62, indicating blockade of the MEK and autophagy pathways in KL lung tumors (Fig. 3G). Less KL tumor cell proliferation was observed when mice were treated with either HCQ or the combination (Fig. 3G), which is consistent with the reduced tumor growth (Fig. 3B–E). These same treatments were applied to the KrasLSL-G12D/+;p53Flox/Flox GEMMs bearing KP lung tumors. The single agents or the combination therapy had no anti-tumor effect on established KP lung tumors (Fig. 3H–N).

A Scheme of KrasLSL_G12D/+;Lkb1Flox/Flox GEMM bearing KL lung tumors treated with vehicle control, HCQ (50 mg/kg, daily, I.P.), Trametinib (1 mg/kg, 5 days/week, oral gavage), or the combination. B Representative gross lung pathology (left panel) and wet lung weight (right panel) of mice bearing KL lung tumors treated with vehicle control, HCQ, Trametinib or the combination for four weeks. C Representative H&E staining of lung tissues of the mice from (B). D, E Quantification of tumor number (D) and tumor burden (E) from (C). F The body weight of the mice bearing KL lung tumor on the day of sacrifice. G IHC for p62, pERK, and Ki67 of KL lung tumors from (B) (left panel) and quantification of pERK and Ki67 (right panel). H Scheme of KrasLSL_G12D/+;p53Flox/Flox GEMM bearing KP lung tumors treated with vehicle control, HCQ (50 mg/kg, daily, I.P.), Trametinib (1 mg/kg, 5 days/week, oral gavage), or the combination. I Representative gross lung pathology (left panel) and wet lung weight (right pane) of mice bearing KP lung tumors treated with vehicle control, HCQ, Trametinib or the combination for four weeks. J Representative H&E staining of lung tissues of the mice from (I). K, L Quantification of tumor number (K) and tumor burden (L) from (J). M The body weight of the mice bearing KP lung tumor on the day of sacrifice. N IHC for p62, pERK, and Ki67 of KP lung tumors from (I) (left panel) and quantification of pERK and Ki67 (right panel). Data are mean ± s.e.m. *P < 0.05; **P < 0.01; ***P < 0.001.
Combination of HCQ and Trametinib synergistically inhibited KL PDX tumor growth, but not KP PDX tumor growth
Patient-derived tumor xenograft models (PDXs) have increasingly become the preferred tool in research to translate findings for optimal treatment of the human diseases. We therefore examined the effect of combination therapy in treating PDX tumors for KL and KP NSCLC. NSG mice bearing established KL (JAX-J000095635) PDX or KP (JAX-TM00233) PDX were treated with the single agents or the combination, and tumor growth was monitored (Fig. 4A). Compared with the single agents, the combination treatment synergistically inhibited KL PDX tumor growth (Fig. 4B–D). Histology analysis found that the combination treatment increased p62 accumulation, decreased pERK and pS6, and inhibited cell proliferation (Ki67) (Fig. 4E). Although Trametinib inhibited the growth of KP PDX tumors, the addition of HCQ did not show further anti-tumor effects (Fig. 4F–I). In conclusion, through different mouse models, we demonstrated that the use of HCQ to inhibit autophagy specifically causes KL tumors, but not KP tumors, to be sensitive to the MEK inhibitor Trametinib.

A Scheme of Patient-Derived Xenograft (PDX) tumor model treated with vehicle control, HCQ (50 mg/kg, daily, I.P.), Trametinib (Trametinib, 1 mg/kg, 5 days/week, oral gavage), or the combination. B Graph of KL PDX tumor growth in NSG mice treated with vehicle control, HCQ, Trametinib or the combination for 10 weeks. C Gross pathology of KL PDX tumors. D KL PDX tumor weight at the end of experiment. E H&E and IHC for p62, pERK, pS6, and Ki67 (left panel) and quantification of pERK, pS6, and Ki67 (right panel) of KL PDX tumors. F Graph of KP PDX tumor growth in NSG mice treated with vehicle control, HCQ, Trametinib, or the combination for 9.5 weeks. G Gross pathology of KP PDX tumors. H. KP PDX tumor weight at the end of experiment. I H&E and IHC for p62, pERK, pS6, and Ki67 (left panel) and quantification of pERK, pS6, and Ki67 (right panel) of KP PDX tumors. Data are mean ± s.e.m. *P < 0.05; **P < 0.01; ***P < 0.001.
Combination of HCQ and Trametinib impaired KL lung tumor energy homeostasis
To understand the mechanism of the combination treatment in inducing KL tumor regression, we dissociated the KL lung tumors into single-cell suspension one week after treatment with the single agents or the combination and examined the mitochondrial function (Fig. 5A). We found that mitochondrial membrane potential was significantly higher in the KL lung tumor cells from mice treated with Trametinib than vehicle control mice, which was significantly reduced when combined with HCQ treatment (Fig. 5B). Compared with Trametinib alone, the combination of HCQ and Trametinib also resulted in a significant decrease in basal respiration and ATP production in KL lung tumor cells (Fig. 5C, D). Hence, as observed in the in vitro experiments (Fig. 1F–K, M), the combination therapy may impair mitochondrial function to prevent the growth of KL lung tumors in vivo.

A Scheme to examine mitochondrial function in KL lung tumors. B Overlap** images of single-cell suspension from KL lung tumors of the mice treated with vehicle control, HCQ, Trametinib, or the combination for one week and stained with MitoTracker Red CMXRos for mitochondrial membrane potential and Mitotracker Green FM for mitochondrial mass (left panel); and graph of the relative mitochondrial membrane potential of single-cell suspension from KL lung tumors (the ratio of red fluorescence intensity and green fluorescence intensity) (right panel). Blue: Hoechst 33342 for nuclear staining. C Basal mitochondrial respiration of single-cell suspension from KL lung tumors of the mice treated with vehicle control, HCQ, Trametinib, or the combination for one week, measured by Seahorse XFe24 Analyzer. D ATP production of single-cell suspension from KL lung tumors of the mice treated with vehicle control, HCQ, Trametinib, or the combination for one week, measured by Seahorse XFe24 Analyzer. E Scheme of in vivo [U13C6]-glucose tracing in mice treated with vehicle control, HCQ, Trametinib, or the combination for two weeks (top panel) and 13C glucose carbons to glycolytic and TCA cycle intermediates (bottom panel). F Normalized labeling fraction of glucose and TCA cycle intermediates of KL lung tumors by infused [U13C6]-glucose in mice for 2.5 h. G Levels of glucose, pyruvate, and lactate of KL lung tumors from mice treated with vehicle control, HCQ, Trametinib, or the combination for two weeks. H Levels of ATP and AMP of KL lung tumors from mice treated with vehicle control, HCQ, Trametinib or the combination for two weeks. Data are mean ± s.e.m. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.001.
LKB1 loss upregulates tumor glycolysis [47]. In addition, glucose is one of the major carbon sources for KL lung tumor TCA cycle intermediates [48]. To determine the impact of the combination treatment on KL lung tumor glycolysis and TCA cycle metabolism, we performed in vivo [U13C6]-glucose tracing and metabolic flux analysis in mice bearing KL lung tumors after two weeks of drug treatment (Fig. 5E). Total glucose enrichment was comparable in KL lung tumors of mice treated with vehicle control, HCQ, Trametinib or the combination. We observed that HCQ significantly inhibited the glucose flux to glycolytic intermediates glucose-6-phosphate, pyruvate and lactate; but had no effect on TCA cycle metabolites; the combination of HCQ and Trametinib significantly suppressed the glucose flux to glycolytic intermediates and TCA cycle intermediates (Fig. 5F). In addition, the levels of glucose and pyruvate in KL lung tumors from combination treatment were significantly lower than the tumors of mice treated with Trametinib (Fig. 5G). Moreover, compared with the vehicle control or the single agents, the ATP level of KL lung tumors in mice treated with the combination was significantly reduced (Fig. 5H). Taken together, our observations indicate that one of the mechanisms by which the combination therapy inhibits the growth of KL tumors might be the impairment of glucose-mediated metabolism, resulting in energy crisis.
Combination of HCQ and Trametinib induced Ferroptosis in KL tumors
Ferroptosis is characterized by ROS production from accumulated lipid and iron peroxidation [49]. Autophagy promotes ferroptosis by regulating cellular iron homeostasis [50, 51]. However, many existing studies suggest that autophagy promotes drug resistance, while ferroptosis reverses drug resistance in cancer treatment [52, 53]. A recent study reported that Atg7 deletion in Cre-inducible Atg7 knockout mice decreases NRF2 levels and enhances ferroptosis in liver [54]. Increased oxidative stress is usually associated with mitochondrial injury with a marked decrease in mitochondrial membrane potential [55]. Indeed, we observed that combination of HCQ and Trametinib led to reduced mitochondrial membrane potential compared to control or Trametinib treatment alone in KL TDCLs (Fig. 1K, M). We therefore hypothesized that combination of HCQ and Trametinib may cause ferroptosis in KL tumors.
As ferroptosis is associated with lipid peroxidation [56], we first examined the lipid peroxidation in KL and KP TDCLs in the absence or presence of the ferroptosis inhibitor Ferrostatin-1 by C-11 BODIPY staining. We found that HCQ combined with Trametinib significantly increased lipid peroxidation of KL TDCLs compared with vehicle control or Trametinib alone, which was significantly inhibited by the ferroptosis inhibitor Ferrostatin-1 (Fig. 6A). In KP TDCLs, although lipid peroxidation was observed when cells were treated with Trametinib or the combination, Ferrostatin-1 treatment had no effect on lipid peroxidation (Fig. 6A). We further found that Ferrostatin-1 successfully prevented cell death and restored the reduced cell proliferation caused by the combination therapy in KL TDCLs (Fig. 6B & C).

A Lipid peroxidation of KL and KP TDCLs was examined by C11-BODIPY (1 μm) staining in cells treated with vehicle control, HCQ (10 μM), Trametinib (2.5 nM) and the combination in the absence or presence of ferroptosis inhibitor ferrostatin-1 (0.5 μM). B Clonogenic survival assay of KL and KP TDCLs treated with vehicle control, HCQ (10 μM), Trametinib (2.5 nM) and the combination in the absence or presence of ferroptosis inhibitor ferrostatin-1 (0.5 μM). C Relative cell proliferation of KL TDCLs treated with vehicle control, HCQ (10 μM), Trametinib (2.5 nM) and the combination in the absence or presence of ferroptosis inhibitor ferrostatin-1 (0.5 μM). D, E IHC (D) and quantification (E) of 4-HNE staining in KL and KP lung tumors from GEMMs as well as PDX tumors. F Graph of KL allograft tumor growth in C57BL/6 mice treated with vehicle control, HCQ (50 mg/kg, daily, I.P.), Trametinib (1 mg/kg, 5 days/week, oral gavage), and the combination in the absence or presence of ferroptosis inhibitor Liproxstatin-1 (10 mg/kg, daily, I.P.). G Gross KL allograft tumor from (F) at the end of experiment. H KL allograft tumor weight from (G) at the end of experiment. I Body weight of the mice on the day of sacrifice from (F). J Representative H&E and IHC for Ki67 of KL allograft tumors from (G) (left panel) and quantification of Ki67 (right panel). Data are mean ± s.e.m. *P < 0.05; **P < 0.01; ***P < 0.001.
Consistent with the in vitro data, KL lung tumors and KL PDX tumors from the mice treated with the combination therapy showed increased expression of ferroptosis marker 4-hydroxynonenal (4-HNE) compared with vehicle control and the single agents (Fig. 6D, E); this difference was not observed in KP lung tumors and PDX tumors (Fig. 6D, E). These prompted us to test the hypothesis that combination of HCQ and Trametinib may cause ferroptosis, thereby suppressing the growth of KL tumors. We therefore induced subcutaneous allograft KL tumors by injecting KL TDCLs in syngeneic C57BL/6 mice. Once the tumor size reached to 200 mm3, we treated the mice with the single agents or the combination in the absence or presence of the ferroptosis inhibitor Liproxstatin-1. The combination of HCQ and Trametinib significantly inhibited KL allograft tumor growth compared with the control or the single agents; while the ferroptosis inhibitor Liproxstatin-1 significantly rescued the KL allograft tumor growth in mice treated with the combination compared with vehicle control (Fig. 6F–H). There was no difference in the body weight between treatment groups (Fig. 6I), indicating lack of drug toxicities. Moreover, decreased cell proliferation was observed in the tumors of mice with combination treatment, which was abolished by the Liproxstatin-1 treatment (Fig. 6J). Taken together, we demonstrated that one of mechanisms for reducing KL tumor growth through the combination of HCQ and Trametinib might be by inducing ferroptotic cell death.
