
Abstract
Cholangiocarcinoma (CCA), a lethal malignancy of the biliary epithelium, is the second most common primary liver cancer. The poor prognosis of CCA is due to the high rate of tumour invasion and distant metastasis. We found that the RNA-binding protein LIN28B, a known regulator of microRNA biogenesis, stem cell maintenance, and oncogenesis, is expressed in a subpopulation of CCA patients. To further investigate the potential role of LIN28B in CCA pathogenesis, we studied the effect of LIN28B overexpression in the cholangiocyte cell line MMNK-1 and cholangiocarcinoma cell lines HuCCT-1 and KKU-214. Here, we show that enhanced LIN28B expression promoted cancer stem cell-like properties in CCA, including enhanced cell migration, epithelial-to-mesenchymal transition (EMT), increased cell proliferation and spheroid formation. Proteomic analysis revealed TGF-β-induced protein (TGFBI) as a novel LIN28B target gene, and further analysis showed upregulation of other components of the TGF-β signalling pathway, including TGF-β receptor type I (TGFBRI) expression and cytokine TGFB-I, II and III secretion. Importantly, the small molecule TGF-β inhibitor SB431542 negated the effects of LIN28B on both cell migration and clonogenic potential. Overexpression of TGFBI alone promoted cholangiocarcinoma cell migration and EMT changes, but not spheroid formation, suggesting that TGFBI partially contributes to LIN28B-mediated aggressive cell behaviour. These observations are consistent with a model in which TGF-β and LIN28B work together to form a positive feedback loop during cholangiocarcinoma metastasis and provide a therapeutic intervention opportunity.
Similar content being viewed by others


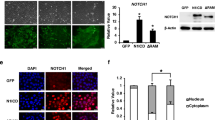
Introduction
Cholangiocarcinoma (CCA) is a biliary tract cancer with one of the highest mortality rates among all cancers and has an increasing incidence rate worldwide [1]. The incidence of CCA is differentially distributed in differing geographical areas, especially in Northeast Thailand, which has the highest incidence and is closely related to liver fluke-induced CCA [2, 3]. CCA is recognised as a devastating cancer with limited therapeutic options and poor prognosis. Molecular profiling of CCA revealed a high degree of intertumour and intratumour heterogeneity [4]. Based on an immunostaining study, it has been suggested that CCA may contain cancer stem cell-like subpopulation switches that have high tumour-initiating potential and chemoresistance and that could be responsible for relapse after treatment [5]. Understanding the mechanisms governing CCA aggressiveness and drug resistance could lead to new therapeutic targets for the treatment of this cancer.
LIN28 is an RNA-binding protein that plays pivotal roles in development, stem cell maintenance, energy metabolism and tumorigenesis [6, 7]. LIN28A and its paralogue, LIN28B, have been known to block let-7 miRNA maturation and modulate mRNA translational efficacy by binding to mRNA targets [8,9,10,11]. The expression of LIN28 is generally found in develo** foetal tissues and then disappears after birth. It has been reported that the LIN28A and LIN28B genes are reactivated in many human malignancies, such as breast cancer, colon cancer, lung cancer, ovarian cancer and hepatocellular carcinoma (HCC), and their expression is correlated with poor clinical disease outcomes [12]. LIN28 overexpression was reported to be sufficient to initiate HCC in transgenic mouse models [13]. Mechanisms proposed to be responsible for oncogenic transformation of LIN28 include derepression of let-7 targets such as cMyc, RAS and HMGA2 oncogenes [14,45]. Whether CAPG is involved in CCA pathogenesis or CSC maintenance requires further investigation.
Materials and methods
Cell lines and cell culture
The human immortalised cholangiocyte cell line MMNK-1, a representative normal bile duct cell line was established as previously described [46], and bile duct cancer HuCCT-1 and KKU-214 cells were obtained from the JCRB Cell Bank, Japan. The HEK293gp cell line was obtained from Stem Cell and Cell Therapy Research Center, Chulalongkorn University, Thailand. MMNK-1, HEK293gp and KKU-214 cells were cultured in high glucose Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% FBS, 1% GlutaMAX and 1% antimycotic–antibiotic reagents (Gibco). HuCCT-1 cells were cultured in RPMI 1640 medium (HyClone) supplemented with 10% FBS, 1% GlutaMAX and 1% antimycotic–antibiotic reagents. All cells were maintained at 37 °C in a humidified 5% CO2 atmosphere.
Stable LIN28B-expressing cell lines
The pBabe-control and pBabe-LIN28B constructs were kindly gifted from the Daley Lab. The VSVG plasmid was obtained from the Stem Cell and Cell Therapy Research Unit, Chulalongkorn University, Thailand. For viral production, the compositions of transfection complex formation were prepared following the manufacturer’s protocol. Briefly, the plasmids encoding LIN28B, VSVG, and X-tremeGENE HP DNA transfection reagent (Roche) were diluted with Opti-MEM I medium. HEK293gp cells were incubated with transfection complex for 24 h, and virus was collected after three days of transfection. The cells were incubated with polybrene (Sigma–Aldrich) and virus containing LIN28B and the control. LIN28B and control-expressing cell lines were selected and maintained in medium containing 1 µg/ml puromycin.
RNA extraction and quantitative real-time PCR
Total RNA was extracted using TRIzol reagent (Ambion). mRNA was reverse transcribed into cDNA using a RevertAidH Minus Kit (Thermo Scientific), and qPCR was performed using SYBR Green master using the set of primers described in Supplementary Table 1. For quantitative analysis of miRNA, cDNA was synthesised using a miScript II RT Kit (Qiagen). The expression of miRNAs was evaluated using miScript Primer Assays (Qiagen) together with the miScript SYBR Green PCR Kit (Qiagen). The relative miRNA and mRNA expression levels were normalised to those of the internal controls, U6 for miRNA and GAPDH for mRNA. The primer sequences are provided in Supplementary Table 1.
Immunohistochemical staining
Paraffin-embedded CCA tissues were obtained with informed consent from 20 CCA patients who received surgical resection at KCMH, Chulalongkorn University, between 2016-2020. The use of tissues for this study was approved by the Ethics Committee of the Faculty of Medicine, Chulalongkorn University (No. 244/58). Paraffin-embedded sections of CCA were deparaffinized in xylene and rehydrated by submerging in stepwise decreasing concentrations of ethanol. The sections were boiled under pressure in 0.01 M citrate buffer pH 6.0 for antigen unmasking and then incubated with 0.3% H2O2 in methanol to neutralise endogenous peroxidase. Sections were then submerged in diluted 5% FBS in PBS to block nonspecific binding, incubated with primary antibody dilution, 1:500 of LIN28B (Cell Signaling Technology) overnight before incubation with secondary antibody conjugated HRP (Dako). Peroxidase activity was determined by adding 3,3-diaminobenzidine (DAB) substrate solution (DAKO) and finally counterstained with Mayer’s haematoxylin. Immunofluorescence in CCA tissues was performed using an Opal™ staining Kit (PerkinElmer).
Western blot
Protein expression was performed by following standard western blotting procedures. Briefly, cellular proteins were extracted from cells using RIPA buffer (Cell Signaling Technology) containing 1X protease and phosphatase inhibitors, and the protein concentration was measured using BCA reagent (Thermo Scientific). Protein was separated with 4–20% Tris-glycine gradient SDS polyacrylamide gel electrophoresis (Bio-Rad) and then transferred onto a nitrocellulose membrane. The membrane was blocked for nonspecific binding in 5% BSA in TBST buffer and subsequently incubated with the following diluted primary antibodies: anti-LIN28B (D4H1) (1:5000, Cell Signaling Technology), total SMAD2 (D43B4) (1:2000, Cell Signaling Technology), phosphoSMAD2 (Ser465/467) (1:1000, Merck Millipore), TGFBRI (H-100) (1:2000, Santa Cruz), TGFBI (1:2000, Abcam), and β-actin (13E5) (1:2000, Cell Signalling Technology) (Supplementary Table 2) overnight at 4 °C. Then, the membrane was incubated for 1 h with the secondary antibody. The level of protein expression was visualised using an Odyssey® CLx imaging system (Li-cor).
Cell viability assay
In brief, cells were seeded onto 96-well plates, and the medium was removed at the indicated time. Cells were incubated with 0.5 mg/ml MTT (Sigma–Aldrich) in PBS for 3 h at 37 °C. MTT-formazan crystals were solubilized by adding DMSO, and the absorbance was then measured at 570 nm using Varioskan Flash (Thermo Scientific).
Cell scratching assay
The cells suspended in serum-free media were seeded into each well of a 12-well plate and incubated overnight. Prior to scratching, the medium was removed, a wound was created using a sterile 200 µl pipette tip, and the cells were washed twice with PBS before adding 10% FBS-supplemented medium. The initial wound area and wound area at the indicated times of experiments were captured under an AxioObserver inverted microscope (Zeiss), and the wound gap was measured using ZEN2 blue2.3 software (Zeiss). The percentage of wound confluence was calculated by following a previous study [47].
Transwell invasion and migration assays
For cell invasion, a Transwell insert 8 µm PET membrane (Corning) was coated with 250 µg/ml Matrigel (Corning). A total of 1 × 104 cells suspended in serum-free media were seeded per well into a Matrigel-coated Transwell insert, and medium containing 10% FBS was added to the lower chamber of a 24-well plate. After the indicated incubation time, the invaded cells were fixed with 4% paraformaldehyde, permeabilized with absolute methanol, and then stained with 0.05% crystal violet in PBS. Non-invaded cells were discarded by cotton swabs. The migration assay procedure was performed as mentioned above, differing only where the cells were seeded on top of an uncoated Matrigel Transwell insert.
Soft agar colony formation assay
In brief, 0.8% base agar was prepared by boiling 0.8 g of agarose in sterile PBS and then mixing with culture medium prior to addition to a six-well plate. A total of 3 × 104 cells were suspended in top agar (1:1 dilution of base agar and culture medium), and 1 ml was added to the top base agar. Cells were cultured for 4 weeks and fed twice per week. Randomised fields of each well were captured under an Axio Observer inverted microscope (Zeiss) with z stack and mosaic programmes. All colonies were counted, and the size of all colonies was measured using ZEN2 blue 2.3 software.
Spheroid formation
A total of 1.5 × 103 cells were suspended in sphere media consisting of DMEM/F12 supplemented with Gluta max, antibiotic-antimycotic B27, 20 ng/ml human recombinant epidermal growth factor (hEGF) and human basic fibroblast growth factor (hbFGF). Cells were then seeded into ultralow attachment 24-well plates. Cells were cultured for 1 week, and all spheroids were captured under an inverted microscope (ZEISS). Spheroids were counted, and the size was measured by ZEN2 blue 2.3 software. In the inhibitor treatment assay, cells were pre-treated with 10 μM SB431542 inhibitor (Tocris Bioscience) for 24 h and then assayed and continuously cultured in media containing SB431542.
Xenograft mice
The protocol of the animal study was approved by Chulalongkorn University Animal Care and Use Committee of Chulalongkorn University Laboratory Animal Center (CULAC) and preceded in accordance with CULAC guidelines. Male NSG mice (Jackson Laboratory) (4–6 weeks) were subcutaneously injected into the left and right lower flanks with 100 µl of MMNK-1 control and LIN28B-overexpressing MMNK-1 cell suspension (1 × 106 cells per site) in Matrigel (Corning). All mice were terminated on day 28 of the experiment, and tumour size was measured. Tumour volume was determined using the formula V = 0.5 × L × W2 (L = long and W = short tumour diameter).
TGF-β cytokine array
After culturing cells for 24 h, the medium was collected and centrifuged at 1000×g for 15 min at 4 °C, and then the supernatant was collected. Differential concentrations of cytokines were evaluated using Bio-Plex Pro™ Human TGF-β cytokines (Bio-Rad) according to the manufacturer’s instructions. The concentrations of cytokines were analysed relative to the cytokine standard curve.
Proteomic analysis
Protein extraction and in-solution digestion
The cells were lysed with a mixture of 5% sodium deoxycholate (SDC) and 1X protease inhibitor. The protein concentrations were measured by using a BCA protein assay. The sample containing 250 µg of protein was prepared for in-solution digestion methods, following a previous study [48]. Peptides in the MMNK-1_control sample were labelled with light isotope reagents (formaldehyde; CH2O and cyanoborohydride; NaBH3CN), and peptides in the MMNK-1_LIN28B sample were labelled with heavy isotope reagents (13C-labelled formaldehyde; 13CD2O and cyanoborodeuteride; NaBD3CN). The samples were fractionated into 10 fractions using a Pierce High pH Reversed-Phase Peptide Fractionation Kit (Thermo Scientific). All eluted fractions were subjected to LC-MS/MS analyses on a Q Exactive Plus mass spectrometer.
Data analysis
Raw mass spectra files were analysed with MaxQuant software (version 1.6.2.10) and searched against the Human Swiss-Prot Database (20,408 proteins, downloaded December 2018) as well as a list of common protein contaminants (http://www.thegpm.org). Trypsin/P was set as the digestion enzyme with a maximum of two missed cleavages allowed. Carbamidomethylation of cysteine and acetylation at the N-terminus were set as fixed modifications, while oxidation of methionine was set as a variable modification. Dimethyl labelling was selected as the quantification mode with dimethyl Lys 0 and N-term 0 as the light labels, dimethyl Lys 4 and N-term 4 as the intermediate labels and Lys 8 and N-term 8 as the intermediate labels. The maximum false discovery rate (FDR) was set at 1% for both peptide and protein levels. Other parameters in MaxQuant were set at their default values. Heavy-to-light protein expression ratios were extracted from the protein group output of MaxQuant, log-transformed, and subjected to one-sample Student’s t test analyses. The list of 234 significant differentially expressed protein groups was selected at a false discovery rate of 5% using the Benjamini-Hochberg procedure. Only 2539 protein groups with at least 2 razor and unique peptides were considered here. Gene set enrichment analysis was performed using the WebGestalt online interface (https://academic.oup.com/nar/article/47/W1/W199/5494758) with Panther pathway annotation (https://academic.oup.com/nar/article/47/D1/D419/5165346).
Statistical analysis
Data are displayed as the mean ± SD using GraphPad Prism version 8. For statistical analysis, Student’s t test was performed, and P values indicated by *P < 0.05, **P < 0.01 and ***P < 0.001 were considered statistically significant.
References
Massarweh NN, El-Serag HB. Epidemiology of hepatocellular carcinoma and intrahepatic cholangiocarcinoma. Cancer Control. 2017;24:1–11.
Banales JM, Cardinale V, Carpino G, Marzioni M, Andersen JB, Invernizzi P, et al. Expert consensus document: cholangiocarcinoma: current knowledge and future perspectives consensus statement from the European Network for the Study of Cholangiocarcinoma (ENS-CCA). Nat Rev Gastroenterol Hepatol. 2016;13:261–80.
Sripa B, Pairojkul C. Cholangiocarcinoma: lessons from Thailand. Curr Opin Gastroenterol. 2008;24:349–56.
Lendvai G, Szekerczes T, Illyes I, Dora R, Kontsek E, Gogl A, et al. Cholangiocarcinoma: classification, histopathology and molecular carcinogenesis. Pathol Oncol Res. 2020;26:3–15.
Wu HJ, Chu PY. Role of cancer stem cells in cholangiocarcinoma and therapeutic implications. Int J Mol Sci. 2019;20:4154.
Shyh-Chang N, Daley GQ. Lin28: primal regulator of growth and metabolism in stem cells. Cell Stem Cell. 2013;12:395–406.
Viswanathan SR, Powers JT, Einhorn W, Hoshida Y, Ng TL, Toffanin S, et al. Lin28 promotes transformation and is associated with advanced human malignancies. Nat Genet. 2009;41:843–8.
Viswanathan SR, Daley GQ, Gregory RI. Selective blockade of microRNA processing by Lin28. Science. 2008;320:97–100.
Newman MA, Thomson JM, Hammond SM. Lin-28 interaction with the Let-7 precursor loop mediates regulated microRNA processing. RNA 2008;14:1539–49.
Heo I, Joo C, Cho J, Ha M, Han J, Kim VN. Lin28 mediates the terminal uridylation of let-7 precursor MicroRNA. Mol Cell. 2008;32:276–84.
Wilbert ML, Huelga SC, Kapeli K, Stark TJ, Liang TY, Chen SX, et al. LIN28 binds messenger RNAs at GGAGA motifs and regulates splicing factor abundance. Mol Cell. 2012;48:195–206.
Viswanathan SR, Daley GQ. Lin28: a microRNA regulator with a macro role. Cell 2010;140:445–9.
Nguyen LH, Robinton DA, Seligson MT, Wu L, Li L, Rakheja D, et al. Lin28b is sufficient to drive liver cancer and necessary for its maintenance in murine models. Cancer Cell. 2014;26:248–61.
Zhou J, Ng SB, Chng WJ. LIN28/LIN28B: an emerging oncogenic driver in cancer stem cells. Int J Biochem Cell Biol. 2013;45:973–8.
Wang H, Zhao Q, Deng K, Guo X, **a J. Lin28: an emerging important oncogene connecting several aspects of cancer. Tumour Biol. 2016;37:2841–8.
Balzeau J, Menezes MR, Cao S, Hagan JP. The LIN28/let-7 pathway in cancer. Front Genet. 2017;8:31.
Chen L, Yan HX, Yang W, Hu L, Yu LX, Liu Q, et al. The role of microRNA expression pattern in human intrahepatic cholangiocarcinoma. J Hepatol. 2009;50:358–69.
Wang M, Wen TF, He LH, Li C, Zhu WJ, Trishul NM. A six-microRNA set as prognostic indicators for bile duct cancer. Int J Clin Exp Med. 2015;8:17261–70.
Yang H, Li TW, Peng J, Tang X, Ko KS, **a M, et al. A mouse model of cholestasis-associated cholangiocarcinoma and transcription factors involved in progression. Gastroenterology 2011;141:378–88, 88 e1-4.
McDaniel K, Hall C, Sato K, Lairmore T, Marzioni M, Glaser S, et al. Lin28 and let-7: roles and regulation in liver diseases. Am J Physiol Gastrointest Liver Physiol. 2016;310:G757–65.
Guo Y, Chen Y, Ito H, Watanabe A, Ge X, Kodama T, et al. Identification and characterization of lin-28 homolog B (LIN28B) in human hepatocellular carcinoma. Gene 2006;384:51–61.
Vaquero J, Guedj N, Claperon A, Nguyen Ho-Bouldoires TH, Paradis V, Fouassier L. Epithelial-mesenchymal transition in cholangiocarcinoma: From clinical evidence to regulatory networks. J Hepatol. 2017;66:424–41.
Liu Y, Li H, Feng J, Cui X, Huang W, Li Y, et al. Lin28 induces epithelial-to-mesenchymal transition and stemness via downregulation of let-7a in breast cancer cells. PLoS ONE. 2013;8:e83083.
Thapa N, Lee BH, Kim IS. TGFBIp/betaig-h3 protein: a versatile matrix molecule induced by TGF-beta. Int J Biochem Cell Biol. 2007;39:2183–94.
**ong H, Zhao W, Wang J, Seifer BJ, Ye C, Chen Y, et al. Oncogenic mechanisms of Lin28 in breast cancer: new functions and therapeutic opportunities. Oncotarget 2017;8:25721–35.
King CE, Wang L, Winograd R, Madison BB, Mongroo PS, Johnstone CN, et al. LIN28B fosters colon cancer migration, invasion and transformation through let-7-dependent and -independent mechanisms. Oncogene 2011;30:4185–93.
Kugel S, Sebastian C, Fitamant J, Ross KN, Saha SK, Jain E, et al. SIRT6 suppresses pancreatic cancer through control of Lin28b. Cell. 2016;165:1401–15.
Lovnicki J, Gan Y, Feng T, Li Y, **e N, Ho CH, et al. LIN28B promotes the development of neuroendocrine prostate cancer. J Clin Invest. 2020;130:5338–48.
Franses JW, Philipp J, Missios P, Bhan I, Liu A, Yashaswini C, et al. Pancreatic circulating tumor cell profiling identifies LIN28B as a metastasis driver and drug target. Nat Commun. 2020;11:3303.
Bergamaschi D, Samuels Y, O’Neil NJ, Trigiante G, Crook T, Hsieh JK, et al. iASPP oncoprotein is a key inhibitor of p53 conserved from worm to human. Nat Genet. 2003;33:162–7.
Bronevetsky Y, Burt TD, McCune JM. Lin28b regulates fetal regulatory t cell differentiation through modulation of TGF-beta signaling. J Immunol. 2016;197:4344–50.
Chen PY, Qin L, Barnes C, Charisse K, Yi T, Zhang X, et al. FGF regulates TGF-beta signaling and endothelial-to-mesenchymal transition via control of let-7 miRNA expression. Cell Rep. 2012;2:1684–96.
Park JT, Kato M, Lanting L, Castro N, Nam BY, Wang M, et al. Repression of let-7 by transforming growth factor-beta1-induced Lin28 upregulates collagen expression in glomerular mesangial cells under diabetic conditions. Am J Physiol Ren Physiol. 2014;307:F1390–403.
Zhang Z, Zhang S, Ma P, **g Y, Peng H, Gao WQ, et al. Lin28B promotes melanoma growth by mediating a microRNA regulatory circuit. Carcinogenesis 2015;36:937–45.
Ottaviani S, Stebbing J, Frampton AE, Zagorac S, Krell J, de Giorgio A, et al. TGF-beta induces miR-100 and miR-125b but blocks let-7a through LIN28B controlling PDAC progression. Nat Commun. 2018;9:1845.
Bellomo C, Caja L, Moustakas A. Transforming growth factor beta as regulator of cancer stemness and metastasis. Br J Cancer. 2016;115:761–9.
Wang Y, Li J, Guo S, Ouyang Y, Yin L, Liu S, et al. Lin28B facilitates the progression and metastasis of pancreatic ductal adenocarcinoma. Oncotarget 2017;8:60414–28.
**awath N, Chamgramol Y, Furukawa Y, Obama K, Tsunoda T, Sripa B, et al. Comparison of gene expression profiles between Opisthorchis viverrini and non-Opisthorchis viverrini associated human intrahepatic cholangiocarcinoma. Hepatology 2006;44:1025–38.
Han B, Cai H, Chen Y, Hu B, Luo H, Wu Y, et al. The role of TGFBI (betaig-H3) in gastrointestinal tract tumorigenesis. Mol Cancer. 2015;14:64.
Costanza B, Rademaker G, Tiamiou A, De Tullio P, Leenders J, Blomme A, et al. Transforming growth factor beta-induced, an extracellular matrix interacting protein, enhances glycolysis and promotes pancreatic cancer cell migration. Int J Cancer. 2019;145:1570–84.
Fico F, Santamaria-Martinez A. TGFBI modulates tumour hypoxia and promotes breast cancer metastasis. Mol Oncol. 2020;14:3198–210.
Zhu J, Nie S, Wu J, Lubman DM. Target proteomic profiling of frozen pancreatic CD24+ adenocarcinoma tissues by immuno-laser capture microdissection and nano-LC-MS/MS. J Proteome Res. 2013;12:2791–804.
Steitz AM, Steffes A, Finkernagel F, Unger A, Sommerfeld L, Jansen JM, et al. Tumor-associated macrophages promote ovarian cancer cell migration by secreting transforming growth factor beta induced (TGFBI) and tenascin C. Cell Death Dis. 2020;11:249.
Corona A, Blobe GC. The role of the extracellular matrix protein TGFBI in cancer. Cell Signal. 2021;84:110028.
Zhaojie L, Yuchen L, Miao C, Yacun C, Shayi W, Anbang H, et al. Gelsolin-like actin-cap** protein has prognostic value and promotes tumorigenesis and epithelial-mesenchymal transition via the Hippo signaling pathway in human bladder cancer. Ther Adv Med Oncol. 2019;11:1–25.
Maruyama M, Kobayashi N, Westerman KA, Sakaguchi M, Allain JE, Totsugawa T, et al. Establishment of a highly differentiated immortalized human cholangiocyte cell line with SV40T and hTERT. Transplantation 2004;77:446–51.
Buachan P, Chularojmontri L, Wattanapitayakul SK. Selected activities of Citrus maxima Merr. fruits on human endothelial cells: enhancing cell migration and delaying cellular aging. Nutrients 2014;6:1618–34.
Makjaroen J, Somparn P, Hodge K, Poomipak W, Hirankarn N, Pisitkun T. Comprehensive proteomics identification of IFN-lambda3-regulated antiviral proteins in HBV-transfected cells. Mol Cell Proteom. 2018;17:2197–215.
Acknowledgements
We thank Dr. Anapat Sanpavat, Dr. Krit Suwannaphom, Dr. Fern Chanduayvit, Dr. Shanop Shuangshoti, Dr. Roongruedee Chaiteerakij, Dr. Naruemon Klaikeaw, Dr. Boonchoo Sirichindakul and Dr. Nattiya Hirankarn for their help with patient tissue samples. We are thankful for Dr. George Daley and Dr. Hao Zhu for LIN28B related plasmid constructs. This project has been funded by Grant no p-15-50269 from the National Science and Technology Development Agency (NSTDA), Thailand, and Ratchadapiseksompotch Fund, Faculty of Medicine, Chulalongkorn University RA(MF)05/59, RA(MF)12/60, and GB-CU_61_26_30_16, Chulalongkorn Academic Advancement into its 2nd Century (CUAASC) project and the Thailand Research Fund (RTA6280004) and Center of Excellence in Hepatitis and Liver Cancer, Chulalongkorn University. Puthdee N is supported by Chulalongkorn University graduate scholarship to commemorate the 72nd anniversary of his majesty king bhumibolAdulyadej.
Author information
Authors and Affiliations
Contributions
N.P. carried out the experiments. S.S. and T.P. contributed to the proteomic study and data analysis. S.R. conceived the original idea and designed the experiments. N.I. was in charge of overall direction, experimental design and data analysis. P.T. supervised the project. N.I. wrote the manuscript with input from all authors. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
COMPETING INTERESTS
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Puthdee, N., Sriswasdi, S., Pisitkun, T. et al. The LIN28B/TGF-β/TGFBI feedback loop promotes cell migration and tumour initiation potential in cholangiocarcinoma. Cancer Gene Ther 29, 445–455 (2022). https://doi.org/10.1038/s41417-021-00387-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41417-021-00387-5
- Springer Nature America, Inc.
This article is cited by
-
Novel prognostic marker TGFBI affects the migration and invasion function of ovarian cancer cells and activates the integrin αvβ3-PI3K-Akt signaling pathway
Journal of Ovarian Research (2024)
-
Modelling the complex nature of the tumor microenvironment: 3D tumor spheroids as an evolving tool
Journal of Biomedical Science (2024)
-
Downregulation of RNA binding protein 47 predicts low survival in patients and promotes the development of renal cell malignancies through RNA stability modification
Molecular Biomedicine (2023)