
Abstract
Tubulointerstitial fibrosis (TIF) is involved in the development of diabetic kidney disease (DKD). Transforming growth factor β1 (TGF-β1) is involved in the extensive fibrosis of renal tissue by facilitating the partial epithelial-mesenchymal transition (EMT), increasing the synthesis of extracellular matrix (ECM), inhibiting degradation, inducing apoptosis of renal parenchyma cells, and activating renal interstitial fibroblasts and inflammatory cells. Recent studies indicated that bone morphogenetic protein-7 (BMP-7) upregulated the expression of endogenous SnoN against renal TIF induced by TGF-β1 or hyperglycemia. Nevertheless, the mechanisms underlying the BMP-7-mediated restoration of SnoN protein level remains elusive. The present study demonstrated the increased expression of BMP-7 in diabetic mellitus (DM) mice by hydrodynamic tail vein injection of overexpressed BMP-7 plasmid, which attenuated the effects of DM on kidney in mice. Partial tubular EMT and the accumulation of Collagen-III were resisted in DM mice that received overexpressed BMP-7 plasmid. Similar in vivo results showed that BMP-7 was competent to alleviate NRK-52E cells undergoing partial EMT in a high-glucose milieu. Furthermore, exogenous BMP-7 activated the Smad1/5 pathway to promote gene transcription of SnoN and intervened ubiquitination of SnoN; both effects repaired the SnoN protein level in renal tubular cells and kidney tissues of DM mice. Therefore, these findings suggested that BMP-7 could upregulate SnoN mRNA and protein levels by activating the classical Smad1/5 pathway to refrain from the partial EMT of renal tubular epithelial cells and the deposition of ECM in DKD-induced renal fibrosis.
Similar content being viewed by others


Introduction
Diabetic kidney disease (DKD) involves glomeruli and tubules, and the common pathological changes include glomerulosclerosis and tubulointerstitial fibrosis (TIF), which eventually develop into renal sclerosis [1, 2]. TIF features interstitial matrix deposition, inflammation, fibroblast activation, microvascular rarefaction, and tubular cell loss. Renal tubular epithelial cells (RTECs), as the main renal parenchyma cells, activate several profibrotic signaling pathways after sustained-stimulation by hyperglycemia, resulting in TIF-associated functional disorders [3]. Hitherto, transforming growth factor β1 (TGF-β1) is considered as the key cytokine to promote renal TIF in DKD [4, 5]. It also partakes in fibrosis of renal tissue by facilitating the partial epithelial-mesenchymal transition (EMT), increasing the synthesis of extracellular matrix (ECM), inhibiting degradation, inducing apoptosis of renal parenchyma cells, as well as activating renal interstitial fibroblasts and inflammatory cells.
Bone morphogenetic protein-7 (BMP-7) is a member of the TGF-β superfamily, which is mainly expressed in podocytes and tubular epithelial cells in the kidney [6, 7]. Intracellular signal transduction of BMP-7 includes a classical pathway mediated by Smad proteins and a nonclassical pathway being independent of Smad proteins. Briefly, BMP-7 rapidly activates Smad1/5/8, which then binds to Smad4 in the cytoplasm to form a transcriptional complex. This complex is a structural requisite for nuclear translocation and mediating transcriptional activation of target genes [6, 8]. BMP-7 also activates a number of nonclassical pathways, such as extracellular signal-regulated kinase (ERK), C-Jun amino-terminal kinase (JNK), and p38 mitogen protein kinase (p38MAPK) [8,9,10]. Our previous results and other studies have confirmed that hyperglycemia decreased the mRNA and protein levels of BMP-7 in RTECs, and it was negatively correlated with renal fibrosis [10,11,38]. However, the level of SnoN protein was also influenced by the degradation of the ubiquitin-proteasome system, and Luo et al. speculated that the expression of SnoN protein in RTECs by BMP-7 was related to the inhibited degradation [14, 16]. Therefore, we examined the ubiquitination of SnoN protein in vivo and in vitro, with an increase in the ubiquitination of SnoN protein level in NRK-52E cells cultured in high-glucose milieu and kidneys of diabetic mice, in which BMP-7 limited the phosphorylation and ubiquitination of SnoN protein in cells and kidneys (Fig. 8a–c).
In conclusion, we speculated that BMP-7 could upregulate SnoN mRNA level by activating the classical Smad1/5 signaling pathway, and it also could reduce the ubiquitination of SnoN protein, finally restoring the SnoN protein level to inhibit hyperglycemia-mediated renal tubular epithelial fibrosis (Fig. 8d). However, further researches are required to indicate how BMP-7 can inhibit the ubiquitination-mediated degradation of SnoN protein.
Materials and methods
Animal model
The number of experimental animals was determined to be 36 mice by the evaluation of the degree of freedom (E) of analysis of variance and the estimation of modeling success rate. A total of 36 healthy C57BL/6 mice (weight: 18 ± 2 g) were obtained from Bei**g Si Bei Fu Bioscience Co., Ltd (Bei**g, China) and housed in the Animal Center of Guizhou Medical University (Guizhou, China). This study was conducted in accordance with the guidelines of the National Health and Medical Research Council of China for the care and use of animals for scientific purposes. Diabetic mice were produced by injecting 0.01 mol/L streptozotocin (STZ, prepared with sterile citric acid–sodium citrate buffer, pH 4.5; Sigma-Aldrich, MO, St. Louis, USA).intraperitoneally (i.p.) at a dose of 55 mg/kg/d for 5 days. Fasting blood glucose levels of mice were detected after 72 h. The blood glucose level ≥16.7 mmol/L indicated that the mouse model of DM was established successfully. All mice were randomly divided into the diabetic group (DM group, n = 18), while normal control (NC) mice were age-matched (n = 18), and an equivalent volume of solvent was injected into each control rat. Mice were given a normal diet and unlimited drinking water. After confirming that the mice have diabetes for 6 weeks, the mice were divided into six groups: (i) vehicle (Ringer’s Solution:NaCl 0.147 M, KCl 4 mM, CaCl2 2.5 mM) injection in normal mice (NC + Control); (ii) vector plasmids injection in normal mice (NC + Vector); (iii) OE-BMP-7 plasmid injection in normal mice (NC + BMP-7); (iv) vehicle injection in diabetic mice (DM + Control); (v) vector plasmid injection in diabetic mice (DM + Vector); (vi) OE-BMP-7 plasmid injection in diabetic mice (DM + BMP-7) (n = 6). The purpose of injecting exogenous OE-BMP-7 plasmids via hydrodynamic tail vein injection, a method to rapidly deliver naked plasmids to mouse tissues, is to enable the high expression of BMP-7 in the mouse kidney. Mice were injected respectively through the tail vein with vehicle-Ringer’s Solution 1.5 ml as control, 1.5 ml Ringer’s Solution with 15 mg vector plasmids as Vector treatment, or OE-BMP-7 plasmids as BMP-7 treatment every week for 6 weeks [27]. At the end of 12 weeks, all mice were sacrificed. Their 24-h urine was collected in metabolic cages before sacrificing the animals. The mice were fasted for 6–8 h before anesthetizing with pentobarbital sodium. The femoral artery was punctured, and serum was separated in blood samples at 4000 rpm for 10 min by centrifugation at 4 °C. Urine and serum were stored at –20 °C for measuring the urine protein and biochemical indices. Subsequently, the kidneys of the mice were harvested; one part of the kidney was fixed with 4% paraformaldehyde for embedding tissue sections into paraffin, and the other part was snap-frozen in liquid nitrogen and stored at –80 °C for RNA and protein extractions.
Evaluation of biochemical markers
The oxidase method was used to measure serum glucose (Nan**g Jiancheng Bioengineering Institute, Nan**g, China) [39]. Immune turbidimetry and the Benedict–Behre method was used to measure urinary microalbumin/urinary creatinine. Urine microalbumin measurement used the enzyme-linked immunosorbent method (Elabscience Biotechnology Co., Ltd., Wuhan, China), and urine creatinine measurement was performed using sarcosine oxidase method (Nan**g Jiancheng Bioengineering Institute). The levels of total cholesterol (TC) and triglycerides (TGs) were determined using enzymatic assay kits, according to the manufacturer’s instructions (Nan**g Jiancheng Bioengineering Institute).
Histopathological analysis
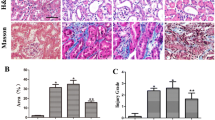
Kidneys fixed in paraformaldehyde were embedded in paraffin, and transverse sections (4 µm) were sliced for hematoxylin-eosin (H&E) staining and Masson’s trichrome staining. The morphology of renal tissue was observed under a light microscope, and renal tissue fibrosis was observed by H&E and Masson’s trichrome staining. H&E’s renal tubular lumen area and Masson’s trichrome-positive area were analyzed relative to the whole area from six random fields (200×).
Cell culture and treatments
RTECs (NRK-52E cells, Conservation Genetics CAS Kunming Cell Bank, Kunming, China) were cultured in normal-glucose (5.5 mmol/L glucose) in a Dulbecco’s modified Eagle’s medium (DMEM; HyClone, Logan, UT, USA) supplemented with 5% fetal bovine serum (FBS; Invitrogen, Carlsbad, CA, USA) and HEK 293 T cells were cultured in Ham’s F 12 nutrient medium (F-12; HyClone, Logan, UT, USA) supplemented with 10% fetal bovine serum (FBS; Invitrogen, Carlsbad, CA, USA) in an incubator with 5% CO2 at 37 °C. The cells were growth-arrested in a serum-free medium for 20 h and then treated with the following media: (1) normal-glucose control group (NG) (5.5 mmol/L glucose); (2) high-glucose control group (HG) (5.5 mmol/L glucose + 19.5 mmol/L d-glucose); (3) high glucose + BMP-7 (12.5, 25, 50, 100, and 200 ng/mL rhBMP-7); and (3) high glucose + MG132(5 μmol/L). Cells in each group were cultured for 48 h for further study.
Transfection
The plasmid was extracted according to the manufacturer’s protocol. Transfection was conducted when the NRK-52E cells reached 50–60% confluency. A DMEM culture medium (no serum, no antibiotics), polyethyleneimine, and plasmid (ski-shRNA (sh-Skil), overexpressed Smad1 plasmid, and overexpressed Smad5 plasmid (OE-Smad1/5), Smad1shRNA plasmid, and Smad5 shRNA plasmid (sh-Smad1/5), Supplementary Figs. 2–4) were placed in a sterile environment for 20 min. The culture medium was discarded. The liquid was put in an Eppendorf tube and blended, and the cells were then incubated at 37 °C in the presence of 5% CO2 for 6 h. Afterward, the DMEM medium (no serum, no antibiotics) was added, and the cells were incubated at 37 °C overnight in the presence of 5% CO2. Then, the culture medium was discarded, and the cells were washed with phosphate-buffered saline (PBS). The culture media of different treatments were added to the cells as needed, and the system was incubated at 37 °C in the presence of 5% CO2 for 48 h.
Immunohistochemistry and immunofluorescence (IF) staining
The biotin-streptavidin-peroxidase method (ZSBio, Bei**g, China) was used to stain tissue sections incubated with mouse-anti-α-SMA (1:100; Santa Cruz Biotechnology Inc., Dallas, TX, USA) and antibodies to show the distribution and expression of the protein in kidney tissues. For the negative control, PBS was substituted for the primary antibody. The secondary antibodies were affinity-purified biotinylated anti-mouse immunoglobulin G (IgG), followed by streptavidin-peroxidase complex. The signals were visualized using 3,3′-diaminobenzidine (DAB) chromogen. The tissue sections were counterstained with H&E.
Cells cultured on coverslips were twice washed with cold PBS and fixed with cold methanol: acetone (1:1) for 10 min on ice. Then, 3-μm-thick cryosections of renal tissue were prepared for IF staining. After extensive washing, the sections were blocked with bovine serum antigen for 30 min at room temperature and incubated with the following specific primary antibodies: rabbit-anti-BMP-7 (1:200; Proteintech, Wuhan, China), rabbit-anti-SnoN (1:200; Proteintech) rabbit-anti-E-cadherin (1:200, Santa Cruz Biotechnology, Inc.), mouse-anti-α-SMA (1:100), and rabbit-anti-Collagen-III (1:200, Sigma-Aldrich) overnight at 4 °C. After that, the cells were stained with cyanine-5 (Cy5)-conjugated goat-anti-rabbit IgG (1:400; Protientech), or cyanine-3 (Cy3)-conjugated goat-anti-rabbit IgG (1:400; Protientech), and cyanine-3(Cy3)-conjugated goat-anti-mouse IgG (1:400; Protientech). After washing, the sections were stained with 4′,6-diamidino-2-phenylindole (DAPI) to visualize the nuclei and observed under an inverted fluorescence microscope. Six visual fields (400×) were randomly selected in each group, and the gray value obtained by positive staining was analyzed by the ImageJ software for statistics.
Western blotting
Protein expression in renal tissue and cultured cells was analyzed by Western blotting. Cells were lysed in ice-cold lysis buffer, and protein concentration was determined using the BCA Protein Assay Kit (Beyotime). The protein samples were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (PAGE) and electrotransferred to polyvinylidene difluoride (PVDF) membrane (Millipore, Germany). Subsequently, the membranes were blocked with 5% non-fat milk for 1 h at room temperature and probed with primary antibodies, such as rabbit-anti-BMP-7 (1:1000), rabbit-anti-Smad1 (1:1000; Proteintech), rabbit-anti-Smad5 (1:1000; Proteintech), rabbit-anti-Phospho- Smad1/5(Ser463/465) (1:1000, Cell Signaling Technology), rabbit-anti-E-cadherin (1:1000), rabbit-anti-Vimentin (1:1000; Santa Cruz), rabbit-anti-Collagen-III (1:1000), rabbit-anti-SnoN (1:1000), and rabbit-anti-β-actin (1:400; Proteintech), for 16 h at 4 °C. The signals were detected by chemiluminescence after incubation with appropriate secondary antibodies (). Images were acquired using the Bio-Rad gel imaging system (Bio-Rad, CA, USA), and the intensities of the immunoreactive bands were quantified using the Quantity One 4.6 software (Bio-Rad).
Immunoprecipitation
Total protein lysates were prepared in 50 mM Tris-HCl (pH 7.4) 0.2 M NaCl, 2 mM ethylene diamine tetraacetic acid (EDTA), 0.5% NP-40, 50 mM NaF, 0.5 mM Na3VO4, 20 mM sodium pyrophosphate, 1 mM phenylmethanesulfonyl fluoride (PMSF), 10 µg/mL aprotinin, 10 µg/mL leupeptin, 1 mM dithiothreitol (DTT), and 150 mM NaCl. Co-immunoprecipitation was performed by adding Dynabeads-Ab complexes to an equivalent amount of protein lysates(5 μg/μl) and incubating the mixture overnight at 4 °C. Finally, the coimmunoprecipitate complexes were washed and subjected to Western blot analysis. The input group was the positive control group, which was the whole protein extracted from the cells.
Real-time quantitative reverse transcription-polymerase chain reaction (RT-qPCR)
Total RNA in the rat renal cortex or NRK-52E cells was extracted using TRIzol (Tiangen, Dalian, China) method, according to the manufacturer’s protocol. First-strand cDNA was synthesized from 5 μg total RNA in renal tissues and 3 μg total RNA in NRK-52E cells using the RevertAid First-Strand cDNA Synthesis Kit (Fermentas, Lithuania) and stored at −20 °C. RT-qPCR was performed using iQSYBR GreenSupermix (Bio-Rad Laboratories Inc., Hercules, CA, USA).
After 45 cycles of amplification, data were collected to plot kinetic curves, and the Ct values were acquired. These steps were repeated three times, and the 2–ΔΔCt method was used to calculate the mRNA level of the target genes and normalized to that of β-actin in each group. Primer sequences for quantitative PCR were shown in Supplementary Table S1.
Luciferase reporter assay
The SnoN-promoter-luciferase reporter was constructed by Longqian Biotech (China). Actively proliferating HEK 293 T cells were trypsinized and seeded in plates at a suitable density for routine culture. Following 24 h of incubation, transfection was carried out with Lipofectamine 2000 (Invitrogen) as directed by the manufacturer for 48 h. This was followed by cell lysis and sample analysis with a Dual-Luciferase Reporter Assay System (E1960; Promega, USA). Firefly and Renilla luciferase activities were measured, and the ratio of Firefly luciferase activity to that of Renilla luciferase was derived. Triplicate experiments were independently repeated three times.
Statistical analysis
All data were expressed as mean ± standard deviation (\({{{\bar{\mathrm X}}}}\) ± SD). SPSS 18.0 software (IBM, Armonk, NY, USA) was used to perform statistical analysis. Statistical analysis between the groups was performed using an unpaired Students t-test, while comparison among multiple groups was performed using one-way analysis of variance(ANOVA) followed by post hoc Bonferroni correction. P < 0.05 was considered statistically significant.
Data availability
All the data generated or analysed during this study are included in this published article and its supplementary files.
References
Colhoun HM, Marcovecchio ML. Biomarkers of diabetic kidney disease. Diabetologia. 2018;61:996–1011.
Azushima K, Gurley SB, Coffman TM. Modelling diabetic nephropathy in mice. Nat Rev Nephrol. 2018;14:48–56.
Milica Bozic, Maite Caus, Raul R Rodrigues-Diez, Neus Pedraza, Marta Ruiz-Ortega, Eloi Garí, et al. Protective role of renal proximal tubular alpha-synuclein in the pathogenesis of kidney fibrosis. Nat Commun. 2020;11:1943.
Huang Fengjuan, Zhao Yanyan, Wang Qingzhu, Hillebrands Jan-Luuk, Born Jacobvanden, Ji Linlin, An Tingting, et al. Dapagliflozin attenuates renal tubulointerstitial fibrosis associated with type 1 diabetes by regulating STAT1/TGFβ1 signaling. Front Endocrinol. 2019;10:441.
Rauchman M, Griggs D. Emerging strategies to disrupt the central TGF-β axis in kidney fibrosis. Transl Res. 2019;209:90–104.
Wen H, Kumar V, Mishra A, Song S, Aslam R, Hussain A, et al. Grem2 mediates podocyte apoptosis in high glucose milieu. Biochimie 2019;160:113–21.
Gravesen E, Lerche Mace M, Nordholm A, Hofman-Bang J, Hruska K, Haagen Nielsen C, et al. Exogenous BMP7 in aortae of rats with chronic uremia ameliorates expression of profibrotic genes, but does not reverse established vascular calcification. PLoS ONE. 2018;13:e0190820. 5
Young IC, Chuang ST, Gefen A, Kuo WT, Yang CT, Hsu CH, Lin FH, et al. A novel compressive stress-based osteoarthritis-like chondrocyte system. Exp Biol Med. 2017;242:1062–71.
Kim CL, Choi SH, Mo JS. Role of the hippo pathway in fibrosis and cancer. Cells. 2019;8:468.
Awazu M, Nagata M, Hida M. BMP7 dose-dependently stimulates proliferation and cadherin-11 expression via ERK and p38 in a murine metanephric mesenchymal cell line. Physiol Rep. 2017;5:e13378.
Tsujimura T, Idei M, Yoshikawa M, Takase O, Hishikawa K. Roles and regulation of bone morphogenetic protein-7 in kidney development and diseases. World J Stem Cells. 2016;8:288–96. 26
Wang Y, **ao Y, Li S, Shi L, Liu L, Zhang Y, et al. BMP-7 enhances SnoN mRNA expression in renal tubular epithelial cells under high-glucose conditions. Mol Med Rep. 2017;16:3308–14.
Manson SR, Song JB, Guo Q, Liapis H, Austin PF. Cell type specific changes in BMP-7 expression contribute to the progression of kidney disease in patients with obstructive uropathy. J Urol. 2015;193:1860–9.
Higgins DF, Ewart LM, Masterson E, Tennant S, Grebnev G, Prunotto M, et al. BMP7-induced-Pten inhibits Akt and prevents renal fibrosis. Biochim Biophys Acta Mol Basis Dis. 2017;1863:3095–104.
Manson SR, Austin PF, Guo Q, Moore KH. BMP-7 signaling and its critical roles in kidney development, the responses to renal injury, and chronic kidney disease. Vitam Horm. 2015;99:91–144.
Luo DD, Phillips A, Fraser D. Bone morphogenetic protein-7 inhibits proximal tubular epithelial cell Smad3 signaling via increased SnoN expression. Am J Pathol. 2010;176:1139–47.
Tang J, Gifford CC, Samarakoon R, Higgins PJ. Deregulation of negative controls on TGF-β1 signaling in tumor progression. Cancers. 2018;25:10.
Zeglinski MR, Hnatowich M, Jassal DS, Dixon IM. SnoN as a novel negative regulator of TGF-β/Smad signaling: a target for tailoring organ fibrosis. Am J Physiol Heart Circ Physio. 2015;308:H75–82. 15
Tang H, Su H, Fan D, Ye C, Lei CT, Jiang HJ, et al. MAD2B-mediated SnoN downregulation is implicated in fibroblast activation and tubulointerstitial fibrosis. Am J Physiol Ren Physiol. 2016;311:F207–16.
Wang Y, Liu L, Peng W, Liu H, Liang L, Zhang X et al. Ski-related novel protein suppresses the development of diabetic nephropathy by modulating transforming growth factor-β signaling and microRNA-21 expression. J Cell Physiol. 2019;234:17925–36.
Wang Y, Mao Y, Zhang X, Liu H, Peng W, Liang L et al. TAK1 may promote the development of diabetic nephropathy by reducing the stability of SnoN protein. 2019;228:1–10.
Vigolo E, Markó L, Hinze C, Müller DN, Schmidt-Ullrich R, Schmidt-Ott KM. Canonical BMP signaling in tubular cells mediates recovery after acute kidney injury. Kidney Int. 2019;95:108–22.
H. William Schnaper. The tubulointerstitial pathophysiology of progressive kidney disease. Adv Chronic Kidney Dis. 2017;24:107–16.
Hsing CH, Lin CF, So E, Sun DP, Chen TC, Li CF, et al. α2-Adrenoceptor agonist dexmedetomidine protects septic acute kidney injury through increasing BMP-7 and inhibiting HDAC2 and HDAC5. Am J Physiol Ren Physiol. 2012;303:F1443–53.
Silva FMO, Costalonga EC, Silva C, Carreira ACO, Gomes SA, Sogayar MC, et al. Tamoxifen and bone morphogenic protein-7 modulate fibrosis and inflammation in the peritoneal fibrosis model developed in uremic rats. Mol Med. 2019;25:41.
Ma T, Huang C, Xu Q, Yang Y, Liu Y, Meng X, et al. Suppression of BMP-7 by histone deacetylase 2 promoted apoptosis of renal tubular epithelial cells in acute kidney injury. Cell Death Dis. 2017;8:e3139.
Liu L, Wang Y, Yan R, Liang L, Zhou X, Liu H, et al. BMP-7 inhibits renal fibrosis in diabetic nephropathy via miR-21 downregulation. Life Sci. 2019;238:116957.
Lv S, Liu G, Sun A, Wang J, Cheng J, Wang W, et al. Mesenchymal stem cells ameliorate diabetic glomerular fibrosis in vivo and in vitro by inhibiting TGF-β signalling via secretion of bone morphogenetic protein 7. Diab Vasc Dis Res. 2014;11:251–61.
Li Z, Liu J, Wang W, Zhao Y, Yang D, Geng X, et al. Investigation of hub genes involved in diabetic nephropathy using biological informatics methods. Ann Transl Med. 2020;8:1087.
Hu HH, Chen DQ, Wang YN, Feng YL, Cao G, Vaziri ND, et al. New insights into TGF-β/Smad signaling in tissue fibrosis. Chem Biol Interact. 2018;292:76–83.
Lovisa S, LeBleu VS, Tampe B, Sugimoto H, Vadnagara K, Carstens JL, et al. Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat Med. 2015;21:998–1009.
Li RX, Yiu WH, Wu HJ, Wong DW, Chan LY, Lin M, et al. BMP7 reduces inflammation and oxidative stress in diabetic tubulopathy. Clin Sci. 2015;128:269–80.
Song Y, Lv S, Wang F, Liu X, Cheng J, Liu S, Wang X, Chen W, Guan G, Liu G, Peng C. Overexpression of BMP‑7 reverses TGF‑β1‑induced epithelial‑mesenchymal transition by attenuating the Wnt3/β‑catenin and TGF-β1/Smad2/3 signaling pathways in HK‑2 cells. Mol Med Rep. 2020;21:833–41.
Meng XM, Nikolic-Paterson DJ, Lan HY. TGF-β: the master regulator of fibrosis. Nat Rev Nephrol. 2016;12:325–38.
Li X, Diao Z, Ding J, Liu R, Wang L, Huang W, et al. The downregulation of SnoN expression in human renal proximal tubule epithelial cells under high-glucose conditions is mediated by an increase in Smurf2 expression through TGF-β1 signaling. Int J Mol Med. 2016;37:415–22.
Liu L, Shi M, Wang Y, Zhang C, Su B, **ao Y, Guo B. SnoN upregulation ameliorates renal fibrosis in diabetic nephropathy. PLoS ONE. 2017;12:e0174471.
Lv W, Booz GW, Wang Y, Fan F, Roman RJ. Inflammation and renal fibrosis: Recent developments on key signaling molecules as potential therapeutic targets. Eur J Pharm. 2018;820:65–76.
Fei T, ** of SMAD target genes reveals the role of BMP signaling in embryonicstem cell fate determination. Genome Res. 2010;20:36–44.
Bankar SB, Bule MV, Singhal RS, Ananthanarayan L. Glucose oxidase-an overview. Biotechnol Adv. 2009;27:489–501.
Funding
This work was supported by the National Natural Science Foundation of China (Grant No. 81760131), the Selection Funding for Innovation and Entrepreneurship of High-level Overseas Study Talents in Guizhou Province (Overseas Study Talent Selection Funding Contract (2018) No. 01), and Graduate Research Foundation of Guizhou Province [Grant No. YJSCXJH(2019)071].
Author information
Authors and Affiliations
Contributions
WP and **ngcheng Zhou performed the experiments and wrote the manuscript. TX, YM, **aohuan Zhang, HL, Luqun Liang, and Lingling Liu analyze the data. Lirong Liu, YX, FZ, SL, MS, YZ, and LT put forward constructive opinions and participated in topic discussions. YW and BG designed the study and revised the MS. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
Collection and usage of clinical specimens were approved by Guizhou Medical University’s Ethical Committee Board. Animal protocols were approved by the Animal Ethic Committee of Guizhou Medical University.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by Professor Alessandro Finazzi-Agrò
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Peng, W., Zhou, X., Xu, T. et al. BMP-7 ameliorates partial epithelial-mesenchymal transition by restoring SnoN protein level via Smad1/5 pathway in diabetic kidney disease. Cell Death Dis 13, 254 (2022). https://doi.org/10.1038/s41419-022-04529-x
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-022-04529-x
- Springer Nature Limited
This article is cited by
-
DNA-dependent protein kinase catalytic subunit (DNA-PKcs) drives chronic kidney disease progression in male mice
Nature Communications (2023)