
Abstract
Tumour-associated macrophages (TAMs) sustain a tumour-supporting and immunosuppressive milieu and therefore aggravate cancer prognosis. To modify TAM behaviour and unlock their anti-tumoural potential, novel TAM-reprogramming immunotherapies are being developed at an accelerating rate. At the same time, scientific discoveries have highlighted more sophisticated TAM phenotypes with complex biological functions and contradictory prognostic associations. To understand the evolving clinical landscape, we reviewed current and past clinically evaluated TAM-reprogramming cancer therapeutics and summarised almost 200 TAM-reprogramming agents investigated in more than 700 clinical trials. Observable overall trends include a high frequency of overlap** strategies against the same therapeutic targets, development of more complex strategies to improve previously ineffective approaches and reliance on combinatory strategies for efficacy. However, strong anti-tumour efficacy is uncommon, which encourages re-directing efforts on identifying biomarkers for eligible patient populations and comparing similar treatments earlier. Future endeavours will benefit from considering the shortcomings of past treatment strategies and accommodating the emerging complexity of TAM biology.
Similar content being viewed by others
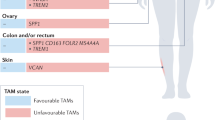
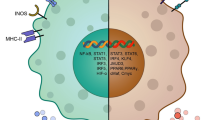
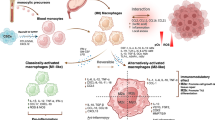
Introduction
Tumour-associated macrophages (TAMs) make an alluring cancer immunotherapy target. For one, their abundance in tumours allows TAMs to greatly influence the interplay between cancer cells and the surrounding tumour microenvironment (TME), resulting in tumour-promoting milieu essential for cancer progression [ A plethora of research has highlighted the various ways in which TAMs promote cancer development and progression, providing ample therapeutic opportunities. In short, TAMs act in every stage of the metastatic cascade by promoting primary tumour growth [24], angiogenesis [25], immune evasion [26], invasion [15, 27, 28] and metastatic spread [15, 29,30,31]. These TAM functions develop gradually when TAMs co-evolve with cancer and are based on homoeostasis-promoting functions of healthy macrophages [4]. Further emphasising their therapeutic potential, TAMs promote cancer immunotherapy resistance by limiting T-cell entry into tumours and suppressing anti-tumour T-cell activation [26, 32, 33]. However, TAMs can fight cancer by phagocytosis [34], killing with reactive radicals [35, 36] and activating anti-tumoural immunity via antigen presentation and cytokine secretion [37]. The long co-evolution with cancer cells ultimately dampens these anti-tumoural TAM properties [4], unless prevented with therapeutics. Considering the tumour-promoting roles of TAMs, it is no surprise that a high abundance of TAMs is associated with poor prognosis in most solid cancers [38]. Colorectal and prostate cancer are exceptions to this, but when immunosuppressive M2 marker-expressing TAMs are quantified instead, a negative prognosis is evident across cancer types [38]. A high abundance of immunosuppressive TAMs is characteristic of so-called non-inflamed tumours that lack T cells and are often resistant to immune checkpoint inhibitor (ICI) therapy, further contributing to poorer prognosis [39]. However, these overall trends give a simplistic view, as the intratumoural localisation of TAMs and selected treatment regimen also affect their prognostic value. For instance, tumour islet TAMs are often associated with a more favourable prognosis than stromal TAMs [47,48,49]. Once monocytes enter the cancer tissue, their phenotype is modified by the cancer type, affected organ and intratumoural localisation, as these determine the characteristics of local tissue niches. Factors such as neighbouring cells, matrix composition, pH and cytokine environment will then further fine-tune TAM phenotypes and functions [10, 26]. Furthermore, monocyte differentiation into macrophages is altered in cancer, resulting in additional accumulation of immature phenotypes, such as MDSCs [50]. Investigating TAM phenotypes in different tumour areas and cancer types is possible using single-cell and spatial analysis technologies. For instance, Ma et al. used existing datasets of single-cell RNA-sequenced TAMs and identified seven TAM phenotypes common to multiple cancers [10]. Interestingly, these identified TAM phenotypes have both pro- and anti-tumoural properties within the same subset, such as interferon (IFN)-primed TAMs that express IFN-regulated mediators contributing to both T-cell activation and exhaustion, and regulatory TAMs that resemble immunosuppressive M2-like macrophages with their PD-L1, IL-10 and MRC1 expression but also express co-stimulatory and major histocompatibility complex (MHC) molecules [10]. As expected, these TAM phenotypes strongly connect TAM functions with their localisation within the tumour. For instance, pro-angiogenic TAMs reside in hypoxic tumour areas, where they promote metastatic spread and angiogenesis [10], whereas immunosuppressive lipid-associated TAMs are located in areas of invasion, where they suppress T-cell responses [10, 51], and IFN-primed TAMs co-localise with CXCL13-expressing T cells to regulate their responses [10]. Clearly, these different and opposing functions of TAM subsets require tailored therapeutic targeting. It is yet to be determined which of these TAM subsets are susceptible to functional reprogramming and which are the most crucial to be reprogrammed. If these TAM subsets possess both anti- and pro-tumoural capacities, successful manipulation would promote their anti-tumoural functions without activating the pro-tumoural ones. Of note, depending on cancer type, the markers for these subsets may differ, and some of the widely investigated TAM molecules are associated with several subsets, such as TREM2 [10]. Nevertheless, uncovering TAM functions at the subset level will provide opportunities for develo** even more sophisticated TAM-reprogramming therapeutic approaches. To obtain a comprehensive understanding of the clinical landscape of TAM-reprogramming strategies, we gathered data from past and present clinical trials investigating TAM-reprogramming agents in cancer. For this, we first searched PubMed for potential TAM targets using the following search conditions: macrophage and (cancer or neoplasm or tumour or tumor or malignancies) and (phase or clinical or trial or target), where ‘macrophage’ and the cancer-related term had to be in the title or abstract. The obtained 17,457 unique articles published before 1.2.2024 were screened by Rayyan [52] using the following exclusion criteria: (1) the paper does not discuss or identify therapeutic targets, specifically for cancer and in macrophages; (2) the therapeutic strategy primarily depletes TAMs or inhibits their recruitment and (3) the therapeutic is re-purposed. Screening was performed mostly manually, with Rayyan-created ratings used to exclude the lowest matching articles only after screening one-third of all articles and confirming the irrelevance of the lowliest rated articles. The identified TAM targets and therapeutics from the obtained 550 review and research articles or the word ‘macrophage’ were then used as the search condition in clinicaltrials.gov to find clinically investigated TAM-reprogramming therapeutics (condition/disease = ‘Cancer’). During the search, we excluded withdrawn trials and therapeutics with first trial start date before 1.1.2000, and expanded our original TAM target list with additional TAM-targeting therapeutics from the company pipelines encountered. In this way, 194 clinically evaluated TAM-reprogramming therapeutics were gathered with information on the clinical phase, number of trials, combinatory regimens, first clinical trial date, investigated cancer types and actual number of treated patients. Clinical development was classified as discontinued if the therapeutic had been removed from the company’s pipeline without information on selling the asset, most recent trials had been terminated, or trials had remained inactive for several years without new trials being launched. Additional information was collected from publicly available sources, including published conference abstracts, press releases, web-based company pipelines and quarterly reports. While we call these therapeutics ‘TAM reprogramming’ for clarity, many of them are not macrophage-specific and may have other functions, as described in the discussion of individual targets. We also acknowledge the presence of other crucial pathways controlling TAM phenotypes not discussed in this review. For instance, cytokines, prostaglandins and other inflammatory mediators have additional broad effects on various immune and non-immune cell types and represent potential strategies for modulating cancer-associated inflammation rather than specifically controlling TAMs. Overall, the past decade has seen a steep increase in the number of TAM-reprogramming therapeutics that have entered clinical trials (Fig. 1a). Therapeutics targeting the CD47–SIRPα axis have particularly contributed to this rapid development. Additionally, several novel macrophage targets have entered the clinics, including members of the LILRB and scavenger receptor families (Fig. 1b). a Number of macrophage-reprogramming therapeutics entering clinical trials each year since 2000, coloured by target group. b A timeline showing when each macrophage-reprogramming therapeutic target was first clinically investigated in cancer. Bubble colours indicate macrophage target groups. Such development is encouraging because currently only a few macrophage-modifying therapies have been approved for clinical use, the number of therapeutics against the same target is high, and not many programmes have proceeded beyond phase 2 (Fig. 2a). The approved therapeutics are duvelisib, a PI3Kγ and PI3Kδ inhibitor for haematological malignancies [53], and imiquimod, a toll-like receptor 7 (TLR7) agonist for topical treatment of basal cell carcinoma [54]. Additionally, approved early activators of pattern recognition receptors include Bacillus Calmette-Guerin, an attenuated bacteria that activates TLR2 and TLR4 receptors in non-invasive bladder cancer [54], and mifamurtide, a synthetic analogue of bacterial cell wall that activates TLR4 and NOD2 receptors in osteosarcoma [55]. a Number of therapeutic agents that have been investigated in clinical trials. Therapeutics are shown by target group and coloured by clinical development phase as of January 2024. b Number of treated patients by target group, coloured by the development status. c Number of therapeutic agents by target group, coloured by development status and highest clinical development phase at the time of discontinuation. Percentages indicate proportions of discontinued therapeutics. d Bar plots of macrophage-reprogramming therapeutics with ongoing clinical development as of January 2024, grouped by molecule type and coloured by clinical development phase. Phase 3-investigated therapeutics comprise additional TLR agonists, CD47–SIRPα axis blockers, IDO1 inhibitors and STAT inhibitors (Fig. 2a), and we report staggeringly high numbers of patients treated with these agents (Fig. 2b). Unfortunately, these advanced targets show a high proportion of discontinued therapeutics, such as 100% for IDO1, 61% for TLRs and 50% for STAT, indicating non-favourable therapeutic properties (Fig. 2c). Surprisingly, some targets with known safety- or efficacy-related issues are still highly investigated, such as CD47–SIRPα axis, STING and CD40 (Fig. 2c), reflecting novel approaches taken to develop next-generation agents with a more favourable therapeutic profile. We will review the properties of each target separately further below, but commonly poor efficacy is caused by on-target side effects that limit dosing, or activation of compensatory/counteracting pathways that enable tumour immune escape. Emerging efficacy- and safety-related challenges have motivated a vast amount of translational research. Novel treatment strategies have already been clinically investigated alongside conventional small molecules and monoclonal antibodies (Fig. 2d), and various approaches have been exploited to circumvent the original challenges. To direct treatment effects, alternative delivery routes (intratumoural vs. systemic) are utilised [56,57,58]. To target systemically administered therapeutics to the TME, antibody–drug-conjugates [59, 60], antibodies with pH-dependent target binding [61] and bi-specific antibodies and fusion proteins have been developed [56, 62, 63]. Although most TAM-targeted bi-specifics bind a second target on cancer cells or the TME, some use the second arm to enhance immune activation by engaging PD-1, PD-L1 or 4-1BB instead (Supplementary Table 1). The efficacy of monoclonal antibodies can be modified by re-engineering the Fc region when the efficacy depends on antibody-induced effector functions or cross-linking [56, 64]. Indeed, we observed that most of the clinical candidates carry an IgG1 Fc region, which has been occasionally modified to decrease or enhance effector functions (data not shown). Finally, more complex delivery systems using exosomes [65, 66], bacteria [67] or viral vectors [68, 69] have also been tested in clinics (e.g. NCT04592484, NCT05375604, NCT04167137, NCT03852511, NCT02654938). Overall, some of the above approaches have demonstrated superior efficacy or safety in preclinical studies [63, 70, 71], supporting the advancement of their clinical development. Because TAM-targeted therapeutics are predicted to support the efficacy of other treatments rather than eradicate cancer on their own [4, 5], we also evaluated the prevalence of combination treatment regimens. Expectedly, less than a quarter of clinically investigated TAM-reprogramming agents have been studied as monotherapy above phase 1, whereas more than half have been studied in combination with ICIs (Fig. 3a). a Dot plot showing how commonly macrophage-reprogramming therapeutics have been investigated in combination with other treatment types. Dot size indicates number and dot colour proportion of macrophage-reprogramming therapeutics investigated with the indicated treatment combinations, separately for each target. ‘Monotherapy phase 1’ indicates therapeutics at phase 1 clinical development not yet investigated in combination with other treatments. Percentages were calculated from all TAM-reprogramming therapeutics (n = 194). b Illustration depicting subcellular localisation of macrophage-reprogramming therapeutic targets. Targets are coloured by clinical development status with coloured areas depicting proportions among therapeutics against the same target. To summarise, a surge of macrophage-reprogramming therapeutics has proceeded to clinical development during the past decade, and while several older targets have faced challenges, novel strategies have been undertaken to improve their therapeutic profiles. Next, we will discuss each TAM-reprogramming target separately, focussing on the mode-of-action, types of clinically investigated therapeutics and clinical results thus far. Firstly, we describe strategies that alter specific TAM functions, such as phagocytosis, scavenging, pattern recognition or interactions with other immune cells, and then proceed to therapeutic strategies that alter the overall TAM phenotype towards pro-inflammatory direction. Subcellular localisation of each target is illustrated in Fig. 3b. As the majority of these therapeutics have been evaluated in (advanced) solid tumours and haematological malignancies, we will only occasionally describe the investigated cancer types and provide a supplementary trial ID table (Supplementary Table 1) for further reference. To escape phagocytosis, healthy and cancerous cells express CD47, which binds macrophage SIRPα to inhibit cytoskeletal rearrangements necessary for phagocytosis [72, 73]. Since the first-in-class monoclonal CD47-targeting antibody magrolimab [74], ~50 therapeutics blocking the CD47–SIRPα interaction have been clinically investigated. A notable portion of these therapeutics are bi-specific antibodies and fusion proteins that mostly recognise another molecule on cancer cells or inhibit PD-L1/PD-1. Overall, blocking CD47 in haematological cancers and SIRPα in solid tumours yields better efficacy [75], but two phase 3 magrolimab trials (NCT4313881 and NCT04778397) were recently terminated because of poor efficacy in acute myeloid leukaemia and myelodysplastic syndrome [76, 77]. CD47 blockade on red blood cells prevents the therapeutic from reaching cancer cells and causes anaemia by inducing red blood cell phagocytosis and agglutination. To circumvent this, newer strategies use antibodies that preferentially bind to cancer cells or bi-specific agents to target cancer cells [63, 75]. Furthermore, enhanced phagocytosis can promote either pro- or anti-inflammatory reprogramming of TAMs, depending on signals from the phagocytosed cells and type of phagocytosis. Anti-inflammatory responses would limit monotherapy efficacy by promoting immunosuppression and tumour growth [78]. Analogous to CD47–SIRPα interaction, CD24 on cancer cells inhibits phagocytosis by binding to macrophage Siglec-10 [79]. CD24-targeting antibodies of the IgG1 subclass (IMM47, ATG-031) are investigated in phase 1 trials, and at least preclinical IMM47 efficacy depends on its Fc-induced effector functions [80]. Further development should consider other immune- and cancer-related functions of CD24 and additional CD24 and Siglec-10 ligands binding to different glycosylated protein forms [79, 81, 82]. Macrophages clear various endogenous and pathogen-related ligands with scavenger receptors that also regulate subsequent immune responses to these ligands [83, 84]. Apart from clinically investigated CD163 and Clever-1, preclinical studies have identified MARCO [85, 86] and CD206 [87] as potential therapeutic targets. Inhibiting MARCO with the monoclonal antibody PY265 supports anti-tumour immunity via pro-inflammatory conversion of TAMs and MDSCs [88], and specific CD206-targeted peptides can likewise support M1-like macrophage phenotype or deliver therapeutics to CD206-expressing TAMs [87, 89]. Under homoeostasis, CD163 scavenges haptoglobin–haemoglobin complexes [90], and in cancer, CD163 is a widely known marker for tumour-promoting M2-like macrophages, generally associated with poor prognosis [83, 91, 92]. CD163 in TAMs is associated with STAT3 activation and anti-inflammatory IL-10 and TGF-β secretion [93]. A single CD163-targeting antibody (OR2805, IgG1 subclass), which activates T-cell responses in preclinical models [94], is being clinically evaluated in a phase 1/2 trial (NCT05094804). Clever-1 (Stabilin-1) is a scavenger and adhesion molecule expressed by monocytes, macrophages and endothelial cells [95]. It regulates macrophage lysosomal acidification to halt antigen presentation and T-cell activation [96]. A Clever-1-blocking antibody, bexmarilimab (IgG4), activates T-cells and IFN responses in advanced solid cancers and elicits cancer type-dependent disease control alongside few objective responses in a phase 1/2 trial [96, 97]. Promising objective responses have been observed in combination with azacytidine in acute myeloid leukaemia and myelodysplastic syndrome in a phase 1/2 trial (NCT05428969) that is also recruiting azacytidine-refractory patients [98]. Pattern recognition receptors alert innate immunity to pathogens and tissue damage, but their systemic administration is often limited by the associated side effects of overt immune activation [99]. TLRs are widely expressed, but their stimulation on antigen-presenting cells activates pro-inflammatory cytokine secretion, expression of co-stimulatory molecules and antigen presentation to support T-cell activation [100,101,102,103]. Depending on the TLR type, the activating therapeutics are either small molecules or larger lipid or nucleic acid derivatives that can be administered systemically or locally [58, 100]. Several therapeutics have faced discontinuation after showing weak monotherapy efficacy [101], possibly due to the induction of tolerance and simultaneous activation of pro-tumoural pathways that support regulatory T cells (Tregs), MDSCs and cancer cell proliferation [104,105,106]. Mostly, these agents are now deemed as boosters for other therapeutics, especially cancer vaccines and ICIs [102]. As an exception, the TLR7 agonist imiquimod and the attenuated TLR2- and TLR4-activating Bacillus Calmette-Guerin bacteria have been approved for skin carcinoma and non-muscle invasive bladder carcinoma, respectively [54]. Upon recognising cytosolic pathogen-derived or damaged endogenous DNA, cGAS produces cyclic GMP–AMP, which activates STING to elicit type I IFN production and NF-κB activation. These pathways promote cancer cell death, anti-inflammatory macrophage polarisation, antigen presentation, T-cell priming and recruitment [107, 108]. However, chronic STING activation may have undesired opposite effects, such as enhanced cancer cell survival and immunosuppressive IDO1 induction [108]. Nevertheless, at least twenty STING-activating therapeutics have been clinically evaluated in phase 1 and 2 trials. Early STING agonists were intratumourally administered synthetic cyclic dinucleotides (e.g. ADU-S100, MK-1454) characterised by insufficient monotherapy efficacy, limited penetration inside the cells and susceptibility to enzymatic degradation [57, 109, 110]. Therefore, alternative delivery methods, such as liposomal formulations, nanoparticles, antibody–drug-conjugates (TAK-500, XMT-2056), exosomes (exoSTING: NCT04592484) and a bacterial vector (SYNB1891: NCT4167137) are being developed [109]. Additionally, compounds that enable intravenous administration (SB11285: NCT04096638) and a polymer that prolongs STING activation (ONM-501: NCT06022029) are under clinical investigation. As a DNA-binding protein, HMGB1 supports chromatin organisation and transcription, but extracellular HMGB1 released from dying cells and activated macrophages stimulates inflammatory responses via TLRs and RAGE [111, 112]. In cancer, some HMGB1-activated pathways aggravate prognosis by supporting invasion and metastasis [112, 113]. The HMGB1-binding prodrug SB17170 modulates myeloid cell cytokine secretion to increase T-cell infiltration [114], and it is being evaluated in a phase 1 trial (NCT05522868). Dectin-2 (CLEC6A) defends against fungi and mycobacteria [115, 116]. Its activation stimulates the secretion of pro-inflammatory chemokines and cytokines, including TNFα and IL-12 [116, 106, 152, 192], tendency to alter TAMs rather than healthy tissue macrophages [193], lack of compensatory pathways in the TME [149] and access for therapeutic manipulation within solid tumours [57]. To direct therapeutic effects away from macrophages in healthy tissues, therapeutic efficacy could depend on tumour-specific expression, ongoing chronic inflammation at tumour sites or monocyte-derived macrophages. Enhancing specificity with such TME-specific targets or TME-directed delivery mechanisms could potentially widen the therapeutic window that is currently limited by side effects [22, 56, 63, 194]. Alternatively, targeting circulating monocytes can affect their differentiation into TAMs and therefore provide access within solid tumours without the need for direct tissue penetration. Fundamental understanding of TAM biology will pave the way to more effective treatment approaches [4, 5, 195]. Unfortunately, TAM subset complexity revealed by single-cell RNA sequencing [Full size tableTAMs as cancer therapy targets
Clinical landscape analysis
Past and present clinical trials



TAM-reprogramming therapeutic targets
Phagocytosis checkpoints
CD47–SIRPα
CD24–Siglec-10
Scavenger receptors
CD163
Clever-1
Pattern recognition
Toll-like receptors (TLRs)
STING
HMGB1
Dectin-2
Conclusions
All in all, past and present trials have proven that complete responses are occasionally attainable with TAM-reprogramming monotherapy, although wider benefits will first be achieved in combination with other treatment modalities. The path forward should emphasise selected data-driven treatment strategies that use state-of-the-art knowledge on TAM biology and patient biomarkers.
Data availability
A Supplementary table of collected therapeutics and corresponding trial IDs can be accessed via the electronic version of the manuscript (Supplementary Table 1).
References
Cheng S, Li Z, Gao R, **ng B, Gao Y, Yang Y, et al. A pan-cancer single-cell transcriptional atlas of tumor infiltrating myeloid cells. Cell. 2021;184:792–809.e23.
Kelly PM, Davison RS, Bliss E, McGee JO. Macrophages in human breast disease: a quantitative immunohistochemical study. Br J Cancer. 1988;57:174–7.
Morantz RA, Wood GW, Foster M, Clark M, Gollahon K. Macrophages in experimental and human brain tumors. Part 2: studies of the macrophage content of human brain tumors. J Neurosurg. 1979;50:305–11.
Kloosterman DJ, Akkari L. Macrophages at the interface of the co-evolving cancer ecosystem. Cell. 2023;186:1627–51.
Mantovani A, Allavena P, Marchesi F, Garlanda C. Macrophages as tools and targets in cancer therapy. Nat Rev Drug Discov. 2022;21:799–820.
Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity. 2014;41:49–61.
Sica A, Larghi P, Mancino A, Rubino L, Porta C, Totaro MG, et al. Macrophage polarization in tumour progression. Semin Cancer Biol. 2008;18:349–55.
Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41:14–20.
Cassetta L, Fragkogianni S, Sims AH, Swierczak A, Forrester LM, Zhang H, et al. Human tumor-associated macrophage and monocyte transcriptional landscapes reveal cancer-specific reprogramming, biomarkers, and therapeutic targets. Cancer Cell. 2019;35:588–602.e10.
Ma RY, Black A, Qian BZ. Macrophage diversity in cancer revisited in the era of single-cell omics. Trends Immunol. 2022;43:546–63.
Guilliams M, Svedberg FR. Does tissue imprinting restrict macrophage plasticity? Nat Immunol. 2021;22:118–27.
Stout RD, Jiang C, Matta B, Tietzel I, Watkins SK, Suttles J. Macrophages sequentially change their functional phenotype in response to changes in microenvironmental influences. J Immunol. 2005;175:342–9.
Subramanian S, Busch CJ, Molawi K, Geirsdottir L, Maurizio J, Vargas Aguilar S, et al. Long-term culture-expanded alveolar macrophages restore their full epigenetic identity after transfer in vivo. Nat Immunol. 2022;23:458–68.
Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature. 2013;496:445–55.
Lopez-Yrigoyen M, Cassetta L, Pollard JW. Macrophage targeting in cancer. Ann N Y Acad Sci. 2021;1499:18–41.
Kumar V, Donthireddy L, Marvel D, Condamine T, Wang F, Lavilla-Alonso S, et al. Cancer-associated fibroblasts neutralize the anti-tumor effect of CSF1 receptor blockade by inducing PMN-MDSC infiltration of tumors. Cancer Cell. 2017;32:654–68.e5.
Ries CH, Cannarile MA, Hoves S, Benz J, Wartha K, Runza V, et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell. 2014;25:846–59.
Nywening TM, Belt BA, Cullinan DR, Panni RZ, Han BJ, Sanford DE, et al. Targeting both tumour-associated CXCR2+ neutrophils and CCR2+ macrophages disrupts myeloid recruitment and improves chemotherapeutic responses in pancreatic ductal adenocarcinoma. Gut. 2018;67:1112–23.
Bonapace L, Coissieux MM, Wyckoff J, Mertz KD, Varga Z, Junt T, et al. Cessation of CCL2 inhibition accelerates breast cancer metastasis by promoting angiogenesis. Nature. 2014;515:130–3.
Sandhu SK, Papadopoulos K, Fong PC, Patnaik A, Messiou C, Olmos D, et al. A first-in-human, first-in-class, phase I study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 in patients with solid tumors. Cancer Chemother Pharm. 2013;71:1041–50.
Argyle D, Kitamura T. Targeting macrophage-recruiting chemokines as a novel therapeutic strategy to prevent the progression of solid tumors. Front Immunol. 2018;9:2629.
Cassetta L, Pollard JW. Targeting macrophages: therapeutic approaches in cancer. Nat Rev Drug Discov. 2018;17:887–904.
Etzerodt A, Tsalkitzi K, Maniecki M, Damsky W, Delfini M, Baudoin E, et al. Specific targeting of CD163+ TAMs mobilizes inflammatory monocytes and promotes T cell-mediated tumour regression. J Exp Med. 2019;216:2394–411.
O’Sullivan C, Lewis CE, Harris AL, McGee JO. Secretion of epidermal growth factor by macrophages associated with breast carcinoma. Lancet. 1993;342:148–9.
Riabov V, Gudima A, Wang N, Mickley A, Orekhov A, Kzhyshkowska J. Role of tumor associated macrophages in tumor angiogenesis and lymphangiogenesis. Front Physiol. 2014;5:75.
DeNardo DG, Ruffell B. Macrophages as regulators of tumour immunity and immunotherapy. Nat Rev Immunol. 2019;19:369–82.
Wyckoff J, Wang W, Lin EY, Wang Y, Pixley F, Stanley ER, et al. A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Res. 2004;64:7022–9.
Bonde AK, Tischler V, Kumar S, Soltermann A, Schwendener RA. Intratumoral macrophages contribute to epithelial-mesenchymal transition in solid tumors. BMC Cancer. 2012;12:35.
Wyckoff JB, Wang Y, Lin EY, Li JF, Goswami S, Stanley ER, et al. Direct visualization of macrophage-assisted tumor cell intravasation in mammary tumors. Cancer Res. 2007;67:2649–56.
Qian B, Deng Y, Im JH, Muschel RJ, Zou Y, Li J, et al. A distinct macrophage population mediates metastatic breast cancer cell extravasation, establishment and growth. PLoS ONE. 2009;4:e6562.
Gil-Bernabé AM, Ferjancic S, Tlalka M, Zhao L, Allen PD, Im JH, et al. Recruitment of monocytes/macrophages by tissue factor-mediated coagulation is essential for metastatic cell survival and premetastatic niche establishment in mice. Blood. 2012;119:3164–75.
Peranzoni E, Lemoine J, Vimeux L, Feuillet V, Barrin S, Kantari-Mimoun C, et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti-PD-1 treatment. Proc Natl Acad Sci USA. 2018;115:E4041–50.
Kryczek I, Zou L, Rodriguez P, Zhu G, Wei S, Mottram P, et al. B7-H4 expression identifies a novel suppressive macrophage population in human ovarian carcinoma. J Exp Med. 2006;203:871–81.
Jaiswal S, Jamieson CH, Pang WW, Park CY, Chao MP, Majeti R, et al. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell. 2009;138:271–85.
Hibbs JB, Taintor RR, Vavrin Z, Rachlin EM. Nitric oxide: a cytotoxic activated macrophage effector molecule. Biochem Biophys Res Commun. 1988;157:87–94.
Hibbs JB, Vavrin Z, Taintor RR. L-arginine is required for expression of the activated macrophage effector mechanism causing selective metabolic inhibition in target cells. J Immunol. 1987;138:550–65.
Perez-Diez A, Liu X, Matzinger P. Neoantigen presentation and IFNγ signaling on the same tumor-associated macrophage are necessary for CD4 T cell-mediated antitumor activity in mice. Cancer Res Commun. 2022;2:316–29.
Fridman WH, Zitvogel L, Sautès-Fridman C, Kroemer G. The immune contexture in cancer prognosis and treatment. Nat Rev Clin Oncol. 2017;14:717–34.
Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. 2017;541:321–30.
Li J, **e Y, Wang X, Li F, Li S, Li M, et al. Prognostic impact of tumor-associated macrophage infiltration in esophageal cancer: a meta-analysis. Future Oncol. 2019;15:2303–17.
Wu P, Wu D, Zhao L, Huang L, Chen G, Shen G, et al. Inverse role of distinct subsets and distribution of macrophage in lung cancer prognosis: a meta-analysis. Oncotarget. 2016;7:40451–60.
Mei J, **ao Z, Guo C, Pu Q, Ma L, Liu C, et al. Prognostic impact of tumor-associated macrophage infiltration in non-small cell lung cancer: a systemic review and meta-analysis. Oncotarget. 2016;7:34217–28.
Di Caro G, Cortese N, Castino GF, Grizzi F, Gavazzi F, Ridolfi C, et al. Dual prognostic significance of tumour-associated macrophages in human pancreatic adenocarcinoma treated or untreated with chemotherapy. Gut. 2016;65:1710–20.
Malesci A, Bianchi P, Celesti G, Basso G, Marchesi F, Grizzi F, et al. Tumor-associated macrophages and response to 5-fluorouracil adjuvant therapy in stage III colorectal cancer. Oncoimmunology. 2017;6:e1342918.
Mulder K, Patel AA, Kong WT, Piot C, Halitzki E, Dunsmore G, et al. Cross-tissue single-cell landscape of human monocytes and macrophages in health and disease. Immunity. 2021;54:1883–900.e5.
Ginhoux F, Schultze JL, Murray PJ, Ochando J, Biswas SK. New insights into the multidimensional concept of macrophage ontogeny, activation and function. Nat Immunol. 2016;17:34–40.
Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate map** analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330:841–5.
Zhu Y, Herndon JM, Sojka DK, Kim KW, Knolhoff BL, Zuo C, et al. Tissue-resident macrophages in pancreatic ductal adenocarcinoma originate from embryonic hematopoiesis and promote tumor progression. Immunity. 2017;47:323–38.e6.
Chen Z, Feng X, Herting CJ, Garcia VA, Nie K, Pong WW, et al. Cellular and molecular identity of tumor-associated macrophages in glioblastoma. Cancer Res. 2017;77:2266–78.
Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12:253–68.
Timperi E, Gueguen P, Molgora M, Magagna I, Kieffer Y, Lopez-Lastra S, et al. Lipid-associated macrophages are induced by cancer-associated fibroblasts and mediate immune suppression in breast cancer. Cancer Res. 2022;82:3291–306.
Ouzzani M, Hammady H, Fedorowicz Z, Elmagarmid A. Rayyan—a web and mobile app for systematic reviews. Syst Rev. 2016;5:210.
Blair HA. Duvelisib: first global approval. Drugs. 2018;78:1847–53.
Vacchelli E, Galluzzi L, Eggermont A, Fridman WH, Galon J, Sautès-Fridman C, et al. Trial watch: FDA-approved Toll-like receptor agonists for cancer therapy. Oncoimmunology. 2012;1:894–907.
Ando K, Mori K, Corradini N, Redini F, Heymann D. Mifamurtide for the treatment of nonmetastatic osteosarcoma. Expert Opin Pharmacother. 2011;12:285–92.
Salomon R, Dahan R. Next generation CD40 agonistic antibodies for cancer immunotherapy. Front Immunol. 2022;13:940674.
Amouzegar A, Chelvanambi M, Filderman JN, Storkus WJ, Luke JJ. STING agonists as cancer therapeutics. Cancers. 2021;13:2695.
Engel AL, Holt GE, Lu H. The pharmacokinetics of Toll-like receptor agonists and the impact on the immune system. Expert Rev Clin Pharm. 2011;4:275–89.
Soomer-James J, Damelin M, Malli N. Abstract 4423: XMT-2056, a HER2-targeted STING agonist antibody-drug conjugate, exhibits ADCC function that synergizes with STING pathway activation and contributes to anti-tumor responses. Cancer Res. 2023;83:4423.
Li BT, Pegram MD, Lee K-W, Sharma M, Lee J, Spira AI, et al. A phase 1/2 study of a first-in-human immune-stimulating antibody conjugate (ISAC) BDC-1001 in patients with advanced HER2-expressing solid tumors. J Clin Oncol. 2023;41:Abstract 2538.
Sen S, Call J, Papadopoulos K, Smith FD, van der Horst EH. Abstract 1532: A phase 1/2 study of safety, tolerability, and pharmacokinetics of SNS-101, a pH-sensitive anti-VISTA mAb, as monotherapy and in combination with cemiplimab in patients with advanced solid tumors. J Immunother Cancer. 2023;11:A1755.
Ye S, Cohen D, Belmar NA, Choi D, Tan SS, Sho M, et al. A bispecific molecule targeting CD40 and tumor antigen mesothelin enhances tumor-specific immunity. Cancer Immunol Res. 2019;7:1864–75.
Chen YC, Shi W, Shi JJ, Lu JJ. Progress of CD47 immune checkpoint blockade agents in anticancer therapy: a hematotoxic perspective. J Cancer Res Clin Oncol. 2022;148:1–14.
White AL, Chan HT, Roghanian A, French RR, Mockridge CI, Tutt AL, et al. Interaction with FcγRIIB is critical for the agonistic activity of anti-CD40 monoclonal antibody. J Immunol. 2011;187:1754–63.
Kamerkar S, Leng C, Burenkova O, Jang SC, McCoy C, Zhang K, et al. Exosome-mediated genetic reprogramming of tumor-associated macrophages by exoASO-STAT6 leads to potent monotherapy antitumor activity. Sci Adv. 2022;8:eabj7002.
Jang SC, Economides KD, Moniz RJ, Sia CL, Lewis N, McCoy C, et al. ExoSTING, an extracellular vesicle loaded with STING agonists, promotes tumor immune surveillance. Commun Biol. 2021;4:497.
Luke JJ, Piha-Paul SA, Medina T, Verschraegen CF, Varterasian M, Brennan AM, et al. Phase I study of SYNB1891, an engineered E. coli nissle strain expressing STING agonist, with and without atezolizumab in advanced malignancies. Clin Cancer Res. 2023;29:2435–44.
Eremina NV, Kazey VI, Mishugin SV, Leonenkov RV, Pushkar DY, Mett VL, et al. First-in-human study of anticancer immunotherapy drug candidate mobilan: safety, pharmacokinetics and pharmacodynamics in prostate cancer patients. Oncotarget. 2020;11:1273–88.
Patel M, Cox C, Krige D, Walker L, Evilevitch V, Carter J, et al. Phase 1 clinical trial results for NG-350A, a novel transgene-armed and tumor-selective vector: differential effects of intravenous (IV) versus intratumoral (IT) dosing on immune pharmacodynamics (PD). J Clin Oncol. 2023;41:Abstract 2572.
Hägerbrand K, Varas L, Deronic A, Nyesiga B, Sundstedt A, Ljung L, et al. Bispecific antibodies targeting CD40 and tumor-associated antigens promote cross-priming of T cells resulting in an antitumor response superior to monospecific antibodies. J Immunother Cancer. 2022;10:e005018.
Dahan R, Barnhart BC, Li F, Yamniuk AP, Korman AJ, Ravetch JV. Therapeutic activity of agonistic, human Anti-CD40 monoclonal antibodies requires selective FcγR engagement. Cancer Cell. 2016;29:820–31.
Feng M, Jiang W, Kim BYS, Zhang CC, Fu YX, Weissman IL. Phagocytosis checkpoints as new targets for cancer immunotherapy. Nat Rev Cancer. 2019;19:568–86.
Tsai RK, Discher DE. Inhibition of “self” engulfment through deactivation of myosin-II at the phagocytic synapse between human cells. J Cell Biol. 2008;180:989–1003.
Sikic BI, Lakhani N, Patnaik A, Shah SA, Chandana SR, Rasco D, et al. First-in-human, first-in-class phase I trial of the anti-CD47 antibody Hu5F9-G4 in patients with advanced cancers. J Clin Oncol. 2019;37:946–53.
Son J, Hsieh RC, Lin HY, Krause KJ, Yuan Y, Biter AB, et al. Inhibition of the CD47-SIRPα axis for cancer therapy: a systematic review and meta-analysis of emerging clinical data. Front Immunol. 2022;13:1027235.
Gilead Sciences, Inc. Gilead to discontinue phase 3 ENHANCE study of magrolimab plus azacitidine in higher-risk MDS. 2023. https://www.gilead.com/news-and-press/press-room/press-releases/2023/7/gilead-to-discontinue-phase-3-enhance-study-of-magrolimab-plus-azacitidine-in-higher-risk-mds.
Gilead Sciences, Inc. Gilead statement on the discontinuation of magrolimab study in AML with TP53 mutations. 2023. https://www.gilead.com/news-and-press/company-statements/gilead-statement-on-the-discontinuation-of-magrolimab-study-in-aml-with-tp53-mutations.
Mehrotra P, Ravichandran KS. Drugging the efferocytosis process: concepts and opportunities. Nat Rev Drug Discov. 2022;21:601–20.
Barkal AA, Brewer RE, Markovic M, Kowarsky M, Barkal SA, Zaro BW, et al. CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature. 2019;572:392–6.
Li S, Chen D, Guo H, Yang Y, Liu D, Yang C, et al. IMM47, a humanized monoclonal antibody that targets CD24, exhibits exceptional anti-tumor efficacy by blocking the CD24/Siglec-10 interaction and can be used as monotherapy or in combination with anti-PD1 antibodies for cancer immunotherapy. Antib Ther. 2023;6:240–52.
Chen W, Hu Z, Guo Z. Targeting CD24 in cancer immunotherapy. Biomedicines. 2023;11:3159.
Chen GY, Tang J, Zheng P, Liu Y. CD24 and Siglec-10 selectively repress tissue damage-induced immune responses. Science. 2009;323:1722–5.
Kazakova E, Iamshchikov P, Larionova I, Kzhyshkowska J. Macrophage scavenger receptors: tumor support and tumor inhibition. Front Oncol. 2022;12:1096897.
Canton J, Neculai D, Grinstein S. Scavenger receptors in homeostasis and immunity. Nat Rev Immunol. 2013;13:621–34.
Georgoudaki AM, Prokopec KE, Boura VF, Hellqvist E, Sohn S, Östling J, et al. Reprogramming tumor-associated macrophages by antibody targeting inhibits cancer progression and metastasis. Cell Rep. 2016;15:2000–11.
Eisinger S, Sarhan D, Boura VF, Ibarlucea-Benitez I, Tyystjärvi S, Oliynyk G, et al. Targeting a scavenger receptor on tumor-associated macrophages activates tumor cell killing by natural killer cells. Proc Natl Acad Sci USA. 2020;117:32005–16.
Jaynes JM, Sable R, Ronzetti M, Bautista W, Knotts Z, Abisoye-Ogunniyan A, et al. Mannose receptor (CD206) activation in tumor-associated macrophages enhances adaptive and innate antitumor immune responses. Sci Transl Med. 2020;12:eaax6337.
Jahchan N, Liang L, Pollack J, Juric V, Yang X, Lacayo S, et al. Tuning the tumor myeloid microenvironment by targeting MARCO positive myeloid cells to unleash anti-tumor immunity. [Poster]. In: Keystone symposia—cancer immunotherapy. Whistler, Canada; 2022. https://www.pionyrtx.com/file.cfm/19/docs/keystone%20py265%20poster-nj%20%20final.pdf.
Scodeller P, Simón-Gracia L, Kopanchuk S, Tobi A, Kilk K, Säälik P, et al. Precision targeting of tumor macrophages with a CD206 binding peptide. Sci Rep. 2017;7:14655.
Kristiansen M, Graversen JH, Jacobsen C, Sonne O, Hoffman HJ, Law SK, et al. Identification of the haemoglobin scavenger receptor. Nature. 2001;409:198–201.
Allison E, Edirimanne S, Matthews J, Fuller SJ. Breast cancer survival outcomes and tumor-associated macrophage markers: a systematic review and meta-analysis. Oncol Ther. 2023;11:27–48.
Troiano G, Caponio VCA, Adipietro I, Tepedino M, Santoro R, Laino L, et al. Prognostic significance of CD68+ and CD163+ tumor associated macrophages in head and neck squamous cell carcinoma: a systematic review and meta-analysis. Oral Oncol. 2019;93:66–75.
Hasita H, Komohara Y, Okabe H, Masuda T, Ohnishi K, Lei XF, et al. Significance of alternatively activated macrophages in patients with intrahepatic cholangiocarcinoma. Cancer Sci. 2010;101:1913–9.
Probst P, Simmons R, Dinh H, Zuck M, Wall V, Bouchlaka M, et al. Abstract 271: Development of OR2805, an anti-CD163 antibody derived from an elite responder to checkpoint inhibitor therapy that relieves immunosuppression caused by M2c macrophages. J Immunother Cancer. 2021;9:A294.
Hollmén M, Figueiredo CR, Jalkanen S. New tools to prevent cancer growth and spread: a ‘Clever’ approach. Br J Cancer. 2020;123:501–9.
Virtakoivu R, Rannikko JH, Viitala M, Vaura F, Takeda A, Lönnberg T, et al. Systemic blockade of Clever-1 elicits lymphocyte activation alongside checkpoint molecule downregulation in patients with solid tumors: results from a phase I/II clinical trial. Clin Cancer Res. 2021;27:4205–20.
Rannikko JH, Verlingue L, de Miguel M, Pasanen A, Robbrecht D, Skytta A, et al. Bexmarilimab-induced macrophage activation leads to treatment benefit in solid tumors: the phase I/II first-in-human MATINS trial. Cell Rep. Med. 2023;4:101307.
Kontro M, Stein AS, Pyörälä M, Rimpiläinen J, Siitonen T, Hollmén M, et al. Encouraging efficacy observed in bexmab study: a phase 1/2 study to assess safety and efficacy of bexmarilimab in combination with standard of care in myeloid malignancies. Blood. 2023;142:2915.
Shekarian T, Valsesia-Wittmann S, Brody J, Michallet MC, Depil S, Caux C, et al. Pattern recognition receptors: immune targets to enhance cancer immunotherapy. Ann Oncol. 2017;28:1756–66.
Rolfo C, Giovannetti E, Martinez P, McCue S, Naing A. Applications and clinical trial landscape using Toll-like receptor agonists to reduce the toll of cancer. NPJ Precis Oncol. 2023;7:26.
Galluzzi L, Vacchelli E, Eggermont A, Fridman WH, Galon J, Sautès-Fridman C, et al. Trial watch: experimental toll-like receptor agonists for cancer therapy. Oncoimmunology. 2012;1:699–716.
Pahlavanneshan S, Sayadmanesh A, Ebrahimiyan H, Basiri M. Toll-like receptor-based strategies for cancer immunotherapy. J Immunol Res. 2021;2021:9912188.
Rodell CB, Arlauckas SP, Cuccarese MF, Garris CS, Li R, Ahmed MS, et al. TLR7/8-agonist-loaded nanoparticles promote the polarization of tumour-associated macrophages to enhance cancer immunotherapy. Nat Biomed Eng. 2018;2:578–88.
Yang Y, Li H, Fotopoulou C, Cunnea P, Zhao X. Toll-like receptor-targeted anti-tumor therapies: advances and challenges. Front Immunol. 2022;13:1049340.
Dajon M, Iribarren K, Cremer I. Toll-like receptor stimulation in cancer: a pro- and anti-tumor double-edged sword. Immunobiology. 2017;222:89–100.
Lu H. TLR agonists for cancer immunotherapy: tip** the balance between the immune stimulatory and inhibitory effects. Front Immunol. 2014;5:83.
Motwani M, Pesiridis S, Fitzgerald KA. DNA sensing by the cGAS-STING pathway in health and disease. Nat Rev Genet. 2019;20:657–74.
Zhao K, Huang J, Zhao Y, Wang S, Xu J, Yin K. Targeting STING in cancer: challenges and emerging opportunities. Biochim Biophys Acta Rev Cancer. 2023;1878:188983.
Huang C, Shao N, Huang Y, Chen J, Wang D, Hu G, et al. Overcoming challenges in the delivery of STING agonists for cancer immunotherapy: a comprehensive review of strategies and future perspectives. Mater Today Bio. 2023;23:100839.
Harrington KJ, Brody J, Ingham M, Strauss J, Cemerski S, Wang M, et al. LBA15: Preliminary results of the first-in-human (FIH) study of MK-1454, an agonist of stimulator of interferon genes (STING), as monotherapy or in combination with pembrolizumab (pembro) in patients with advanced solid tumors or lymphomas. Ann Oncol. 2018;29:VIII712.
Chen R, Kang R, Tang D. The mechanism of HMGB1 secretion and release. Exp Mol Med. 2022;54:91–102.
Idoudi S, Bedhiafi T, Pedersen S, Elahtem M, Alremawi I, Akhtar S, et al. Role of HMGB1 and its associated signaling pathways in human malignancies. Cell Signal. 2023;112:110904.
Huber R, Meier B, Otsuka A, Fenini G, Satoh T, Gehrke S, et al. Tumour hypoxia promotes melanoma growth and metastasis via High Mobility Group Box-1 and M2-like macrophages. Sci Rep. 2016;6:29914.
Choi YI, Kim KY, Jung NJ, Lee JJ, Park SB. Abstract 5599: A novel orally available anti-cancer drug candidate, SB17170, represses myeloid-derived suppressor cells by targeting HMGB1. Cancer Res. 2022;82:5599.
Kerscher B, Willment JA, Brown GD. The Dectin-2 family of C-type lectin-like receptors: an update. Int Immunol. 2013;25:271–7.
Sato K, Yang XL, Yudate T, Chung JS, Wu J, Luby-Phelps K, et al. Dectin-2 is a pattern recognition receptor for fungi that couples with the Fc receptor gamma chain to induce innate immune responses. J Biol Chem. 2006;281:38854–66.
Kenkel JA, **ao F, Ho PY, Nolin JL, Gadkari RK, Torrez LB, et al. Abstract 2964: Targeting tumor-associated macrophages to enhance anti-tumor immunity with the Dectin-2 agonistic antibody BDC-3042. Cancer Res. 2023;83:2964. https://www.boltbio.com/wp-content/uploads/2023/04/Kenkel-et-al-AACR-2023-Poster-v3.pdf.
Li J, Liu XG, Ge RL, Yin YP, Liu YD, Lu WP, et al. The ligation between ERMAP, galectin-9 and dectin-2 promotes Kupffer cell phagocytosis and antitumor immunity. Nat Immunol. 2023;24:1813–24.
Vonderheide RH. CD40 agonist antibodies in cancer immunotherapy. Annu Rev Med. 2020;71:47–58.
Luke JJ, Barlesi F, Chung K, Tolcher AW, Kelly K, Hollebecque A, et al. Phase I study of ABBV-428, a mesothelin-CD40 bispecific, in patients with advanced solid tumors. J Immunother Cancer. 2021;9:e002015.
Lakhani NJ, Stewart DB, Richardson DL, Dockery DE, Van Le L, Call JA, et al. Phase 1 dose escalation study of SL-172154 (SIRPα-Fc-CD40L) in platinum-resistant ovarian cancer. J Clin Oncol. 2023;41:Abstract 5544.
Alligator Bioscience. Alligator Bioscience announces positive mitazalimab OPTIMIZE-1 phase 2 results meeting primary endpoint and demonstrating clinically relevant survival benefits in 1st line pancreatic cancer. 2024. https://alligatorbioscience.se/en/news/alligator-bioscience-announces-positive-mitazalimab-optimize-1-phase-2-results-meeting-primary-endpoint-and-demonstrating-clinically-relevant-survival-benefits-in-1st-line-pancreatic-cancer/.
Weiss SA, Sznol M, Shaheen M, Berciano-Guerrero M, Couselo EM, Rodríguez-Abreu D, et al. A phase II trial of the CD40 agonistic antibody sotigalimab (APX005M) in combination with nivolumab in subjects with metastatic melanoma with confirmed disease progression on anti-PD-1 therapy. Clin Cancer Res. 2024;30:74–81.
Deng M, Chen H, Liu X, Huang R, He Y, Yoo B, et al. Leukocyte immunoglobulin-like receptor subfamily B: therapeutic targets in cancer. Antib Ther. 2021;4:16–33.
Barkal AA, Weiskopf K, Kao KS, Gordon SR, Rosental B, Yiu YY, et al. Engagement of MHC class I by the inhibitory receptor LILRB1 suppresses macrophages and is a target of cancer immunotherapy. Nat Immunol. 2018;19:76–84.
Chen HM, van der Touw W, Wang YS, Kang K, Mai S, Zhang J, et al. Blocking immunoinhibitory receptor LILRB2 reprograms tumor-associated myeloid cells and promotes antitumor immunity. J Clin Invest. 2018;128:5647–62.
Siu LL, Wang D, Hilton J, Geva R, Rasco D, Perets R, et al. First-in-class anti-immunoglobulin-like transcript 4 myeloid-specific antibody MK-4830 abrogates a PD-1 resistance mechanism in patients with advanced solid tumors. Clin Cancer Res. 2022;28:57–70.
DiNardo C, Pollyea D, Aribi A, Jonas B, Jeyakumar D, Roboz G, et al. A first-in-human phase 1 study of IO-202 (anti-LILRB4 mAb) in acute myeloid leukemia (AML) with monocytic differentiation and chronic myelomonocytic leukemia (CMML) patients. [Poster]. In: 28th annual congress of EHA. Frankfurt, Germany; 2023. https://static1.squarespace.com/static/60dc8b9325bba74a54bb572f/t/648f46d45216c12186dabea0/1687111382762/P536-EHA2023-poster+final.pdf.
Taylor MH, Patel MR, Powderly JD, Woodard P, Chung L, Tian H, et al. Abstract CT040: A first-in-human phase 1 trial of IO-108, an antagonist antibody targeting LILRB2 (ILT4), as monotherapy and in combination with pembrolizumab in adult patients with advanced relapsed or refractory solid tumors: dose escalation study. Cancer Res. 2023;83:CT040.
Van Laethem F, Donaty L, Tchernonog E, Lacheretz-Szablewski V, Russello J, Buthiau D, et al. LAIR1, an ITIM-containing receptor involved in immune disorders and in hematological neoplasms. Int J Mol Sci. 2022;23:16136.
He S, Huang J, Rodriguez L, Cortez C, Li B, Ho C, et al. Abstract LB219: Preclinical development of NGM438, a novel anti-LAIR1 antagonist monoclonal antibody for the treatment of collagen-rich solid tumors. Cancer Res. 2022;82:LB219.
Lebbink RJ, van den Berg MC, de Ruiter T, Raynal N, van Roon JA, Lenting PJ, et al. The soluble leukocyte-associated Ig-like receptor (LAIR)-2 antagonizes the collagen/LAIR-1 inhibitory immune interaction. J Immunol. 2008;180:1662–9.
Myint H, Tian L, Shaik J, Barbu E, Zhou Q, Morawski A. Abstract 487: NC410, a fusion protein of LAIR-2 (Leukocyte Associated Immunoglobulin-like Receptor) with human IgG1 Fc, is safe & tolerable with evidence of immune modulation in subjects with advanced solid tumors. J Immunother Cancer. 2021;9:A516.
Keerthivasan S, Şenbabaoğlu Y, Martinez-Martin N, Husain B, Verschueren E, Wong A, et al. Homeostatic functions of monocytes and interstitial lung macrophages are regulated via collagen domain-binding receptor LAIR1. Immunity. 2021;54:1511–26.e8.
Wang J, Sun J, Liu LN, Flies DB, Nie X, Toki M, et al. Siglec-15 as an immune suppressor and potential target for normalization cancer immunotherapy. Nat Med. 2019;25:656–66.
Takamiya R, Ohtsubo K, Takamatsu S, Taniguchi N, Angata T. The interaction between Siglec-15 and tumor-associated sialyl-Tn antigen enhances TGF-β secretion from monocytes/macrophages through the DAP12-Syk pathway. Glycobiology. 2013;23:178–87.
Shum E, Myint H, Shaik J, Zhou Q, Barbu E, Morawski A, et al. Abstract 490: Clinical benefit through Siglec-15 targeting with NC318 antibody in subjects with Siglec-15 positive advanced solid tumors. J Immunother Cancer. 2021;9:A520.
NextCure. NextCure provides update and reports third quarter 2022 financial results. 2022. https://www.globenewswire.com/news-release/2022/11/03/2548354/0/en/NextCure-Provides-Update-and-Reports-Third-Quarter-2022-Financial-Results.html.
Juric V, Mayes E, Binnewies M, Lee T, Canaday P, Pollack JL, et al. TREM1 activation of myeloid cells promotes antitumor immunity. Sci Transl Med. 2023;15:eadd9990.
Fang HY, Hughes R, Murdoch C, Coffelt SB, Biswas SK, Harris AL, et al. Hypoxia-inducible factors 1 and 2 are important transcriptional effectors in primary macrophages experiencing hypoxia. Blood. 2009;114:844–59.
Raggi F, Bosco MC. Targeting mononuclear phagocyte receptors in cancer immunotherapy: new perspectives of the triggering receptor expressed on myeloid cells (TREM-1). Cancers. 2020;12:1337.
Kirschenbaum D, **e K, Ingelfinger F, Katzenelenbogen Y, Abadie K, Look T, et al. Time-resolved single-cell transcriptomics defines immune trajectories in glioblastoma. Cell. 2024;187:149–65.e23.
Molgora M, Esaulova E, Vermi W, Hou J, Chen Y, Luo J, et al. TREM2 modulation remodels the tumor myeloid landscape enhancing anti-PD-1 immunotherapy. Cell. 2020;182:886–900.e17.
Patnaik A, Hamilton EP, Winer IS, Tan W, Hubbard JM, Schenk EL, et al. A phase 1a dose-escalation study of PY314, a TREM2 (Triggering Receptor Expressed on Macrophages 2) targeting monoclonal antibody. J Clin Oncol. 2022;40:Abstract 2648.
Xu W, Hiếu T, Malarkannan S, Wang L. The structure, expression, and multifaceted role of immune-checkpoint protein VISTA as a critical regulator of anti-tumor immunity, autoimmunity, and inflammation. Cell Mol Immunol. 2018;15:438–46.
Dharmadhikari B, Thakkar D, Zharkova O, Ray D, Tirado-Magallanes R, Lai J, et al. Anti-VISTA antibody HMBD-002 reprograms tumor associated macrophages and promotes cytotoxic T cell response. [Poster]. In: 37th annual meeting (SITC 2022); Boston, MA; 2022. https://hummingbirdbioscience.com/wp-content/uploads/2022/11/Anti-VISTA-antibody-HMBD-002-reprograms-tumor-associated-macrophages-and-promotes-cytotoxic-T-cell-response.pdf.
Johnson M, Lines JL, Carriere C, Molloy M, Martell R, Von Roemeling R, et al. Abstract 392: Phase 1 study of CI-8993 anti-VISTA antibody in patients with advanced solid tumor malignancies. J Immunother Cancer. 2020;8:A238. https://www.curis.com/wp-content/uploads/2020/11/Curis-CI-8993-SITC-TIP-Poster-2020.pdf.
Iadonato S, Ovechkina Y, Lustig K, Cross J, Eyde N, Frazier E, et al. A highly potent anti-VISTA antibody KVA12123—a new immune checkpoint inhibitor and a promising therapy against poorly immunogenic tumors. Front Immunol. 2023;14:1311658.
Opitz CA, Somarribas Patterson LF, Mohapatra SR, Dewi DL, Sadik A, Platten M, et al. The therapeutic potential of targeting tryptophan catabolism in cancer. Br J Cancer. 2020;122:30–44.
Zeitler L, Murray PJ. IL4i1 and IDO1: oxidases that control a tryptophan metabolic nexus in cancer. J Biol Chem. 2023;299:104827.
Long GV, Dummer R, Hamid O, Gajewski TF, Caglevic C, Dalle S, et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): a phase 3, randomised, double-blind study. Lancet Oncol. 2019;20:1083–97.
Moyer BJ, Rojas IY, Murray IA, Lee S, Hazlett HF, Perdew GH, et al. Indoleamine 2,3-dioxygenase 1 (IDO1) inhibitors activate the aryl hydrocarbon receptor. Toxicol Appl Pharm. 2017;323:74–80.
Rodríguez PC, Ochoa AC. Arginine regulation by myeloid derived suppressor cells and tolerance in cancer: mechanisms and therapeutic perspectives. Immunol Rev. 2008;222:180–91.
Thomas AC, Mattila JT. “Of mice and men”: arginine metabolism in macrophages. Front Immunol. 2014;5:479.
Zhang H, Zhu X, Friesen TJ, Kwak JW, Pisarenko T, Mekvanich S, et al. Annexin A2/TLR2/MYD88 pathway induces arginase 1 expression in tumor-associated neutrophils. J Clin Invest. 2022;132:e153643.
Steggerda SM, Bennett MK, Chen J, Emberley E, Huang T, Janes JR, et al. Inhibition of arginase by CB-1158 blocks myeloid cell-mediated immune suppression in the tumor microenvironment. J Immunother Cancer. 2017;5:101.
Zhang I, Alizadeh D, Liang J, Zhang L, Gao H, Song Y, et al. Characterization of arginase expression in glioma-associated microglia and macrophages. PLoS ONE. 2016;11:e0165118.
Zea AH, Rodriguez PC, Atkins MB, Hernandez C, Signoretti S, Zabaleta J, et al. Arginase-producing myeloid suppressor cells in renal cell carcinoma patients: a mechanism of tumor evasion. Cancer Res. 2005;65:3044–8.
Li HS, Watowich SS. Innate immune regulation by STAT-mediated transcriptional mechanisms. Immunol Rev. 2014;261:84–101.
Hashimoto S, Hashimoto A, Muromoto R, Kitai Y, Oritani K, Matsuda T. Central roles of STAT3-mediated signals in onset and development of cancers: tumorigenesis and immunosurveillance. Cells. 2022;11:2618.
Hong D, Kurzrock R, Kim Y, Woessner R, Younes A, Nemunaitis J, et al. AZD9150, a next-generation antisense oligonucleotide inhibitor of STAT3 with early evidence of clinical activity in lymphoma and lung cancer. Sci Transl Med. 2015;7:314ra185.
Proia TA, Singh M, Woessner R, Carnevalli L, Bommakanti G, Magiera L, et al. STAT3 antisense oligonucleotide remodels the suppressive tumor microenvironment to enhance immune activation in combination with anti-PD-L1. Clin Cancer Res. 2020;26:6335–49.
Reilley MJ, McCoon P, Cook C, Lyne P, Kurzrock R, Kim Y. STAT3 antisense oligonucleotide AZD9150 in a subset of patients with heavily pretreated lymphoma: results of a phase 1b trial. J Immunother Cancer. 2018;6:119.
Mackert JR, Qu P, Min Y, Johnson PF, Yang L, Lin PC. Dual negative roles of C/EBPα in the expansion and pro-tumor functions of MDSCs. Sci Rep. 2017;7:14048.
Hashimoto A, Sarker D, Reebye V, Jarvis S, Sodergren MH, Kossenkov A, et al. Upregulation of C/EBPα inhibits suppressive activity of myeloid cells and potentiates antitumor response in mice and patients with cancer. Clin Cancer Res. 2021;27:5961–78.
Sarker D, Plummer R, Meyer T, Sodergren MH, Basu B, Chee CE, et al. MTL-CEBPA, a small activating RNA therapeutic upregulating C/EBP-α, in patients with advanced liver cancer: a first-in-human, multicenter, open-label, phase I trial. Clin Cancer Res. 2020;26:3936–46.
Zhou J, Li H, **a X, Herrera A, Pollock N, Reebye V, et al. Anti-inflammatory activity of MTL-CEBPA, a small activating RNA drug, in LPS-stimulated monocytes and humanized mice. Mol Ther. 2019;27:999–1016.
Rothhammer V, Quintana FJ. The aryl hydrocarbon receptor: an environmental sensor integrating immune responses in health and disease. Nat Rev Immunol. 2019;19:184–97.
Murray IA, Patterson AD, Perdew GH. Aryl hydrocarbon receptor ligands in cancer: friend and foe. Nat Rev Cancer. 2014;14:801–14.
McGovern K, Castro AC, Cavanaugh J, Coma S, Walsh M, Tchaicha J, et al. Discovery and characterization of a novel aryl hydrocarbon receptor inhibitor, IK-175, and its inhibitory activity on tumor immune suppression. Mol Cancer Ther. 2022;21:1261–72.
Aggen DH, McKean M, Lakhani NJ, Bashir B, Hoffman-Censits J, Alhalabi O, et al. Abstract 661: Initial results from a phase 1a/b study of IK-175, an oral AHR inhibitor, as single agent and in combination with nivolumab in patients with advanced solid tumors and urothelial cancer. J Immunother Cancer. 2022;10:A691. https://ikenaoncology.com/wp-content/uploads/2022/11/IK175-001-SITC-Clinical-Poster-FINAL-November-2022.pdf.
Falkenberg KJ, Johnstone RW. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat Rev Drug Discov. 2014;13:673–91.
Ho TCS, Chan AHY, Ganesan A. Thirty years of HDAC inhibitors: 2020 insight and hindsight. J Med Chem. 2020;63:12460–84.
Ossenkoppele GJ, Lowenberg B, Zachee P, Vey N, Breems D, Van de Loosdrecht AA, et al. A phase I first-in-human study with tefinostat—a monocyte/macrophage targeted histone deacetylase inhibitor—in patients with advanced haematological malignancies. Br J Haematol. 2013;162:191–201.
Guerriero JL, Sotayo A, Ponichtera HE, Castrillon JA, Pourzia AL, Schad S, et al. Class IIa HDAC inhibition reduces breast tumours and metastases through anti-tumour macrophages. Nature. 2017;543:428–32.
Das Gupta K, Shakespear MR, Iyer A, Fairlie DP, Sweet MJ. Histone deacetylases in monocyte/macrophage development, activation and metabolism: refining HDAC targets for inflammatory and infectious diseases. Clin Transl Immunol. 2016;5:e62.
Lanahan SM, Wymann MP, Lucas CL. The role of PI3Kγ in the immune system: new insights and translational implications. Nat Rev Immunol. 2022;22:687–700.
Kaneda MM, Messer KS, Ralainirina N, Li H, Leem CJ, Gorjestani S, et al. PI3Kγ is a molecular switch that controls immune suppression. Nature. 2016;539:437–42.
Ali K, Soond DR, Pineiro R, Hagemann T, Pearce W, Lim EL, et al. Inactivation of PI(3)K p110δ breaks regulatory T-cell-mediated immune tolerance to cancer. Nature. 2014;510:407–11.
Hong DS, Postow M, Chmielowski B, Sullivan R, Patnaik A, Cohen EEW, et al. Eganelisib, a first-in-class PI3Kγ inhibitor, in patients with advanced solid tumors: results of the phase 1/1b MARIO-1 trial. Clin Cancer Res. 2023;29:2210–9.
Mishra AK, Malonia SK. Advancing cellular immunotherapy with macrophages. Life Sci. 2023;328:121857.
Andreesen R, Hennemann B, Krause SW. Adoptive immunotherapy of cancer using monocyte-derived macrophages: rationale, current status, and perspectives. J Leukoc Biol. 1998;64:419–26.
Klichinsky M, Ruella M, Shestova O, Lu XM, Best A, Zeeman M, et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat Biotechnol. 2020;38:947–53.
Gerber M, Prod’homme T, Wand Y, Kieffer-Kwon KR, Diwanji N, D’Alessandro J, et al. Abstract CT131: Initial preclinical and clinical experience of autologous engineered monocytes in T cell lymphoma. Cancer Res. 2023;83:CT131.
Sloas C, Gill S, Klichinsky M. Engineered CAR-macrophages as adoptive immunotherapies for solid tumors. Front Immunol. 2021;12:783305.
Strati P, Feldman T, Querfeld C, Jain S, McCurley N, Bàrcia R, et al. Phase 1 study of autologous Sirpα-low macrophages (SIRPant-M) administered by intratumoral injection alone or in combination with external-beam radiotherapy in patients with relapsed or refractory non-Hodgkin lymphoma (NCT05967416). Blood. 2023;142:4856.
Green DS, Ning F, Duemler A, Myers TG, Trewhitt K, Ekwede I, et al. Intraperitoneal monocytes plus IFNs as a novel cellular immunotherapy for ovarian cancer: mechanistic characterization and results from a phase I clinical trial. Clin Cancer Res. 2023;29:349–63.
Ferris RL, Saba NF, Gitlitz BJ, Haddad R, Sukari A, Neupane P, et al. Effect of adding motolimod to standard combination chemotherapy and cetuximab treatment of patients with squamous cell carcinoma of the head and neck: the Active8 randomized clinical trial. JAMA Oncol. 2018;4:1583–8.
Lang C, Roy S, Wang Y, Graves D, Xu Y, Serezani CH, et al. Efferocytosis drives myeloid NLRP3 dependent inflammasome signaling secretion of IL-1β to promote tumor growth. Front Immunol. 2022;13:993771.
Ridker PM, MacFadyen JG, Thuren T, Everett BM, Libby P, Glynn RJ, et al. Effect of interleukin-1β inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet. 2017;390:1833–42.
Gocher AM, Workman CJ, Vignali DAA. Interferon-γ: teammate or opponent in the tumour microenvironment? Nat Rev Immunol. 2022;22:158–72.
Lemos H, Ou R, McCardle C, Lin Y, Calver J, Minett J, et al. Overcoming resistance to STING agonist therapy to incite durable protective antitumor immunity. J Immunother Cancer. 2020;8:e001182.
Siwicki M, Gort-Freitas NA, Messemaker M, Bill R, Gungabeesoon J, Engblom C, et al. Resident Kupffer cells and neutrophils drive liver toxicity in cancer immunotherapy. Sci Immunol. 2021;6:eabi7083.
Bissinger S, Hage C, Wagner V, Maser IP, Brand V, Schmittnaegel M, et al. Macrophage depletion induces edema through release of matrix-degrading proteases and proteoglycan deposition. Sci Transl Med. 2021;13:eabd4550.
Cassetta L, Pollard JW. A timeline of tumour-associated macrophage biology. Nat Rev Cancer. 2023;23:238–57.
Laviron M, Petit M, Weber-Delacroix E, Combes AJ, Arkal AR, Barthélémy S, et al. Tumor-associated macrophage heterogeneity is driven by tissue territories in breast cancer. Cell Rep. 2022;39:110865.
Matusiak M, Hickey JW, van IJzendoorn DGP, Lu G, Kidzinski L, Zhu S, et al. Spatially segregated macrophage populations predict distinct outcomes in colon cancer. Cancer Discov. 2024. https://doi.org/10.1158/2159-8290.CD-23-1300.
Yang M, McKay D, Pollard JW, Lewis CE. Diverse functions of macrophages in different tumor microenvironments. Cancer Res. 2018;78:5492–503.
Yoshida S, Shime H, Takeda Y, Nam JM, Takashima K, Matsumoto M, et al. Toll-like receptor 3 signal augments radiation-induced tumor growth retardation in a murine model. Cancer Sci. 2018;109:956–65.
Malfitano AM, Pisanti S, Napolitano F, Di Somma S, Martinelli R, Portella G. Tumor-associated macrophage status in cancer treatment. Cancers. 2020;12:1987.
Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol. 2017;14:399–416.
Wanderley CW, Colón DF, Luiz JPM, Oliveira FF, Viacava PR, Leite CA, et al. Paclitaxel reduces tumor growth by reprogramming tumor-associated macrophages to an M1 profile in a TLR4-dependent manner. Cancer Res. 2018;78:5891–900.
Klug F, Prakash H, Huber PE, Seibel T, Bender N, Halama N, et al. Low-dose irradiation programs macrophage differentiation to an iNOS+/M1 phenotype that orchestrates effective T cell immunotherapy. Cancer Cell. 2013;24:589–602.
Dijkgraaf EM, Heusinkveld M, Tummers B, Vogelpoel LT, Goedemans R, Jha V, et al. Chemotherapy alters monocyte differentiation to favor generation of cancer-supporting M2 macrophages in the tumor microenvironment. Cancer Res. 2013;73:2480–92.
Seifert L, Werba G, Tiwari S, Giao Ly NN, Nguy S, Alothman S, et al. Radiation therapy induces macrophages to suppress T-cell responses against pancreatic tumors in mice. Gastroenterology. 2016;150:1659–72.e5.
Germano G, Frapolli R, Belgiovine C, Anselmo A, Pesce S, Liguori M, et al. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell. 2013;23:249–62.
Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J Exp Med. 2013;210:1695–710.
Weiskopf K, Weissman IL. Macrophages are critical effectors of antibody therapies for cancer. MAbs. 2015;7:303–10.
Yu J, Green MD, Li S, Sun Y, Journey SN, Choi JE, et al. Liver metastasis restrains immunotherapy efficacy via macrophage-mediated T cell elimination. Nat Med. 2021;27:152–64.
Chamseddine AN, Assi T, Mir O, Chouaib S. Modulating tumor-associated macrophages to enhance the efficacy of immune checkpoint inhibitors: a TAM-pting approach. Pharm Ther. 2022;231:107986.
Quail DF, Bowman RL, Akkari L, Quick ML, Schuhmacher AJ, Huse JT, et al. The tumor microenvironment underlies acquired resistance to CSF-1R inhibition in gliomas. Science. 2016;352:aad3018.
Acknowledgements
We thank Juho Jalkanen and Elina Louramo for their valuable feedback on the content of the manuscript and Joe Hettinger for proofreading the manuscript.
Funding
This study was supported by the Cancer Foundations and the Research Council of Finland. Open Access funding provided by University of Turku (including Turku University Central Hospital).
Author information
Authors and Affiliations
Contributions
JHR and MH designed the study. JHR performed the literature (data) search and drafted the first version of the manuscript. MH supervised the work and gave input on the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
MH is currently employed by and own shares of Faron Pharmaceuticals.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rannikko, J.H., Hollmén, M. Clinical landscape of macrophage-reprogramming cancer immunotherapies. Br J Cancer (2024). https://doi.org/10.1038/s41416-024-02715-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41416-024-02715-6
- Springer Nature Limited