
Abstract
Circular RNAs (circRNAs) are a recently discovered kind of regulatory RNAs that have emerged as critical biomarkers of various types of cancers. Metabolic reprogramming has gradually been identified as a distinct hallmark of cancer cells. The pentose phosphate pathway (PPP) plays an indispensable role in satisfying the bioenergetic and biosynthetic demands of cancer cells. However, little is known about the role of circRNAs and PPP in colorectal cancer (CRC). The novel circ_0003215 was identified at low levels in CRC and was negatively correlated with larger tumor size, higher TNM stage, and lymph node metastasis. The decreased level of circ_0003215 was resulted from the RNA degradation by the m6A reader protein YTHDF2. A series of functional assays demonstrated that circ_0003215 inhibited cell proliferation, migration, invasion, and CRC tumor metastasis in vivo and in vitro. Moreover, circ_0003215 regulated the expression of DLG4 via sponging miR-663b, thereby inducing the metabolic reprogramming in CRC. Mechanismly, DLG4 inhibited the PPP through the K48-linked ubiquitination of glucose-6-phosphate dehydrogenase (G6PD). Taken together, we have identified m6A-modified circ_0003215 as a novel regulator of metabolic glucose reprogramming that inhibited the PPP and the malignant phenotype of CRC via the miR-663b/DLG4/G6PD axis.
Similar content being viewed by others

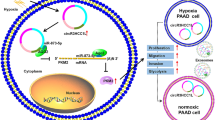

Background
Colorectal cancer (CRC) is the second and third most frequently diagnosed cancer in females and males worldwide, respectively [1]. Despite significant advancements in surgical techniques, chemotherapy, immunotherapy, and molecular-targeted treatments, the overall prognosis and 5-year survival rate of CRC patients remain poor, particularly for those with distant metastatic CRC [2, 3]. Thus, it is crucial to understand the potential mechanisms underlying CRC progression, which may contribute to the enlargement of the molecular pool for the diagnosis and therapy of CRC.
CircRNAs, a newly recognized type of noncoding RNAs (ncRNAs), are generally characterized by the covalently closed loop structure without a 3′-poly-A tail or 5′-cap [ Two circRNA expression profiles (GSE121895 and GSE142837) and a miRNA expression profile (GSE115513) were downloaded from the Gene Expression Omnibus (GEO) database. The CRC RNA sequencing data (COAD, READ) were obtained from The Cancer Genome Atlas (TCGA) database. The “limma” package in R software was used to explore the significantly differentially expressed circRNAs or miRNAs. One hundred pairs of fresh human CRC specimens and their adjacent normal tissues were obtained from Wuhan University’s Zhongnan Hospital. The study participants provided their informed consent before the study began. The clinical information and pathological features of these patients were collected and are presented in Supplementary Table S1. The CRC cell lines (SW620, HCT116, SW480, DLD-1, LOVO, and HT29) and the human embryonic kidney epithelial cell line (HEK-293T) were purchased from the American Type Culture Collection (Manassas, VA, USA). HCT-116, DLD-1, HT29, and LOVO were grown in RPMI-1640 medium (Gibco; Thermo Fisher Scientific, Waltham, MA, USA) with 10% fetal bovine serum (FBS) (HyClone Laboratories Inc, Logan, UT, USA). SW480, SW620, and HEK-293T were cultured in Dulbecco’s Modified Eagle Medium (DMEM) (Gibco; Thermo Fisher Scientific) containing 10% FBS. Total RNA from cells or CRC tissues was extracted using TRIzol reagent (Invitrogen, Waltham, MA, USA). cDNA was synthesized with random primers using HiScript II Q RT SuperMix (Vazyme, Nan**g, China) or miRNA stem-loop RT primers using a miRNA 1st Strand cDNA Synthesis Kit (Vazyme). Subsequently, RT-qPCR was performed using ChamQ Universal SYBR qPCR Master Mix (Vazyme). The primer sequences are listed in Supplementary Table S2 and were synthesized by Tsingke (Nan**g, China). For the RNase R degradation assay, total RNA (2 µg) was administrated with 3 U/µg RNase R (Epicenter). For the actinomycin D assay, SW480 and HT29 cells were cultured with 5 mg/L actinomycin D (Sigma-Aldrich, St. Louis, MO, USA) and incubated for indicated periods. Thereafter, RNA was extracted at each time point and subjected to RT-qPCR. The CRC tissue samples or cells were lysed and denatured in loading buffer. Subsequently, the total proteins were resolved on sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gel and then transferred onto polyvinylidene difluoride (PVDF) membranes. After blocking in 5% non-fat milk, the PVDF membranes were reacted with primary antibodies overnight and then hybridized with the corresponding secondary antibodies. Finally, the protein bands were visualized by chemiluminescence using an ECL system. All primary antibodies are listed in Supplementary Table S3. Specific probes for miR-663b and circ_0003215 were synthesized by GenePharma (Shanghai, China) and RiboBio (Guangzhou, China). All probe sequences are listed in Supplementary Table S4. Briefly, SW480 and HT29 cells were seeded in confocal dishes (Nest Scientific Inc, Woodbridge, NJ, USA), washed, fixed, and then permeabilized with 0.1% Triton X-100. For probe detection, CRC cells were pre-hybridized for 40 min and subsequently hybridized with the specific probe mix overnight at 37 °C. Thereafter, the CRC cells were stained with DAPI at 37 °C for 30 min after washing with a hybridization solution. Finally, the fluorescence images were acquired using a Dragonfly 200 High-Speed Confocal Microscope (Andor Technology Ltd, Belfast, UK). MiR-663b mimics, inhibitors, and small interfering RNAs (siRNAs) targeting circ_0003215 were synthesized by RiboBio. The pLCDH-circ_0003215 overexpression vector was constructed by cloning the cDNA of circ_0003215 into the specific pLCDH-circ expression vector. The short hairpin RNA (shRNA) of DLG4 was constructed by cloning the DNA sequence targeting DLG4 into the pLKO.1 plasmid. All the sequences are listed in Supplementary Table S4. Cytoplasmic and nuclear fractions of CRC cells were separated using a Cytoplasmic and Nuclear RNA Purification kit (Norgen Biotek Corp, Thorold, ON, Canada). Briefly, CRC cells were harvested and lysed on ice for 15 min. After centrifugation, the nuclear and cytoplasmic fractions were collected separately. Afterward, the isolated RNAs in the nucleus and cytoplasm were extracted using TRIzol and subjected to RT-qPCR. Cell proliferative ability was determined using a CCK-8 assay kit (Beyotime Biotech, Bei**g, China). Briefly, CRC cells were seeded in a 96-well plate and then cultured overnight. At 24, 48, 72, and 96 h, CRC cells with different transfections were supplemented with CCK-8 solution (10 μL/well) and cultured for an additional 2 h. Finally, the resulting optical density at 450 nm was measured using a microplate reader (Bio-Rad Laboratories, Hercules, CA, USA). The transwell assays were conducted using transwell chambers (Corning Life Sciences, Corning, NY, USA) and matrigel (BD Biosciences, Franklin Lakes, NJ, USA). For the transwell migration assay, approximately 5 × 104 transfected CRC cells suspended in 200 µL of medium without FBS were seeded in the upper chamber, while the lower chamber contained culture medium with 20% FBS as a chemoattractant. For cell invasion assay, cells were also seeded in the upper chamber precoated with matrigel. Next, the CRC cells with different transfections subjected to migration assay for 24 h and invasion assay for 48 h assay After removal of the cells remaining on the upper surface of the membrane, the cells migrated or invaded undersurface of the membrane were fixed. After staining with 0.1% crystal violet, the cells were observed and counted under a microscope. For the colony formation assay, 5 × 102 transfected CRC cells were seeded into a six-well plate and cultured for 14 d. Afterward, the cell colonies were fixed with paraformaldehyde and stained with 0.1% crystal violet. Next, the plate was washed with PBS, and the total number of cell colonies of more than 50 cells was counted and photographed. Transfected CRC cells were seeded in 6-well culture plates in complete DMEM/RPMI-1640 media and grown to 80% confluence in a monolayer overnight. Next, scratch wounds were inflicted along the diameter of each well using a 100-µL pipette tip. Thereafter, cell debris and suspended cells were removed by extensive washing, and then culture medium with 1% FBS was added. Finally, wound healing was measured and photographed under a microscope at 0 and 24 h within the wound line and the healing area was calculated using Image J software (NIH, Bethesda, MD, USA). A BeyoClick™ EdU-594 Cell Proliferation Kit (Beyotime Biotech) was used for the EdU assay. Briefly, the EdU reagent was directly dissolved into the culture medium at a 10-μM final concentration. After incubation for another 2 h, the CRC cells were fixed with paraformaldehyde for 15 min and stained with Click Additive Solution in the dark. Thereafter, Hoechst staining was used to counterstain nuclei, and EdU-positive cells were photographed and counted under a fluorescence microscope. The targeted binding sites among circ_0003215, miR-663b, and DLG4 were confirmed by the dual-luciferase reporter assay. The binding sites between circ_0003215 and miR-663b, DLG4 and miR-663b were predicted by bioinformatics analysis. Luciferase vectors with circ_0003215 or the 3′-UTR of DLG4 and mutant types containing the firefly and Renilla luciferase genes were obtained from Promega (Madison, WI, USA). Next, HEK-293T cells were transfected with psiCHECK-circ_0003215 vectors (wild-type [WT] or mutant [MUT]) and miR-663b mimics, or psiCHECK-DLG4 vectors (WT1, WT2, or MUT1, MUT2) and miR-663b mimics. Relative luciferase activities were determined according to the corresponding instructions for the Dual-Luciferase Reporter Assay kit (Promega). The RIP assay was conducted using the EZ-Magna RIP kit (MilliporeSigma, Burlington, MA, USA). Briefly, CRC cells were incubated with RIP lysis buffer containing RNase and protease inhibitors on ice for 60 min. Next, the cell lysates were incubated overnight with RIP buffer containing magnetic beads conjugated to IP-grade antibodies. Subsequently, the immune complexes were washed five times and treated with proteinase K. Finally, the immune precipitated RNA was isolated and subjected to RT-qPCR analysis. The biotinylated-circ_0003215 and NC probes were obtained from GenePharma (Shanghai, China). Briefly, CRC cells (1.5 × 107) were fixed and lysed in lysis buffer. Afterward, the supernatant was collected, and 50 μL was pipetted for input after centrifugation, with the remainder incubated with biotin-labeled probes. Subsequently, the complexes were incubated with streptavidin-coupled Dynabeads overnight. Next, the beads that captured the immune complex were washed twice and then treated with proteinase K at 25 °C for 1 h. Finally, the bound RNAs were extracted using TRIzol, followed by RT-qPCR analysis. All probe sequences are listed in Supplementary Table S4. Oxidative PPP flux was determined by measuring 14CO2 release. Briefly, a 10-cm dish with two sealed pinholes containing a 6-cm Petri dish was seeded with CRC cells. Afterward, the cells were cultured for 4 h with medium supplemented with [1-14C] or [6-14C]glucose until 80% confluence. The 14CO2 released from the cells was harvested by sealing the 10-cm dish. Oxidative PPP flux was stopped by injecting 2 N HCl through one of the holes, and the released 14CO2 was harvested for 12 h by injecting hyamine hydroxide into a cup placed on the 10-cm dish through the other hole. Subsequently, the hyamine hydroxide in the cup was dissolved in 60% methanol and subjected to scintillation counting. The 14CO2 content released was obtained by subtracting the scintillation count of the [6-14C] glucose treatment from that of the [1-14C]glucose treatment. The amount of intracellular ROS was quantified using dichlorodihydrofluorescein diacetate (DCFH‐DA; Invitrogen) oxidation. CRC cells transfected with specified plasmids were seeded into 6-well plates. After 24 h, the cells were washed and subsequently treated with 10 μM DCFH-DA for 30 min. Finally, the cells were harvested, resuspended in 500 µL FACS buffer, and subjected to FACS analysis (BD Biosciences). The NADPH/NADP+ ratio was determined using a Colorimetric Assay Kit (Sigma-Aldrich). Briefly, 3 × 106 transfected cells were harvested and lysed with 200 μL NADP+/NADPH extraction buffer. Thereafter, the lysed cells were incubated for 5 min at 60 °C, and then 20 μL assay buffer and 200 μL counter NADPH/NADP+ extraction buffer were added. Next, the samples were centrifuged for 20 min, and the supernatants were used to determine the NADPH/NADP+ ratio. Finally, the absorbance at 565 nm was determined using a plate reader at 0 and 30 min. Cells were seeded in 6-well plates, and when they reached approximately 70% confluence, they were treated with the protein synthesis inhibitor cycloheximide at the indicated times. Afterward, the cells were harvested and analyzed by immunoblotting with specific antibodies. For immunoprecipitation, transfected cells were lysed using RIPA lysis buffer containing protease inhibitors on ice for 2 h. Next, 900 μL samples were incubated with protein A/G magnetic beads conjugated with immunoprecipitation antibodies at 4°C overnight. Finally, the bead-antibody complexes were washed thrice with RIPA lysis buffer, and the immuno-complexes were subjected to western blotting analysis. Cells were transfected with specific plasmids (HA-WT Ub, HA-K6O Ub, HA-K11O Ub, HA-K27O Ub, HA-K29O Ub, HA-K33O Ub, HA-K48O Ub, HA-K63O Ub, etc.). Forty-eight hours later, MG132 (10 μM; Sigma-Aldrich) was directly added to the culture media, and the cells were incubated for an additional 6h. Thereafter, the cells were collected, and the target protein were immunoprecipitated using A/G beads and IP-grade antibodies. Finally, the eluted proteins were analyzed by western blotting. Animal studies were approved by the Institutional Animal Care and Use Committee. All BALB/c mice were kept at the Wuhan Institute of Virology, Chinese Academy of Sciences (Wuhan, China), and were strictly and equally randomized. For the tumorigenesis assay, CRC cells with circ_0003215 overexpression or the respective control cells were suspended in 100 µL PBS and injected into the back of each mouse. Subcutaneous tumor volumes were estimated every 3d using the formula: volume = 1/2 × length × width2. Three weeks later, all mice were sacrificed, and subcutaneous tumors were excised and weighed. For the lung metastasis model, CRC cells (5 × 105) with different transfections were injected via the caudal vein. All tail vein-injected mice were sacrificed after 6 weeks. Subsequently, their lungs were extracted and weighed, and the number of metastatic nodules was carefully calculated, experimenters were blinded from the treatment paradigm and where not involved in data analysis. The sequence data of all circRNAs were searched and downloaded from circBank. The target miRNAs that directly interacted with circ_0003215 were predicted using circBank. Target miRNA genes were predicted using Targetscan, miRWalk, and miBase. All experiments were carried out independently at least thrice. The data are presented as mean ± standard deviation. Statistical analysis were conducted using SPSS 19.0 (SPSS Inc, Chicago, IL, USA) and GraphPad Prism v8.3.0 (GraphPad Software, San Diego, CA, USA). Before statistical analysis, the homogeneity of variance between groups was tested. The correlation between clinicopathologic parameters and circ_0003215 expression was analyzed using the chi-square test. Two-group comparisons of continuous variables were analyzed using the paired or unpaired Student’s t-test, all data followed a normal distribution. P < 0.05 was considered statistically significant.Methods
Microarray and RNA sequencing data
Tissue specimens and ethical approval
Cell culture
RNA extraction and reverse transcription quantitative PCR (RT-qPCR)
RNase R treatment assay and actinomycin D assay
Western blot
Fluorescence in situ hybridization (FISH)
miRNAs, siRNAs, plasmid, and transfection
Nuclear and cytoplasmic extraction
Cell counting kit-8 (CCK-8) assay
Transwell assay
Colony formation assay
Scratch wound healing assay
5-Ethynyl-2′-deoxyuridine (EdU) assay
Luciferase reporter assay
RNA immunoprecipitation (RIP) assay
RNA pull-down assay
Oxidative PPP flux assay
ROS
NADPH/NADP+ ratio
Protein half‑life assay
Immunoprecipitation
Ubiquitination assay
Xenograft nude mouse model
Bioinformatics analysis
Statistical analysis
Data availability
The datasets analyzed during the current study were available from the corresponding authors.
Change history
18 October 2022
A Correction to this paper has been published: https://doi.org/10.1038/s41419-022-05332-4
References
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209–49.
Evelien D, Pieter JT, Jasper LAV, Pashtoon MK, Michael BW. Colorectal cancer. Lancet 2019;394:1467–80.
Puccini A, Lenz HJ. Colorectal cancer in 2017: Practice-changing updates in the adjuvant and metastatic setting. Nat Rev Clin Oncol. 2018;15:77–8.
Su M, **ao YH, Ma JL, Tang YY, Tian B, Zhang YQ, et al. Circlar RNAs in Cancer: emerging functions in hallmarks, stemness, resistance and roles as potential biomarkers. Mol Cancer. 2019;18:90.
Goodall GJ, Wickramasinghe VO. RNA in cancer. Nat Rev Cancer. 2021;21:22–36.
Li J, Xu Q, Huang ZJ, Mao N, Lin ZT, Cheng L, et al. CircRNAs: a new target for the diagnosis and treatment of digestive system neoplasms. Cell Death Dis. 2021;12:205.
Jian XY, He H, Zhu JH, Zhang Q, Zheng ZX, Liang XJ, et al. Hsa_circ_001680 affects the proliferation and migration of CRC and mediates its chemoresistance by regulating BMI1 through miR-340. Mol Cancer. 2020;19:20.
Fang L, Lv JL, Xuan Z, Li BW, Li Z, He ZY, et al. Circular CPM promotes chemoresistance of gastric cancer via activating PRKAA2-mediated autophagy. Clin Transl Med. 2022;12:e708.
Zhang CJ, Zhou XL, Geng XG, Zhang Y, Wang JY, Wang YN, et al. Circular RNA hsa_circ_0006401 promotes proliferation and metastasis in colorectal carcinoma. Cell Death Dis. 2021;12:443.
Dai DJ, Wang HY, Zhu L, ** HC, Wang X. N6-methyladenosine links RNA metabolism to cancer progression. Cell Death Dis. 2018;9:124.
Zhang LL, Hou CF, Chen C, Guo YX, Yuan WT, Yin DT, et al. The role of N-methyladenosine (m6A) modification in the regulation of circRNAs. Mol Cancer. 2020;19:105.
Vander HMG, DeBerardinis RJ. Understanding the intersections between metabolism and cancer biology. Cell 2017;168:657–69.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144:646–74.
Vander HMG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 2009;324:1029–33.
Levine AJ, Puzio-Kuter AM. The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science 2010;330:1340–44.
DeNicola GM, Cantley LC. Cancer’s fuel choice: new flavors for a picky eater. Mol Cell. 2015;60:514–23.
Cheng J, Huang Y, Zhang XH, Yu Y, Wu SM, Jiao J, et al. TRIM21 and PHLDA3 negatively regulate the crosstalk between the PI3K/AKT pathway and PPP metabolism. Nat Commun. 2020;11:1880.
Li QJ, Qin T, Bi ZF, Hong HM, Ding L, Chen JW, et al. Rac1 activates non-oxidative pentose phosphate pathway to induce chemoresistance of breast cancer. Nat Commun. 2020;11:1456.
Ma HH, Zhang FQ, Zhou L, Cao TY, Sun DC, Wen SX, et al. c-Src facilitates tumorigenesis by phosphorylating and activating G6PD. Oncogene 2021;40:2567–80.
Lin RT, Elf S, Shan CL, Kang HB, Ji QJ, Zhou L, et al. 6-Phosphogluconate dehydrogenase links oxidative PPP, lipogenesis and tumour growth by inhibiting LKB1-AMPK signalling. Nat Cell Biol. 2015;17:1484–96.
Kong RR, Yi FS, Wen PS, Liu JH, Chen XP, Ren JQ, et al. Myo9b is a key player in SLIT/ROBO-mediated lung tumor suppression. J Clin Invest. 2015;125:4407.
Park OH, Ha H, Lee YJ, Boo SH, Kwon DH, Song HK, et al. Endoribonucleolytic cleavage of mA-Containing RNAs by RNase P/MRP complex. Mol Cell. 2019;74:494–507.
** XL, Sun BF, Wang L, **ao W, Yang X, Wang WJ, et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 2014;24:177–89.
Fu Y, Dominissini D, Rechavi G, He C. Gene expression regulation mediated through reversible m6A RNA methylation. Nat Rev Genet. 2014;15:293–306.
Li J, Chen K, Dong X, Xu YT, Sun Q, Wang HH, et al. YTHDF1 promotes mRNA degradation via YTHDF1-AGO2 interaction and phase separation. Cell Prolif. 2022;55:e13157.
**a TL, Li XY, Wang XP, Zhu YJ, Zhang H, Cheng WS, et al. N(6)- methyladenosine-binding protein YTHDF1 suppresses EBV replication and promotes EBV RNA decay. EMBO Rep. 2021;22:e50128.
Li JF, **e HY, Ying YF, Chen H, Yan HQ, He LJ, et al. YTHDF2 mediates the mRNA degradation of the tumor suppressors to induce AKT phosphorylation in N6- methyladenosine-dependent way in prostate cancer. Mol Cancer. 2020;19:152.
Shi HL, Wang X, Lu ZK, Zhao BS, Ma HH, Hsu PJ, et al. YTHDF3 facilitates translation and decay of N-methyladenosine-modified RNA. Cell Res. 2017;27:315–28.
Shima H, Matsumoto M, Ishigami Y, Ebina M, Muto A, Sato Y, et al. S-adenosylmethionine synthesis is regulated by selective N-adenosine methylation and mRNA degradation involving METTL16 and YTHDC1. Cell Rep. 2017;21:3354–63.
Nguyen LH, Goel A, Chung DC. Pathways of colorectal carcinogenesis. Gastroenterology 2020;158:291–302.
Long F, Lin Z, Li L, Ma M, Lu ZX, **g L, et al. Comprehensive landscape and future perspectives of circular RNAs in colorectal cancer. Mol Cancer. 2021;20:26.
Liang M, Yao WX, Shi BY, Zhu XJ, Cai R, Yu ZJ, et al. Circular RNA hsa_circ_ 0110389 promotes gastric cancer progression through upregulating SORT1 via sponging miR-127-5p and miR-136-5p. Cell Death Dis. 2021;12:639.
Chen YH, Ling ZN, Cai XL, Xu YF, Lv Z, Man D, et al. Activation of YAP1 by N6-methyladenosine-modified circCPSF6 drives malignancy in hepatocellular carcinoma. Cancer Res. 2022;82:599–614.
Chen RX, Chen X, **a LP, Zhang JX, Pan ZP, Ma XD, et al. N-methyladenosine modification of circNSUN2 facilitates cytoplasmic export and stabilizes HMGA2 to promote colorectal liver metastasis. Nat Commun. 2019;10:4695.
Pamudurti NR, Bartok O, Jens M, Ashwal-Fluss R, Stottmeister C, Ruhe L, et al. Translation of CircRNAs. Mol Cell. 2017;66:9–21.
Zhong YX, Du YJ, Yang X, Mo YZ, Fan CM, **ong F, et al. Circular RNAs function as ceRNAs to regulate and control human cancer progression. Mol Cancer. 2018;17:79.
Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495:333–8.
Li Y, Zheng QP, Bao CY, Li SY, Guo WJ, Zhao J, et al. Circular RNA is enriched and stable in exosomes: a promising biomarker for cancer diagnosis. Cell Res. 2015;25:981–4.
Liu ST, Chen L, Chen H, Xu KK, Peng X, Zhang MC. Circ_0119872 promotes uveal melanoma development by regulating the miR-622/G3BP1 axis and downstream signalling pathways. J Exp Clin Cancer Res. 2021;40:66.
Winter JL, Jung S, Keller S, Gregory RI, Diederichs S. Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat Cell Biol. 2009;11:228–34.
Pellatt DF, Stevens JR, Wolff RK, Mullany LE, Herrick JS, Samowitz W, et al. Expression profiles of miRNA subsets distinguish human colorectal carcinoma and normal colonic mucosa. Clin Transl Gastroenterol. 2016;7:e152.
Li Q, Pan XX, Zhu DM, Deng ZM, Jiang RQ, Wang XH. Circular RNA MAT2B promotes glycolysis and malignancy of hepatocellular carcinoma through the miR-338-3p/PKM2 axis under hypoxic stress. Hepatology 2019;70:1298–316.
Zhou XX, Liu K, Cui J, **ong JX, Wu HS, Peng T, et al. Circ-MBOAT2 knockdown represses tumor progression and glutamine catabolism by miR-433-3p/GOT1 axis in pancreatic cancer. J Exp Clin Cancer Res. 2021;40:124.
Mo YZ, Wang YM, Zhang S, **ong F, Yan QJ, Jiang XJ, et al. Circular RNA circRNF13 inhibits proliferation and metastasis of nasopharyngeal carcinoma via SUMO2. Mol Cancer. 2021;20:112.
Acknowledgements
This work was supported by Engineering construction project of improving the diagnosis and treatment ability of difficult diseases(oncology) (Grant No. ZLYNXM202012), Joint Foundation of Health Commission of Hubei Province (Grant No. znpy2019086), and Wu Jie** Medical Research Foundation(No.320.6750.2021-11-8). The authors would like to thank Editage (http://www.editage.co.kr) for the professional language editing service.
Author information
Authors and Affiliations
Contributions
CBX and HYT designed the study; CBX, HYT, and GR carried out the most experiments. TSH and ZX contributed patient samples and clinical information. CBX and HYT performed bioinformatic analysis. ZX and TSH conducted the statistical analysis. CBX and HYT wrote the manuscript. XXY, CQJ, QQ, RXH, FLF and JCQ revised the paper. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
All experiments were conducted in accordance with the ethical standards of the ethics committee of Zhongnan Hospital of Wuhan University and Wuhan Institute of Virology, Chinese Academy of Sciences.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by Dr Maurizio Fanciulli
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Chen, B., Hong, Y., Gui, R. et al. N6-methyladenosine modification of circ_0003215 suppresses the pentose phosphate pathway and malignancy of colorectal cancer through the miR-663b/DLG4/G6PD axis. Cell Death Dis 13, 804 (2022). https://doi.org/10.1038/s41419-022-05245-2
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-022-05245-2
- Springer Nature Limited