
Abstract
Five new pteridic acids C–G (1–5) were isolated from a culture broth of the marine-derived actinomycete Streptomyces sp. SCSGAA 0027. Their complete structures were elucidated on the basis of NMR data, modified Mosher’s method and quantum chemical calculations. Furthermore, their cytotoxicity, antiviral and antimicrobial activities were also evaluated.
Similar content being viewed by others
Introduction
Spiroketal-containing natural products are an important family of bioactive chemical entities, including benzannulated spiroketals1 and non-benzannulated spiroketals such as spirangiens,2 neomaclafungins,3 leonuketal,4 spirastrellolide A,5 reveromycins6 and phorbaketals,7 which display intriguing scaffolds and diverse biological activities, for example, cytotoxicity, antimicrobial, vasorelaxant and enzyme inhibition. Especially, this group of compounds have attracted many attentions and have become the hotspots of synthesis due to their characteristic structure features and remarkable bioactivities.8, 9, 10, 11 Marine actinomycetes are regarded as an important resource in the quest for new bioactive natural products, including alkaloids, peptides, amides and quinones, which show antimicrobial, antioxidant, anti-inflammatory and cytotoxic activities.12, 13 Our previous study on the secondary metabolites of the marine actinomycete Streptomyces sp. SCSGAA 0027 led to the obtainment of five novel polyhydroxy polyketides nahuoic acids A–E.14 Recently, we further investigated on the strain and succeeded in obtaining five new spirocyclic polyketides pteridic acids C–G (1–5) (Figure 1). Herein, we describe the isolation, structure elucidation and bioactivities of 1–5.

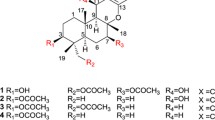
Structures of the isolated compounds 1−5.
Results and discussion
Structural elucidation of compounds 1–5
The molecular formula of pteridic acid C (1) was assigned as C19H32O6 by HR–ESI–MS with four degrees of unsaturation. An initial examination of the 1H NMR spectral data of 1 (Table 1) displayed obvious proton signals for one primary methyl group H3-19 (δH 0.76); four secondary methyl groups H3-14 (δH 1.03), H3-16 (δH 0.92), H3-17 (δH 0.80) and H3-18 (δH 0.84); and two olefin protons H-2 (δH 5.74) and H-3 (δH 6.83). The 13C NMR spectral data of 1 (Table 2) showed carbon signals for a carboxyl group C-1 (δC 167.2); two olefinic methines C-2 (δC 121.4) and C-3 (δC 152.4), eight methines including four oxymethines C-5 (δC 75.3), C-7 (δC 71.4), C-11 (δC 63.7) and C-13 (δC 67.4); two methylenes C-10 (δC 32.8) and C-15 (δC 18.9); and five methyl groups C-14 (δC 19.4), C-16 (δC 15.0), C-17 (δC 5.0), C-18 (δC 12.5) and C-19 (δC 9.2), as evidenced by HSQC spectrum. The planar structure of 1 was established by the analysis of its 2D-NMR data. The COSY cross-peaks [H-2/H-3/H-4/H-5/H-6/H-7/H-8/H3-18, H-4/H3-16, H-6/H3-17, H-7/7-OH] permitted the assignment of the 10-carbon fragment possessing a double bond, whereas the additional cross-peaks [H2-10/H-11/H-12/H-13/H3-14, H-11/11-OH, H-12/H2-15/H3-19] (Figure 2) provided the second spin system with 7-carbon unit. A final exchangeable proton resonance at δH 12.01 was assigned to a terminal carboxylic acid, whose carbon appeared at δC 167.2 (C-1). Furthermore, the observation of HMBC correlations from H-2 and H-3 to C-1, and from methyl doublet H3-16 to C-3/C-4/C-5 indicated the carboxylic acid group attached on C-2. The carboxylic acid and one alkene accounted for only two of the four sites of unsaturation required by the molecular formula, requiring that 1 had to contain two rings. Considering the non-protonated carbon atom C-9 with a chemical shift value of δC 101.3 and the observable HMBC correlations showing methyl doublet H3-18 to C-7/C-8/C-9, and both H-8 and H2-10 to C-9, which suggested a 6,6-spiroketal pattern formed through the ketal carbon C-9.

Key HMBC (↷) and COSY (—) correlations of 1 and 4.
The partial relative configuration of 1 was determined by the analysis of its NOESY spectrum and J-value data. The NOE correlations between H3-18 and H-7, between H3-17 and H-8, and the vicinal coupling constant value of J7–8 (11.5 Hz), indicated the six-membered O-heterocyclic ring encompassing C-5 to C-9 was in a chair conformation where the protons H-7/H-8 were in axial positions, whereas H3-18/7-OH were in equatorial positions. Additional observation of the NOE correlation (Figure 3) between H3-17 and 7-OH, and the relative small J-values for JH-5, H-6 (2.0 Hz) and JH-6,H-7 (4.5 Hz), suggested H-5 and H3-17 orientated axially, whereas H-6 orientated equatorially. Hence, the relative configuration of the aforementioned six-membered ring was defined. In a similar way, the relative configuration of the six-membered ring possessing C-9 to C-13 was established. Analysis of the 1H NMR data of 1 showing the coupling constants of JH-10ax, H-11 and JH-10eq, H-11 with values of 11.5 and 4.0 Hz, respectively, and the observation of NOE correlation of H-10 and H-5, demonstrated that the six-membered ring composed of C-9 to C-13 also preferred a chair conformation where the protons H-10ax (δH 1.15) and H-11 positioned at axial positions while the H-10eq (δH 2.10) and 11-OH were in equatorial positions. The cross-peak between H-12 and H3-14 in the NOESY spectrum of 1 was obscured by noise. However, the coupling constant value of J12–13 (10.5 Hz) clearly suggested that H-12/H3-14 were on the same side with H-12 axial and H3-14 equatorial, whereas H-13 was on the other side with H-13 axial. Further the relative configuration of the spiro junction of the two six-membered rings was established from the key NOE correlation between H-5 and H-10eq. Most notably, although the NOE correlation from H3-17 to H-4 was observable and the coupling constant between H-4 and H-5 (JH-4, H-5=10.0 Hz) was large, the relative configuration of C-4 was still not determined due to the presence of a rotatable side chain. This issue is further addressed below. Thus, the relative configuration of spiral core was preliminarily defined as 5S*, 6S*, 7R*, 8S*, 9S*, 11R*, 12S* and 13R*.

Key NOESY correlations for compound 1. A full color version of this figure is available at The Journal of Antibiotics journal online.
The molecular formula of pteridic acid D (2) was assigned as C18H30O6 by HR–ESI–MS. The 1H and 13C NMR spectral data of 2 (Tables 1 and 2) were very similar to those of 1 with the only obvious difference of the absence of a shielded methylene. Detailed analysis of 2D NMR spectra revealed that the planar structure of 2 was a methyl substituted analogue of 1 at C-12. This was supported by the observation of HMBC correlations from H3-15 (δH 0.80) to C-11 (δC 67.1)/C-12 (δC 45.5)/C-13 (δC 70.1). For determining the relative configuration of spiral core of 2, a NOESY experiment was performed. Unfortunately, the cross-peaks shown in the NOESY spectrum of 2 (see Supplementary Information) were concealed by noise. However, the coupling constants of protons in the 1H NMR spectrum of 2 were closely resembled to those of corresponding protons of 1 (Table 1), which indicated the relative configuration of the spiro core of 2 was the same as that of 1.
The molecular formula of pteridic acids E (3) was assigned as C20H32O6 by HR–ESI–MS. Careful comparison of the 1H and 13C NMR spectroscopic data of 3 (Tables 1 and 2) and 2 revealed that their structures were highly similar, except the additional presence of two deshielded methines in the side chain of 3. The COSY cross-peaks of H-2/H-3/H-4/H-5/H-6/H-7 indicated the connectivity of C-2/C-3/C-4/C-5/C-6/C-7 at the long side chain. This was proved by the HMBC correlations of H-2 (δH 5.82) with C-1 (δC 167.6)/C-4 (δC 128.0), H-3 (δH 7.11) with C-1, H-5 with C-3 (δC 144.7) and H3-17 with C-5 (δC 147.8)/C-6 (δC 38.6)/C-7 (δC 75.2). In addition, the 1H and 13C NMR of 3 were also similar to those of pteridic acids A and B,15 and the specific difference between them was the double bond between C-12 and C-13 in pteridic acids A and B was reduced to be a single bond in 3.
Pteridic acid F (4) had a molecular formula of C21H34O6 as determined by HR–ESI–MS. The 1H and 13C NMR spectroscopic data of 4 (Tables 1 and 2 showed close similarity to those of 3. Detailed analysis of 2D NMR) data demonstrated that the only difference between them was an ethyl group substituted at C-14 of 4 instead of a methyl group. This was confirmed by the HMBC correlations from H3-21 (δH 0.71) to C-14 (δC 50.4)/C-17 (δC 18.6).
Pteridic acid G (5) had a molecular formula of C22H36O6 as determined by HR–ESI–MS. The 1H and 13C NMR spectroscopic data of 5 (Tables 1 and 2) were resembled to those of 4, except for the additional appearance of an oxygenated methyl group (δH 3.62, δC 51.6). The HMBC spectrum showed correlation from methyl singlet with chemical shift value of δH 3.62 to C-1 (δC 166.7), suggesting the methyl esterification of carboxylic acid group in 5. To assess whether 5 was an artifact of isolation, 4 was dissolved in MeOH solvent and then cultured at room temperature for more than 40 days. Monitoring by HPLC indicated a few conversion yields of 4–5, which indicated that 5 might be an artifact of 4.
The relative configurations of spiro core for 3–5 were determined to be the same as that of 1 by their consistent NOE data and coupling constants.
For 1–5, the geometries of the double bond Δ2, 3 for 1–2 and Δ2, 3 and Δ4, 5 for 3−5 were assigned as E configuration on the basis of the vicinal coupling constant with the value(s) of J2–3 (15.5 Hz)/J4–5 (15.0 Hz), respectively.
The determinations of the absolute configurations of 1–5 were separated into two distinct parts of the spiro core and the lone chiral center at the side chain. The absolute configuration of the spiro core was established using the modified Mosher’s ester method. Compound 5 was selected to perform a chemical derivatization of bis-MTPA esters as its carboxyle group was esterified. Treatment of 5 with (R)-MTPA-Cl and (S)-MTPA-Cl, under standard acylation conditions, afforded 9,13-bis-(S)-MTPA-ester (5a) and 9,13-bis-(R)-MTPA-ester (5b), respectively. The 1H chemical shifts around the C-9 and C-13 were assigned by 1H NMR spectroscopic analysis (see Supplementary Information), and the analysis of ΔδS-R values allowed the assignment of the absolute configurations of C-9 and C-13 to be R (Figure 4). Correspondingly, the absolute configurations of C-7/C-8/C-10/C-11/C-14/C-15 were established as 7S, 8S, 10S, 11S, 14S, 15R. Some efforts were made to protect the carboxylic acid group followed by the modified Mosher’s ester derivatization for 1–4. However, due to the low yields of products in the methyl esterification reaction that the samples were treated with 1% sulfuric acid in MeOH and incubated for 5 h at 70 °C, there were limited amount of the samples to perform Mosher’s ester experiments. Thus, the absolute configurations of the spiro cores for 1–4 were considered to be the same as that of 5 considering their identical biosynthesis pathways and their accordant directions of specific rotations.

ΔδS-R values for MTPA esters of 5a and 5b in MeOH.
The absolute configurations of the lone chiral center at the side chain for 1–5 were determined by comparing their experimental electronic circular dichroism (ECD) spectra with calculated ECD spectra. For 1 and 2, two possible stereoisomers at C-4 of the (4S, 5S, 6S, 7R, 8S, 9S)-1/2 and (4R, 5S, 6S, 7R, 8S, 9S)-1/2, in which the configurations for C-11/C-12/C-13 were disregarded, were built and separately subjected to the Molecular Merck force field conformational search followed by geometry optimization using the density functional theory (DFT) method. The resulting low-energy conformers were applied to time-dependent DFT calculations to give two mirror-imaged ECD spectra as shown in Figure 5, which showed a high similarity between the calculated spectrum of the enantiomer (4S, 5S, 6S, 7R, 8S, 9S)-1/2 and the experimental spectra of 1 and 2. Both the calculated and experimental data showed a negative Cotton effect near 210 nm. Therefore, the absolute configurations of C-4 for 1 and 2 were defined as S together. In a similar pattern, the calculated ECD spectrum generated by (6S, 7S, 8S, 9R, 10S, 11S)-3–5 was in agreement with the experimental ones showing a negative Cotton effect near 260 nm, which permitted the absolute configuration of C-6 to be S as depicted in Figure 6. Based on the above results, the complete structures of 1–5 were established. In summary, The reports about 6,6-spiroketal compounds with so much more chiral centers on their O-heterocyclic ring were relatively less and were found in enteridinines,16 oligomycin A,17 streptospirodienoic acids18 and spirangiens.2 Compounds 1–5 extended the members of this family by derivation of different substituents.

Comparison of CAM-B3LYP/TZVP calculated ECD spectra for (4S, 5S, 6S, 7R, 8S, 9S)-1 and 2, and (4R, 5S, 6S, 7R, 8S, 9S)-1 and 2 with the experimental spectra of 1 and 2 in MeOH. A full color version of this figure is available at The Journal of Antibiotics journal online.

Comparison of PBE1PBE/TZVP calculated ECD spectra for (6S, 7S, 8S, 9R, 10S, 11S)-3–5 and (6R, 7S, 8S, 9R, 10S, 11S)-3–5 with the experimental spectra of 3–5 in MeOH. A full color version of this figure is available at The Journal of Antibiotics journal online.
Bioassays
The isolated compounds were tested for their antimicrobial activities against two gorgonian pathogenic fungal strains Aspergullus versicolor SCSGAF 0096 and Aspergullus sydowii SCSGAF 0035. A human pathogenic fungal strain Candida albicans SC5314 and two bacterial strains Escherichia coli and Bacillus subtilis were also tested. The results showed that only compounds 3–5 had weak antibacterial activity against B. subtilis with inhibition zones of 8, 7 and 7 mm, respectively, at the equal concentration of 50 μg per disc, whereas the positive control penicillin showed a inhibition zone of 20 mm at the same dosage. Their cytotoxicity toward the human carcinoma A549, HL-60, K562 and MCF-7 cell lines, and the antiviral activity against herpes simplex virus type 1 were also evaluated. Furthermore, none of these compounds exhibited evident activity. It is notable that the structurally similar pteridic acids A and B had been reported as potent plant growth promoters.15 During the current study, the above activity was not performed due to the lack of relevant experimental condition.
Experimental procedures
General methods
Optical rotations were measured with a MCP 500 polarimeter (Anton Paar, Graz, Austria). UV spectra were recorded using a UV-2600 spectrophotometer (Shimadzu, Kyoto, Japan). ECD spectra were measured with a Chirascan circular dichroism spectrometer (Applied Photophysics, Ltd, London, UK). IR spectra were determined with a FT-IR-NICOLET spectrophotometer. 1H, 13C NMR and 2D NMR spectra were acquired with a Bruker AV-500/700 MHz NMR spectrometer (Bruker, Switzerland) with TMS as reference. ESI–MS and HR–ESI–MS spectroscopic data were acquired with an amaZon SL ion trap mass spectrometer and Ma**s quadrupole-time-of-flight MS (Bruker Daltonics Inc, Bill Ricard, MA, USA), respectively. Semi-preparative reversed-phase HPLC was performed on a Shimadzu LC-20 A preparative liquid chromatography system with an YMC-Pack octadecyl silane column, 250 × 20 mm i.d., S-5 μm. Reversed-phase medium-pressure preparative LC was carried out using the CHEETAH MP200 system (Agela Technologies Ltd, Tian**, China) and Claricep Flash columns filled with octadecyl silane (40–63 μm, YMC). Sephadex LH-20 (GE Healthcare, London, UK) was used for column chromatography column. Silica gel (200−300 mesh) for chromatography column and GF254 for TLC were obtained from Yantai Jiangyou Silica Gel Development Co., Ltd, (Qingdao, China). OD measurements were determined on an enspire multimode microplate reader (varioskan flash).
Fungal material
Strain SCSGAA0027 was isolated from the South China Sea (18°11′ N, 109°25′ E) gorgonian coral Melitodes squamata. The strain (GenBank accession JN049466) was identified as a Streptomyces sp. based on NCBI BLAST analysis of its partial 16S rRNA sequence, which is 99% identical with that of the strain Streptomyces fradiae NBRC 12773 (NR112270). Strain SCSGAA0027 was deposited in the RNAM center, South China Sea Institute of Oceanology, Chinese Academy of Sciences.
Fermentation and extraction
Strain Streptomyces sp. SCSGAA0027 was cultured in 2500 replicate 500 ml Erlenmeyer flasks each containing 120 ml of modified ISP2 fermentation medium (yeast extract 0.4%, malt extract 0.5%, glucose 0.4%, artificial sea salt 1.5%, vitamin B1 0.01%, vitamin B6 0.01%, vitamin lactoflavin 0.01%, nicotinic acid 0.01%, biotin 0.01%, phenylalanine 0.01%, alanine 0.03%, pH 7.2−7.4). These cultures were grown on a rotary shaker (200 r.p.m.) at 28 °C for 5 days. At the end of the fermentation period, the culture broth was separated from the mycelium by filtration. The mycelium was extracted with acetone, while the culture filtrate was extracted with EtOAc. A combined organic extract was acquired.
Isolation and purification
The total extract (50 g) was adsorbed on silica gel (YMC-gel ODS-A, 30 g) and subjected to a reversed-phase medium-pressure LC, eluting with a step gradient of MeOH/H2O solvent mixtures (increasing the MeOH by 20% per 3000 ml from 30% to 100% MeOH, 40 ml min−1 flow rate). The 50% MeOH/H2O fraction (8 g) was further fractionated by reversed-phase medium pressure LC with a MeCN/H2O gradient solvent system (from 20 to 60% acetonitrile, 20 ml min−1 flow rate) to obtain four subfractions (F-1, F-2, F-3 and F-4). Fraction F-1 was further purified by repeated semi-preparative reversed-phase HPLC (YMC-Pack octadecyl silane column, 250 × 20 mm i.d., S-5 μm; 5 ml min−1 flow rate) with MeCN/H2O (28: 72) to obtain 1 (3 mg, tR=30 min), 2 (2.3 mg, tR=24 min). Fraction F-2 (800 mg) was sub-fractionated by a normal-phase medium pressure liquid chromatography with a step gradient of chloroform/MeOH mobile phase (increasing the MeOH by 5% per 200 ml from 0 to 50% MeOH, 10 ml min−1 flow rate), affording four subfractions (F-2-1, F-2-2, F-2-3 and F-2-4). Compounds 3 (2 mg, tR=28 min) and 4 (3 mg, tR=40 min) were obtained from fraction F-2-2 by repeated semi-preparative reversed-phase HPLC with MeCN/H2O (31: 69). Compound 5 (3.3 mg, tR=35 min) was obtained from fraction F-2-3 by repeated semi-preparative reversed-phase HPLC with MeCN/H2O (45: 55).
Syntheses and derivatizations
Preparation of Mosher’s Esters 5a and 5b
Compound 5 (0.8 mg) was dissolved in 600 μl of pyridine, and dimethylaminopyridine (1 mg) and (R)-MTPA-Cl (20 μl) were then added in sequence. The reaction mixture was stirred at rt for 24 h and afterward quenched with two drops of water. The solution was purified by C-18 RP-HPLC (90:10 MeCN/H2O) to obtain a 9,13-bis-(S)-MTPA ester derivative, 5a (1 mg, tR=26 min). In the manner described for 5a, another portion of 5 (0.8 mg) was treated with 600 μl of pyridine, dimethylaminopyridine (1 mg) and (S)-MTPA-Cl (20 μl). The reaction was performed at the same conditions as above and the major product was purified by C-18 RP-HPLC (90:10 MeCN/H2O) to yield the 9,13-bis-(R)-MTPA ester derivative 5b (1 mg, tR=25 min).
Compound characterization
Actinospiroketal A (1)
Pale yellow oil; [α]25D−14 (c 0.43, MeOH); UV (MeOH) λmax (log ɛ) 225 (3.81) nm; ECD (1.19 mM, MeOH) λmax (Δɛ) 213 (−4.51) nm; IR (MeOH) νmax: 3410, 2970, 1967 and 1014 cm−1; 1H and 13C NMR data, see Tables 1 and 2; HRESIMS m/z 379.2097 [M+Na]+ (calcd for C19H32O6Na, 379.2091).
Actinospiroketal B (2)
Pale yellow oil; [α]25D−32 (c 0.12, MeOH); UV (MeOH) λmax (log ɛ) 207 (4.02) nm; ECD (1.12 mM, MeOH) λmax (Δɛ) 212 (−5.22) nm; IR (MeOH) νmax: 3392, 2970, 1967 and 1014 cm−1; 1H and 13C NMR data, see Tables 1 and 2; HRESIMS m/z 365.1925 [M+Na]+ (calcd for C18H30O6Na, 365.1935).
Actinospiroketal C (3)
Pale yellow oil; [α]25D−8 (c 0.10, MeOH); UV (MeOH) λmax (log ɛ) 260 (4.06), 202 (6.66) nm; ECD (0.72 mM, MeOH) λmax (Δɛ) 262 (−1.30) nm; IR (MeOH) νmax: 3365, 2974, 1967, 1670, 1004 and 667 cm−1; 1H and 13C NMR data, see Tables 1 and 2; HRESIMS m/z 391.2091 [M+Na]+ (calcd for C20H32O6Na, 391.2091).
Actinospiroketal D (4)
Pale yellow oil; [α]25D−14 (c 0.25, MeOH); UV (MeOH) λmax (log ɛ) 258 (4.21), 202 (7.06) nm; ECD (0.93 mM, MeOH) λmax (Δɛ) 262 (−1.96) nm; IR (MeOH) νmax: 3334, 1635, 1014, 667 cm−1; 1H and 13C NMR data, see Tables 1 and 2; HRESIMS m/z 405.2247 [M+Na]+ (calcd for C21H34O6Na, 405.2248).
Actinospiroketal E (5)
Pale yellow oil; [α]25D−26 (c 0.17, MeOH); UV (MeOH) λmax (log ɛ) 258 (4.41), 202 (6.17) nm; ECD (0.64 mM, MeOH) λmax (Δɛ) 262 (−2.93) nm; IR (MeOH) νmax: 3345, 3410, 2974, 1697, 1970 and 1004 cm−1; 1H and 13C NMR data, see Tables 1 and 2; HRESIMS m/z 419.2421 [M+Na]+ (calcd for C22H36O6Na, 419.2404).
(S)-MTPA ester of 5 (5a)
1H NMR (methanol-d4, 700 MHz) δH 7.361 (1H, dd, J=10.5, 15.0 Hz, H-3), 6.413 (1H, dd, J=9.0, 15.0 Hz, H-5), 6.361 (1H, dd, J=10.5, 15.0 Hz, H-4), 5.880 (1H, d, J=15.0 Hz, H-2), 5.118 (1H, m, H-13), 5.094 (1H, dd, J=4.0, 12.0 Hz, H-9), 3.924 (1H, dq, J=6.0, 10.0 Hz, H-15), 3.569 (1H, dd, J=1.0, 10.0 Hz, H-7), 2.519 (1H, m, H-6), 2.557 (1H, dd, J=4.0, 13.0 Hz, H-12eq), 2.354 (1H, m, H-8), 1.908 (1H, dd, J=6.0, 12.0 Hz, H-10), 1.499 (1H, dd, J=12.0, 13.0 Hz, H-12ax), 1.258 (1H, overlapped, H-14), 1.080 (3H, d, J=6.0 Hz, H-16), 1.062 (1H, overlapped, H-17α), 1.059 (3H, d, J=7.0 Hz, H-18), 0.951 (3H, d, J=7.0 Hz, H-19), 0.902 (1H, overlapped, H-17β), 0.724 (3H, d, J=7.0 Hz, H-20), 0.461 (3H, t, J=7.0 Hz, H-21).
(R)-MTPA ester of 5 (5b)
1H NMR (methanol-d4, 700 MHz) δH 7.288 (1H, dd, J=10.0, 15.0 Hz, H-3), 6.340 (1H, overlapped, H-5), 6.345 (1H, overlapped, H-4), 5.855 (1H, d, J=15.0 Hz, H-2), 5.107 (1H, dt, J=3.5, 11.0 Hz, H-13), 5.017 (1H, dd, J=5.0, 12.0 Hz, H-9), 3.963 (1H, dq, J=6.0, 10.0 Hz, H-15), 3.541 (1H, dd, J=1.0, 10.0 Hz, H-7), 2.492 (1H, m, H-6), 2.533 (1H, dd, J=4.0, 13.0 Hz, H-12eq), 2.332 (1H, m, H-8), 1.934 (1H, dd, J=6.0, 12.0 Hz, H-10), 1.365 (1H, overlapped, H-12ax), 1.348 (1H, overlapped, H-14), 1.131 (3H, d, J=6.0 Hz, H-16), 1.315 (2H, overlapped, H-17α), 1.032 (3H, d, J=7.0 Hz, H-18), 0.709 (3H, d, J=7.0 Hz, H-19), 0.902 (1H, overlapped, H-17β), 0.877 (3H, d, J=7.0 Hz, H-20), 0.699 (3H, t, J=7.5 Hz, H-21).
Biological assays
Antimicrobial assay
The isolated compounds were tested for their antimicrobial activities against two gorgonian pathogenic fungal strains A. versicolor SCSGAF 0096, A. sydowii SCSGAF 0035, a human pathogenic fungal strain C. albicans SC5314 and two bacterial strains E. coli and B. subtilis using a standard disc diffusion assay.19 The equal one hundred-microliter volumes of fungal spore suspension (containing 1 × 106 spores ml−1) or bacterium solution (108 CFU ml−1) were tiled on 100 mm sterile Petri plates with 20 ml corresponding nutrient agar. Sterile 6 mm diameter circular discs of filter paper were loaded with 50 μg of tested compounds dissolved in dimethyl sulfoxide onto the fungal and bacterial lawn, respectively. An additional set of discs containing 50 μg of standard fungicides amphotericin B or ketoconazole and standard antibiotic penicillin were used as the positive controls. The fungal agar plates were incubated for 48 h at 30 °C while the bacterial plates were cultured for 24 h at 30 °C, and then the inhibition zones were measured at the end of the incubation period. Each test was performed in three independent replicates.
Cytotoxic assay
Cytotoxic activity was evaluated using human carcinoma A549, HL-60, K562 and MCF-7 cell lines by the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) methods.20
Antiviral assay
Antiviral assay was performed with minor modifications herpes simplex virus type 1 (15577 strain) suspension (4 × 104 plaque-forming unit) pre-incubated with or without various concentrations of samples at 37 °C for 2 h. Then the mixtures were diluted (1:1000) and their residual infectivity was determined by plaque reduction assay.21
Computational methods
Molecular Merck force field and DFT/time-dependent DFT calculations were performed with Spartan'14 software package (Wavefunction, Inc., Irvine, CA, USA) and Gaussian09 program package,22 respectively, using default grids and convergence criteria. Molecular Merck force field conformational search generated low-energy conformers within a 10 kcal mol−1 energy window were subjected to geometry optimization using DFT method at the B3LYP/6-31G (d) level. Frequency calculations were run at the same level to estimate their relative thermal free energies (ΔG) at 298.15 K. Energies of the low-energy conformers in MeOH were re-calculated at the B3LYP/def2-TZVP level. Solvent effects were taken into account by using polarizable continuum model. The time-dependent DFT calculations were performed using the hybrid M06,23 PBE1PBE24, 25 and the long-range corrected hybrid CAM-B3LYP26 functionals, and the Ahlrichs’ basis set TZVP (triple zeta valence plus polarization).27 The number of excited states per each molecule was 8 or 12. The ECD spectra were generated by the program SpecDis28 using a Gaussian band shape with 0.30 eV exponential half-width from dipole-length dipolar and rotational strengths. The equilibrium population of each conformer at 298.15 K was calculated from its relative free energies using Boltzmann statistics. The calculated spectra of compounds were generated from the low-energy conformers according to the Boltzmann weighting of each conformer in MeOH solution. In order to reduce computational cost, the structures of 1–5 was truncated by replacement of both 12-Et and 13-Me by a hydrogen, and the truncated structures were used for theoretical calculations.
References
Sperry, J., Wilson, Z. E., Rathwellab, D. C. K. & Brimble, M. A. Isolation, biological activity and synthesis of benzannulated spiroketal natural products. Nat. Prod. Rep. 27, 1117–1137 (2010).
Bruns, N. et al. Spirangien derivatives from the myxobacterium sorangium cellulosum: isolation, structure elucidation, and biological activity. Eur. J. Org. Chem. 4, 847–857 (2015).
Sato, S., Iwata, F., Yamada, S. & Katayama, M. Neomaclafungins A–I: oligomycin-class macrolides from a marine-derived actinomycete. J. Nat. Prod. 75, 1974–1982 (2012).
**ong, L. et al. Leonuketal, a spiroketal diterpenoid from Leonurus japonicus. Org. Lett. 17, 6238–6241 (2015).
Williams, D. E. et al. Spirastrellolide A: revised structure, progress toward the relative configuration, and inhibition of protein phosphatase 2A. Org. Lett. 6, 2607–2610 (2004).
Fremlin, L. et al. Reveromycins revealed: new polyketide spiroketals from Australian marine-derived and terrestrial Streptomyces spp. A case of natural products vs. artifacts. Org. Biomol. Chem. 9, 1201–1211 (2011).
Rho, J. R. et al. Phorbaketals A, B, and C, sesterterpenoids with a spiroketal of hydrobenzopyran moiety isolated from the marine sponge Phorbas sp. Org. Lett. 11, 5590–5593 (2009).
Frank, B. et al. Spiroketal polyketide formation in sorangium: identification and analysis of the biosynthetic gene cluster for the highly cytotoxic spirangienes. Chem. Biol. 14, 221–233 (2007).
Guérinot, A., Lepesqueux, G., Sablé, S., Reymond, S. & Cossy, J. Synthetic efforts toward the spiroketal core of spirangien A. J. Org. Chem. 75, 5151–5163 (2010).
Yadav, J. S., Reddy, N. M., Rahman, M. A. & Prasad, A. R. Synthesis of the spiroketal fragment of (–)-ushikulide. Eur. J. Org. Chem. 25, 5574–5581 (2014).
Butler, B. B., Manda, J. N. & Aponick, A. Synthesis of the spirastrellolide A, B/C spiroketal: enabling solutions for problematic Au(I)-catalyzed spiroketalizations. Org. Lett. 17, 1902–1905 (2015).
Blunt, J. W., Copp, B. R., Keyzers, R. A., Munro, M. H. G. & Prinsep, M. R. Marine natural products. Nat. Prod. Rep. 33, 382–431 (2016).
Vinothkumar, S. & Parameswaran, P. S. Recent advances in marine drug research. Biotechnol. Adv. 31, 1826–1845 (2013).
Nong, X. H., Zhang, X. Y., Xu, X. Y., Wang, J. & Qi, S. H. Nahuoic acids B–E, polyhydroxy polyketides from the marine-derived Streptomyces sp. SCSGAA 0027. J. Nat. Prod. 79, 141–148 (2016).
Igarashi, Y., Iida, T., Yoshida, R. & Furumai, T. Pteridic acids A and B, novel plant growth promoters with auxin-like activity from Streptomyces hygroscopicus TP-A0451. J. Antibiot. 55, 764–767 (2002).
Řezanka, T., Dvořáková, R., Hanuš, L. O. & Dembitsky, V. P. Enteridinines A and B from slime mold Enteridium lycoperdon. Phytochemistry 65, 455–462 (2004).
Wagenaar, M. M., Williamson, R. T., Ho, D. M. & Carter, G. T. Structure and absolute stereochemistry of 21-hydroxyoligomycin A. J. Nat. Prod. 70, 367–371 (2007).
Chen, Y. P. et al. Streptospirodienoic acids A and B, 6,6-spiroketal polyketides from Streptomyces sp. RSC Adv. 4, 63324–63327 (2014).
Acar, J. F., Goldstein, F. W., Kitzis, M. D. & Eyquem, M. T. Resistance pattern of anaerobic bacteria isolated in a general hospital during a two-year period. J. Antimicrob. Chemother. 8, 9–16 (1981).
Pauwels, R. et al. Rapid and automated tetrazolium-based colorimetric assay for the detection of anti-HIV compounds. J. Virol. Methods 20, 309–321 (1988).
Ma, F. et al. Anti-HSV activity of kuwanon X from mulberry leaves with genes expression inhibitory and HSV-1 induced NF-κB deactivated properties. Biol. Pharm. Bull. 39, 1667–1674 (2016).
Frisch, M. J. et al Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford, CT, USA, (2010).
Zhao, Y. & Truhlar, D. G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 120, 215–241 (2008).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Adamo, C. & Barone, V. Toward reliable density functional methods without adjustable parameters: the PBE0 model. J. Chem. Phys. 110, 6158–6170 (1999).
Yanai, T., Tew, D. P. & Handy, N. C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 393, 51–57 (2004).
Schäfer, A., Huber, C. & Ahlrichs, R. J. Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr. Chem. Phys. 100, 5829–5835 (1994).
Bruhn, T., Schaumlöffel, A., Hemberger, Y. & Bringmann, G. SpecDis: quantifying the comparison of calculated and experimental electronic circular dichroism spectra. Chirality 25, 243–249 (2013).
Acknowledgements
We are grateful for the financial support provided by the National Natural Science Foundation of China (41376160 and 81673326), National Natural Science Foundation of China (41606100), Strategic Leading Special Science and Technology Program of Chinese Academy of Sciences (XDA100304002), Natural Science Foundation of Guangdong Province, China (2015A030310349) and Regional Innovation Demonstration Project of Guangdong Province Marine Economic Development (GD2012-D01-002). We appreciate the contribution of the analytical facility center (**ao Zhihui, Li Chuanrong, Sun Aijun and Zhang Yun) of South China Sea Institute of Oceanology, Chinese Academy of Sciences for acquiring NMR, HRESIMS and experimental CD data. We are also grateful for the support given by the Guangzhou Branch of the Supercomputing Center of Chinese Academy of Sciences.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on The Journal of Antibiotics website
Supplementary information
Rights and permissions
About this article

Cite this article
Nong, XH., Wei, XY. & Qi, SH. Pteridic acids C–G spirocyclic polyketides from the marine-derived Streptomyces sp. SCSGAA 0027. J Antibiot 70, 1047–1052 (2017). https://doi.org/10.1038/ja.2017.105
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ja.2017.105
- Springer Japan KK
This article is cited by
-
Development of a drug discovery approach from microbes with a special focus on isolation sources and taxonomy
The Journal of Antibiotics (2023)
-
Iseoic acids and bisiseoate: three new naphthohydroquinone/naphthoquinone-class metabolites from a coral-derived Streptomyces
The Journal of Antibiotics (2023)
-
Streptomyces alleviate abiotic stress in plant by producing pteridic acids
Nature Communications (2023)
-
Isolation and structure determination of allopteridic acids A–C and allokutzmicin from an unexplored actinomycete of the genus Allokutzneria
The Journal of Antibiotics (2023)