
Abstract
Endoplasmic reticulum (ER) stress is involved in inflammation-induced neurotoxicity. Mitofusin 2 (Mfn2), a member of the GTPase family of proteins, resides in the ER membrane and is known to regulate ER stress. However, the potential role and underlying mechanism of Mfn2 in inflammation-induced neuronal dysfunction is unknown. In our study, we explored the potential of Mfn2 to attenuate inflammation-mediated neuronal dysfunction by inhibiting ER stress. Our data show that Mfn2 overexpression significantly ameliorated tumor necrosis factor alpha (TNFα)-induced ER stress, as indicated by the downregulation of the ER stress proteins PERK, GRP78 and CHOP. Mfn2 overexpression also prevented the TNFα-mediated activation of caspase-3, caspase-12 and cleaved poly (ADP-ribose) polymerase (PARP). Cellular antioxidant dysfunction and reactive oxygen species overproduction were also improved by Mfn2 in the setting of TNFα in mouse neuroblastoma N2a cells in vitro. Similarly, disordered calcium homeostasis, indicated by disturbed levels of calcium-related proteins and calcium overloading, was corrected by Mfn2, as evidenced by the increased expression of store-operated calcium entry (SERCA), decreased levels of inositol trisphosphate receptor (IP3R), and normalized calcium content in TNFα-treated N2a cells. Mfn2 overexpression was found to elevate Yes-associated protein (Yap) expression; knockdown of Yap abolished the regulatory effects of Mfn2 on ER stress, oxidative stress, calcium balance, neural death and inflammatory injury. These results lead us to conclude that re-activation of the Mfn2–Yap signaling pathway alleviates TNFα-induced ER stress and dysfunction of mouse neuroblastoma N2a cells. Our findings provide a better understanding of the regulatory role of Mfn2–Yap–ER stress in neuroinflammation and indicate that the Mfn2–Yap axis may be a focus of research in terms of having therapeutic value for the treatment of neurodegenerative diseases.
Similar content being viewed by others


Introduction
Huntington’s disease (HD), a neurodegenerative disorder primarily caused by instable and abnormally long expansion of the polyglutamine (polyQ) tract, is characterized by a chronic loss of neurons in the basal ganglia [1]. Motor ability and cognitive function are impaired during the progression of HD. Emerging evidence suggests that neuroinflammation due to neuro-immune interactions plays a pivotal role in the pathogenesis of intractable neurogenic disorders, especially in the development of HD. Uncontrolled inflammation evokes reactive oxygen species (ROS) overloading which in turn mediates oxidative stress in neurons, an effect that is accompanied with neuronal dysfunction and/or death. Inflammatory cytokines are also released from inflammatory cells and enhance the inflammation-mediated damage in the brain. Given the functional significance of inflammatory injury in the pathogenesis of HD, investigation of the pathophysiological mediator(s) of neuroinflammation is a primary focus of researchers .
Mitofusin 2 (Mfn2), which resides in the outer membrane of the endoplasmic reticulum (ER), plays a pivotal role in ER function, such as calcium balance and protein synthesis. Mfn2 is also involved in mitochondrial regulation, such as mitochondrial fusion, mitophagy and mitochondrial fission, and it has been shown to have anti-inflammatory actions in several disease models [2, 9]. For example, Mfn2 activation attenuates tumor necrosis factor alpha (TNFα)-mediated hair follicle stem cell apoptosis [2, 3]; Mfn2 overexpression sustains mitochondrial function and elevates the cellular survival rate in lipopolysaccharide-treated human kidney-2 cells [4, 5]; in hyperglycemic stress, increased Mfn2 interrupts inflammatory signals in pancreatic β-cells [6]. In addition, the anti-inflammatory property of Mfn2 has also been noted in pulmonary arterial hypertension [7], airway smooth muscle [8] and alveolar epithelial cells [9]. Although a number of studies have indicated that Mfn2 is associated with inflammation-mediated cell stress, the potential role and underlying mechanism of Mfn2 in neuronal dysfunction resulting from inflammation are not yet fully characterized.
The ER, an intracellular organelle, executes several essential functions in cellular pathophysiological processes [10]. The primary action of the ER is to modulate calcium balance via the recycling and releasing of calcium into the cytoplasm. ER also exerts a critical effect on protein generation, synthesis, modification and transport [11,12,13]. The primary result of ER dysfunction is ER stress, an effect that is accompanied with an accumulation of misfolded proteins and calcium overloading [14, 15]. Subsequently, disruption of calcium homeostasis has been found to be associated with oxidative stress that contributes to the activation of apoptosis in a mechanism through ER stress [3, 16]. The molecular features of ER stress-mediated apoptosis is associated with the upregulation of CHOP, an ER stress protein, and activation of caspase-12, a cysteine protease [17]. Based on previous studies, ER stress has been identified as a potential molecular mechanism that is involved the initiation and progression of several neurodegenerative disorders, such as HD [18, 19]. Given the beneficial effects of Mfn2 on ER function and the inflammation response, we investigated whether Mfn2 could attenuate inflammation-mediated neuronal dysfunction by inhibiting ER stress.
The Yes-associated protein (Yap)–Hippo signaling pathway is a novel pathway that is responsible for cell survival. Specifically, Yap regulates cerebral hypoxia–reoxygenation injury by modulating the ROCK1/F-actin pathways [20, 21]. Yap activation modulates the activity of glioblastoma A172 cells in a manner dependent on the MAPK–ERK axis [22, 23] and is also associated with microglial BV-2 cell survival through attenuation of Bnip3-related mitophagy in the setting of an inflammatory microenvironment [24, 25]. Similar results have also been found in gastric cancer [26] and rectal cancer [27]. In the study reported here, we investigated whether Yap is augmented by Mfn2 and promotes the survival of neurons in the context of inflammatory injury.
Materials and methods
Cell culture and treatments
Mouse neuroblastoma N2a cells (ATCC® CCL-131™; American Type Culture Collection, Manassas, VA, USA) were cultured in high-glucose Dulbecco's Modified Eagle's medium (DMEM; Gibco, Grand Island, NY, USA) containing 10% fetal bovine serum (Gibco). After the cells reached 80% confluence, different doses of TNFα were added into the medium for 12 h based on a previous report [28].
Cell viability and TUNEL staining
The MTT colormetric assay was used to assess metabolic activity and cellular viability. Cells were seeded onto a 96-well plate, and the MTT tetrazolium compound was then added to the medium (2 mg/ml; Sigma-Aldrich, St. Louis, MO, USA); the cells were then cultured in the dark for 4 h and then DMSO was added to the medium. The optical density of each well was observed at 490 nm using a spectrophotometer (Epoch 2; BioTek Instruments, Inc., Winooski, VT, USA) [29]. The TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling) assay was used to detect apoptotic cells. Cells were fixed in 4% paraformaldehyde at room temperature for 30 min and the tissue sliced into sections. A TUNEL kit (Roche Apoptosis Detection kit; Roche, Mannheim, Germany) was used to stain the slices according to the instructions of the manufacturer. Finally, the sections were amplified to 400×, and the apoptotic cells in at least ten fields were randomly chosen for detailed examination. The apoptotic index was taken to be the proportion of apoptotic cells to total cells, in accordance to a previous study [30].
Enzyme-linked immunosorbent assay
Mouse neuroblastoma N2a cells (1.5 × 103/well) were seeded into 24-well plates. After incubation with TNFα, the protein was isolated, and the contents of cellular antioxidants, including superoxide dismutase (SOD), glutathione (GSH), and glutathione peroxidase (GPX), were analyzed using enzyme-linked immunosorbent assay (ELISA) kits. In addition, lactose dehydrogenase content and caspase-3 activity were determined using ELISA kits purchased from Beyotime Biotechnology (Nan**g, China: LDH Cytotoxicity Assay Kit, Cat. No.: C0016; Caspase-3 Activity Assay Kit, Cat. No.: C1115), according to a previous study [31]. Caspase-12 activity was determined using the ELISA kit purchased from Abcam (Cambridge, UK: Cat. No.: #ab65664).
Transfection
Small, interfering RNA (siRNA) against the macrophage-stimulating 1 (Mst1) gene and the pDC315-Mst1 vector were obtained from GenePharm (Shanghai, China). Transfection was performed using Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA) following the manufacturer’s instructions [32]. After 6 h, the cells were transferred to complete growth medium, and 48 h later, the cells were harvested and used in subsequent experiments. The siRNA knockdown efficiency and overexpression efficiency were confirmed by western blotting [33]. Western blots were used to verify the overexpression and knockdown efficiency of recombinant human adenovirus ad-Mfn2 infection and Yap siRNA transfection, respectively.
Western blotting
Total protein was extracted by RIPA (R0010; Solarbio Science and Technology, Bei**g, China), and the protein concentration of each sample was detected using a bicinchoninic acid (BCA) kit (20201ES76; Yeasen Biotech Co., Ltd, Shanghai, China). Deionized water was added to generate 30-µg protein samples for each lane. A 10% sodium dodecyl sulphate (SDS) separation gel and a concentration gel were prepared [34]. The following diluted primary antibodies were added to the membrane and incubated overnight at 4 °C: Bcl2 (1:1000; Cell Signaling Technology Inc., Danvers, MA, USA; Cat. No: 3498), Bax (1:1000; Cell Signaling Technology; Cat. No.: 2772), pro-caspase3 (1:1000; Abcam; Cat. No.: 13847), cleaved caspase3 (1:1000; Abcam; Cat. No.: ab49822), cyt-c (1:1000; Abcam; Cat. No.: ab90529), Tom20 (1:1000; Abcam; Cat. No.: ab186735), JNK (1:1000; Cell Signaling Technology; Cat. No.: 4672), p-JNK (1:1000; Cell Signaling Technology; Cat. No.: 9251), Mst1 (1:1000; Cell Signaling Technology; Cat. No.: 3682). The membranes were washed three times with phosphate-buffered saline (PBS) (5 min each time), supplemented with horseradish peroxidase-marked second antibody (1:200; Bioss, Bei**g, China), oscillated and then incubated at 37 °C for 1 h. After incubation, each membrane was washed three times with PBS (5 min each time) and reacted with enhanced chemiluminescence solution (ECL808-25; Biomiga Inc., San Diego, CA, USA) at room temperature for 1 min; then, the extra liquor was removed, and the membranes were covered with preservative film [35]. Each membrane was observed with an X-ray machine (36209ES01; Qian Chen Biological Technology Co. Ltd., Shanghai, China) to visualize protein expression. Glyceraldehyde 3-phosphate dehydrogenase was used as the internal reference. The relative protein expression was the ratio of the gray value of the target band to the inner reference band.
Immunofluorescence
Cells were plated on glass slides in a 6-well plate at a density of 1 × 106 cells per well, following which the cells were fixed in ice-cold 4% paraformaldehyde for 30 min, permeabilized with 0.1% Triton X-100 and blocked with 2% gelatin in PBS at room temperature [36]. The cells were then incubated with the primary antibodies overnight at 4 °C: cyt-c (1:1000; Abcam; Cat. No.: ab90529), p-JNK (1:1000; Cell Signaling Technology; Cat. No.: 9251), Mst1 (1:1000; Cell Signaling Technology; Cat. No.: 3682). After being washed with PBS, the cells were incubated with secondary antibody and 4′,6-diamidino-2-phenylindole (DAPI; 1:1000 dilution in PBS) for 1 h at room temperature. Images were obtained using a fluorescence microscope [37].
Flow cytometry analysis for ROS
Cell suspensions were collected. The liquor (50 g, digested two times) was collected, centrifuged for 2 min with the supernatant removed, supplemented with the superoxide indicator dihydroethdium, incubated at room temperature for 10 min, centrifuged, and washed with PBS [38]. The cells were resuspended by adding binding buffer (1×) in the dark; then, the cells were incubated at room temperature for 30 min and filtered through a nylon mesh (40-µm well). ROS production was measured by fluorescence-activated cell sorting (FACS) [39].
Statistical analysis
The software program SPSS version 17 (IBM Corp., Armonk, NY, USA) was used for statistical analysis. The results are presented as the mean ± standard error of the mean. Data for analysis consisted of three replications, and statistical analyses were conducted using one-way analysis of variance followed by Tukey’s test to compare variable groups. p < 0.05 was considered to be statistically significant.
Results
Mfn2 and Yap are repressed by TNFα in mouse neuroblastoma N2a cells
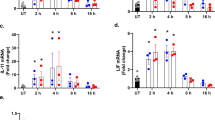
In the present study, we used TNFα (10 ng/ml for 24 h) to establish the inflammatory environment and then determined cell viability using the MTT assay. As shown in Fig. 1a, compared to the control group TNFα treatment reduced the cellular viability of mouse neuroblastoma N2a cells, indicating that inflammation injury had developed in the TNFα-treated mouse neuroblastoma N2a cells. This model was used in all subsequent functional assays. Western blotting was used to observe the changes in Mfn2 and Yap in the setting of the TNFα treatment. As shown in Fig. 1b–d, compared to the control group, Mfn2 expression was downregulated in response to TNFα treatment and Yap expression was significantly repressed. These results indicate that Mfn2 and Yap were both inhibited by the TNFα-mediated inflammatory microenvironment. This finding was further supported by the results of the immunofluorescence assay. As shown in Fig. 1e–h, compared to the control group, the fluorescence intensity of Mfn2 was obviously suppressed in TNFα-treated cells, an effect that was accompanied with a drop in the levels of Yap (Fig. 1e–h). Taken together, the above data provide evidence that TNFα-induced inflammation stress promoted the decrease in both Mfn2 and Yap expression in mouse neuroblastoma N2a cells.

Tumor necrosis factor alpha (TNFα) reduces the levels of mitofusin 2 (Mfn2) and Yes-associated protein (Yap) in mouse neuroblastoma N2a cells. a N2a cells were treated with TNFα and then the viability of the treated N2a cells was determined using the MTT assay. TNFα treatment reduced the viability of the N2a cells. b–d Proteins were isolated from N2a cells and then western blotting was used to confirm alterations in the expression of Yap and Mfn2. e–h Immunofluorescence assay was used to analyze the expression of Mfn2 and Yap in N2a cells. TNFα treatment reduced the levels of Mfn2 and Yap in N2a cells. Data are presented as the mean of three replications with the standard error of the mean (SEM). Asterisk above columns linked by horizontal line indicates a significant difference between treatments at p < 0.05. Cont Control, DAPI 4′,6-diamidino-2-phenylindole, GAPDH glyceraldehyde 3-phosphate dehydrogenase (internal control)
Overexpression of Mfn2 reverses Yap expression and the survival of mouse neuroblastoma N2a cells in the setting of TNFα-induced inflammation stress
The following set of experiments was conducted to verify the roles of Mfn2 downregulation and Yap inactivation in TNFα-mediated damage to mouse neuroblastoma N2a cells. First, adenovirus-loaded Mfn2 was transfected into mouse neuroblastoma N2a cells to reverse the expression of Mfn2; the overexpression efficiency was confirmed by western blotting. As shown in Fig. 2a–c, compared to the control group TNFα reduced the levels of Mfn2 in the N2a cells transfected with the adenovirus vector, and this effect was occurred concomitantly with a decline in Yap expression. Interestingly, overexpression of Mfn2 upregulated Yap expression in TNFα-treated mouse neuroblastoma N2a cells, indicating that Mfn2 would appear to be the upstream mediator of Yap in mouse neuroblastoma N2a cells (Fig. 2a–c). The MTT assay was then performed to observe cell viability in response to Mfn2 overexpression. As shown in Fig. 2d, compared to the control group TNFα treatment reduced the cell viability of mouse neuroblastoma N2a cells, and this effect could be reversed by Mfn2 overexpression, indicative of the anti-apoptotic effect of Mfn2 on inflammation-attacked mouse neuroblastoma N2a cells. Caspase-3 activity was also measured as a reflection of the activation of the caspase family during cell apoptosis. As shown in Fig. 2e, compared to the control group TNFα elevated the activity of caspase-3, illustrating that TNFα induced the death of mouse neuroblastoma N2a cells in an apoptosis-dependent manner. Interestingly, Mfn2 overexpression attenuated TNFα-mediated caspase-3 expression upregulation (Fig. 2e). These results were further supported by the results of the immunofluorescence assay. As shown in Fig. 2f, g, compared to the control group the fluorescence intensity of cleaved caspase-3 was augmented in response to TNFα treatment. However, Mfn2 overexpression attenuated TNFα-mediated casapse-3 activation (Fig. 2f, g). Finally, to determine which protein was the upstream mediator, siRNA against Yap was tested, and the knockdown efficiency is shown in Fig. 2h, i. Compared to the control group, Yap knockdown had no effect on the expression of Mfn2. Based on this result, combined with the data shown in Fig. 2a, we conclude that Mfn2 can be considered to be a primary upstream mediator of the Yap–Hippo pathway. Taken together, the above data verify that Mfn2 was the upstream mediator of Yap and increased Mfn2 sustained mouse neuroblastoma N2a cell viability in the setting of TNFα-mediated inflammation stress.

Overexpression of Mfn2 abolishes TNFα-mediated apoptosis in mouse neuroblastoma N2a cells. a–c Proteins were isolated from N2a cells and the expressions of Mfn2 and Yap were determined by western blotting. Adenovirus-loaded Mfn2 (Ad-Mfn2) was transfected into N2a cells to overexpress Mfn2. d Cell viability was determined by the MTT assay. Overexpression of Mfn2 reversed cell viability in TNFα-treated N2a cells. e An enzyme-linked immuosorbent assay (ELISA) was used to observe the changes in caspase-3 in response to TNFα treatment and/or Mfn2 overexpression. Mfn2 overexpression reduced TNFα-mediated capsase-3 upregulation. f, g The immunofluorescence assay was used to observe the changes in caspase-3 in N2a cells. h–j The knockdown efficiency of small interfering RNA (siRNA) against Yap (si-Yap) was determined using western blotting. Mfn2 expression was also measured in response to Yap siRNA transfection. Data are presented as the mean of three replications with the SEM. Asterisk above columns linked by horizontal line indicates a significant difference between treatments at p < 0.05. Cont Control
Knockdown of Yap abolishes the protective effect of Mfn2 overexpression in TNFα-treated mouse neuroblastoma N2a cells
Having found that Mfn2 overexpression was beneficial to N2a cells in the setting of the TNFα-mediated inflammation microenvironment [40, 41], we next looked at whether Yap, the downstream effector of Mfn2, was also involved in the survival of mouse neuroblastoma N2a cells. To this end, siRNA against Yap was transfected into Mfn2-overexpressed cells, and then cell viability was determined by the MTT assay. As shown in Fig. 3a, compared to the control group TNFα reduced cell viability, and this effect could be reversed by Mfn2 overexpresison. Interestingly, the loss of Yap was able to abolish the pro-survival action of Mfn2 overexpression on mouse neuroblastoma N2a cells in the setting of TNFα stress (Fig. 3a). This finding was further supported by the results of TUNEL staining. As shown in Fig. 3c, compared to the control group the number of TUNEL-positive cells increased in response to TNFα treatment. However, Mfn2 overexpression suppressed the ratio of TUNEL-positive cells (Fig. 3b, c), an effect that was negated by Yap siRNA transfection, as evidenced by the increased number of TUNEL-positive cells in Yap-deleted cells. These results indicate that Mfn2 overexpression alleviated TNFα-mediated death in mouse neuroblastoma N2a cells via the upregulation of Yap.

Yap is activated by Mfn2 and contributes to the survival of mouse neuroblastoma N2a cells under TNFα treatment. a Cell viability was measured by the MTT assay. siRNA against Yap (si-Yap) was transfected into N2a cells to repress Yap expression. b, c Cellular death was measured by the terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay. The number of TUNEL-positive cells was evaluated to reflect the role of Yap in TNFα-mediated N2a cell death. d–f Proteins were isolated from N2a cells and then western blotting was used to observe the changes in caspase-3 and cleaved poly (ADP-ribose) polymerase (PARP). siRNA against Yap was transfected into N2a cells to repress Yap expression. Data are presented as the mean of three replications with the SEM. Asterisk above columns linked by horizontal line indicates a significant difference between treatments at p < 0.05. Pro pro-form of caspase-3, Cle. cleaved, Cont control
Furthermore, to confirm whether apoptosis was modulated by Mfn2 in a Yap-dependent manner, western blotting was used to observe the alterations in caspase-3 and its substrate PARP. As shown in Fig. 3d–f, compared to the control group TNFα enhanced the expression of cleaved caspase-3 and its substrate cleaved PARP. Interestingly, Mfn2 overexpression repressed the levels of cleaved caspase-3 and its substrate cleaved PARP (Fig. 3d–f), and these alterations could be reversed by Yap deletion. These data indicate that Yap, as a downstream effector of Mfn2, was significantly required for Mfn2-mediated mouse neuroblastoma N2a cell survival in the setting of TNFα-mediated inflammatory injury.
Mfn2 attenuates ER stress in mouse neuroblastoma N2a cells
Endoplasmic reticulum stress has been shown to be an early hallmark of neuronal dysfunction in mouse neuroblastoma N2a cells [42, 43]. One of the aims of our study was to determine whether apoptosis in TNFα-mediated mouse neuroblastoma N2a cells was activated by ER stress and whether the Mfn2–Yap signaling pathway protected mouse neuroblastoma N2a cells against TNFα stress through an inhibition of ER stress. Western blotting revealed that the expression of ER stress markers such as PERK, CHOP and GRP78 were elevated in response to TNFα treatment (Fig. 4a–d). Interestingly, ER stress parameters were significantly repressed by Mfn2 overexpression (Fig. 4a–d), indicative of the protective effects of Mfn2 overexpression on ER function in the setting of the inflammatory microenvironment. However, loss of Yap re-activated ER stress in Mfn2-overexpressed cells, suggesting that Yap was required for Mfn2-mediated ER protection (Fig. 4a–d). These data support the notion that ER stress, activated by TNFα, was markedly inhibited by Mfn2 in a Yap-dependent manner in mouse neuroblastoma N2a cells.

The Mfn2–Yap signaling pathway modulates endoplasmic reticulum stress in N2a cells subjected to TNFα treatment. a–d Proteins were isolated from N2a cells and the expressions of the ER stress proteins PERK, CHOP and GRP78 were measured by western blotting. siRNA against Yap (si-Yap) and adenovirus-loaded Mfn2 (Ad-Mfn2) were transfected into N2a cells. e, f Immunofluorescence assay was used to observe the changes in caspase-12 in N2a cells. Mfn2 overexpression attenuated caspase-12 activity and this effect could be negated by Yap deletion. g ELISA assay was used to verify the activity of caspase-12 in response to Yap deletion and/or Mfn2 overexpression. Data are presented as the mean of three replications with the SEM. Asterisk above columns linked by horizontal line indicates a significant difference between treatments at p < 0.05. Cont Control
Excessive ER stress is known to activate the caspase-12-related apoptotic pathway [44, 45]. To confirm this, we used the immunofluorescence assay to observe caspase-12 expression in response to TNFα treatment and/or Mfn2 overexpression. As shown in Fig. 4e, f, compared to the control group the intensity of caspase-12 fluorescence was increased by TNFα, i.e. there was an upregulation of caspase-12 expression, whereas Mfn2 overexpression suppressed the TNFα-mediated caspase-12 augmented expression. Interestingly, loss of Yap abolished the regulator effects of Mfn2 overexpression on caspase-12 modification (Fig. 4e, f). The ELISA assay was then used to observe the activity of caspase-12 in response to Mfn2 overexpression and Yap deletion. As shown in Fig. 4g, compared to the control group TNFα enhanced the activity of caspase-12, and this effect could be attenuated by Mfn2 overexpression. Loss of Yap was able to prevent Mfn2-mediated caspase-12 downregulation (Fig. 4g), as evidenced by increased caspase-12 activity in Yap-deleted cells. Taken together, this information indicates that TNFα-activated ER stress was negatively modulated by the Mfn2–Yap signaling pathway.
Cellular oxidative stress and calcium balance are modulated by Mfn2 in mouse neuroblastoma N2a cells
At the molecular level, ER stress is associated with maintenance of calcium homeostasis and modification of the redox balance [46, 47]. Previous studies have reported that ER stress would induce the disorder of calcium-related proteins, contributing to the oxidative stress through the upregulation of calcium-related ROS overproduction [48, 49]. In the present study, western blotting was used to observe the changes in calcium-related proteins. As shown in Fig. 5a–d, compared to the control group TNFα increased the expression of the inositol trisphosphate receptor (IP3R), which modulates calcium release from the ER, whereas it reduced the levels of store-operated calcium entry (SERCA); this result is indicative of the imbalance in calcium-related proteins. Interestingly, Mfn2 overexpression was able to correct the disorder between calcium-related proteins, and this effect was dependent on Yap, as evidenced by the loss of Yap abolishing the regulatory effects of Mfn2 on calcium-related proteins regulation (Fig. 5a–d). Cytoplasmic calcium content was then measured by immunofluorescence. As shown in Fig. 5e, f, compared to the control group TNFα increased the content of calcium in mouse neuroblastoma N2a cells, indicating that TNFα interrupted calcium recycling in mouse neuroblastoma N2a cells. Interestingly, Mfn2 overexpression reversed the calcium balance, and this effect was negated in Yap-deleted cells (Fig. 5e, f), indicating that the Mfn2–Yap axis was involved in calcium regulation in the setting of TNFα-induced inflammatory injury.

The Mfn2–Yap signaling pathway affects calcium overloading and oxidative stress in N2a cells. a–d Immunofluorescence assay was used to observe the alterations in store-operated calcium entry (SERCA) and the inositol trisphosphate receptor (IP3R). siRNA against Yap (si-Yap) and adenovirus-loaded Mfn2 (Ad-Mfn2) were transfected into N2a cells. e, f Cellular calcium was labeled with Fure-2AM and then the fluorescence intensity of calcium was measured as a reflection of calcium overloading. g, h Reactive oxygen species (ROS) production was measured by the immunofluorescence assay. siRNA against Yap and adenovirus-loaded Mfn2 were transfected into N2a cells. i–k ELISA assay was used to observe the changes in cellular antioxidants. Mfn2 overexpression attenuated TNFα-mediated ROS overproduction and this effect could be negated by Yap deletion. Data are presented as the mean of three replications with the SEM. Asterisk above columns linked by horizontal line indicates a significant difference between treatments at p < 0.05. Cont Control
Oxidative stress was also measured by immunofluorescence. As shown in Fig. 5g, h, compared to the control group TNFα increased the production of ROS in mouse neuroblastoma N2a cells, whereas this effect was significantly repressed by Mfn2 overexpression, as evidenced by the decreased ROS production. Interestingly, loss of Yap abolished the anti-oxidative impact of Mfn2 overexpression on TNFα-treated mouse neuroblastoma N2a cells (Fig. 5g, h). This result was further validated by analyzing the alterations in the levels of cellular antioxidants. As shown in Fig. 5i–k, compared to the control group, TNFα reduced the levels of SOD, GSH and GPX, which is indicative of the inactivation of the anti-oxidative system in the inflammation microenvironment. In contrast, Mfn2 overexpression reversed the content of SOD, GSH and GPX, and this effect could be abolished by Yap deletion, suggesting that Mfn2 reversed the cell anti-oxidative capacity in a mechanism involving the upregulation of Yap (Fig. 5i–k). Taken together, the above data support the functional importance of the Mfn2–Yap cascade in the regulation of TNFα-mediated calcium imbalance and oxidative stress in mouse neuroblastoma N2a cells.
Discussion
Neuroinflammation has been identified as the primary pathogenesis of HD, a neurodegenerative disorder, and anti-inflammatory drugs have been found to be effective in the treatment of patients with HD. Consequently, in attempts to reduce the progression of HD, the authors of several carefully thought-out studies have tried to determine the molecular mechanism by which inflammation exacerbates neuronal dysfunction [50]. In the present study, we used TNFα to mimic neuroinflammation in vitro and observed molecular alterations in response to TNFα treatment. We found that Mfn2 expression was downregulated, an effect that was followed by a decrease in Yap expression. Interestingly, Mfn2 overexpression upregulated Yap expression, and the activated Mfn2–Yap signaling pathway promoted neuron survival in the TNFα-induced microenvironment. To our knowledge, this is the first investigation to identify the Mfn2–Yap axis as a novel signaling pathway involved in the progression of HD.
We found that ER stress was activated by inflammation injury, as evidenced by an increase in the level of markers related to ER stress, elevated ROS production and activated caspase-12-related apoptotic pathway. These results indicate that ER stress could be regarded as a primary downstream effector of inflammation injury, and they are in accordance with previously reported results [51, 52]. For example, in a colonic epithelial cell line, inhibition of ER stress attenuated inflammation injury [53]. ER stress has also been observed to modulate the viability of pancreatic β-cells [54], human monocytic leukemia cell [55], alveolar epithelium [56] and bronchial epithelial cells [57] in the context of an inflammation-induced microenvironment. These findings, together with our results, illustrate the functional importance of ER stress in initiating and transmitting the inflammation signals. Our data also indicate that the protective mechanism of anti-inflammation therapy may be associated with the inhibition of ER stress.
We observed that ER stress resulted in the downregulation of the Mfn2 level after exposure to TNFα treatment. However, Mfn2 overexpression attenuated the expression of ER stress-related proteins, and this effect ultimately repressed the activation of caspase-12, sending pro-survival signals to mouse neuroblastoma N2a cells in the inflammatory microenvironment. This finding is consistent with those from previous studies [51, 58]. For example, in HepG2 cells, increased Mfn2 expression attenuates ER stress, sustaining cellular viability and mitochondrial function [59]. In addition, in Parkinson’s disease, Mfn2-related ER stress also modulates neurodegeneration in a manner dependent on the PINK1/Parkin pathway [60, 61]. Moreover, in patients with remethylation defects, ER stress is also modified via Mfn2 through autophagy [62]. In brain endothelial cells, Mfn2 affects ER stress and mitochondrial homeostasis through cytoskeletal alterations [34]. To that end, Mfn2 in proopiomelanocortin (POMC) neurons connects ER stress with leptin resistance and energy imbalance. Therefore, we consider that these findings verify that ER stress is a targetable pathogenetic component of the phenotypes caused by Mfn2 downregulation in the setting of neuroinflammation. Not only ER stress but mitochondrial fission also seems to be affected by Mfn2 downregulation in the setting of neuroinflammation. For example, an imbalance in Sirt3 deficiency-mediated brain mitochondrial dynamics seems to be associated with Mfn2 downregulation [63]. Mfn2 dysfunction also contributes to the development of neurodegenerative disorders by inducing mitochondrial damage [64]. Accordingly, more studies are necessary to puzzle out whether the Mfn2–Yap signaling pathway is also associated with a disordering of mitochondrial dynamics in response to TNFα stress.
Yap is a pro-survival signal for cells subjected to various stress environments. For example, Yap modulates the viability of thoracic cancer by affecting the activity of programmed death-ligand 1 (PD-L1) [65]. The inhibition of Yap expression can activate breast cancer cell apoptosis; and in cardiac reperfusion injury, the Yap–Hippo pathway is inhibited and contributes to the progression of cardiac dysfunction by augmenting cardiomyocytes [66]. Moreover, in prostate cancer, Yap activation modulates docosahexaenoic acid-induced apoptosis in a manner dependent on the FFAR4 pathway [67]. Notably, the beneficial effects of Yap in brain tissues have also been widely explored. The Yap–Hippo pathway is inactivated in cerebral ischemia reperfusion injury, thereby contributing to mitochondrial dysfunction and neuron death. The integrity of the blood–brain barrier is also sustained by Yap and its substrate PIK3CB in subarachnoid hemorrhage. Moreover, neuron differentiation of human pluripotent stem cells is closely affected by Yap. Notably, several studies have indicated that the inflammation response modulates the transcription of Yap and Mfn2, leading to a drop in the expression of Yap and Mfn2. This mechanism may contribute to the downregulation of Mfn2 and Yap in the setting of TNFα stress.
Overall, our findings define a mechanism by which the Mfn2–Yap pathway mediates protection against inflammation-induced apoptosis of mouse neuroblastoma N2a cells in vitro. However, there are some limitations to our results. First, only in vitro cell experiments were performed, and thus more animal studies and/or animal primary neuron assays are necessary to further support our observations. Second, we did not address the regulatory mechanisms by which Mfn2 modulates the expression of Yap. More studies are required to explore whether Mfn2 affects the transcription of Yap. In addition, although we observed an association between Yap deficiency and increased N2a cell death, it remains unknown whether Yap overexpression is sufficient to confer neuroprotective effects against TNFα similar to those achieved by Mfn2 overexpression.
References
Giampà C, Alvino A, Magatti M, Silini AR, Cardinale A, Paldino E, Fusco FR, Parolini O (2018) Conditioned medium from amniotic cells protects striatal degeneration and ameliorates motor deficits in the R6/2 mouse model of Huntington’s disease. J Cell Mol Med 23:1581–1592
Liu J, Xu Y, Wu Q, Ding Q, Fan W (2018) Sirtuin1 protects hair follicle stem cells from TNFalpha-mediated inflammatory stress via activating the MAPK-ERK-Mfn2 pathway. Life Sci 212:213–224
Bocci M, Sjolund J, Kurzejamska E, Lindgren D, Marzouka NA, Bartoschek M, Hoglund M, Pietras K (2019) Activin receptor-like kinase 1 is associated with immune cell infiltration and regulates CLEC14A transcription in cancer. Angiogenesis 22:117–131
Liu N, Jiang Z, Liu Y, Nie Y, Chen J, Ouyang B, Guan X, Chen M (2017) Human trypsin inhibitor reduces the apoptosis of lipopolysaccharide-induced human kidney2 cells by promoting mitochondrial fusion. Mol Med Rep 16:2899–2906
Moore JBT, Tang XL, Zhao J, Fischer AG, Wu WJ, Uchida S, Gumpert AM, Stowers H, Wysoczynski M, Bolli R (2018) Epigenetically modified cardiac mesenchymal stromal cells limit myocardial fibrosis and promote functional recovery in a model of chronic ischemic cardiomyopathy. Basic Res Cardiol 114:3
Boga JA, Caballero B, Potes Y, Perez-Martinez Z, Reiter RJ, Vega-Naredo I, Coto-Montes A (2018) Therapeutic potential of melatonin related to its role as an autophagy regulator: A review. J Pineal Res 66:e12534
Biernacki M, Ambrozewicz E, Gegotek A, Toczek M, Bielawska K, Skrzydlewska E (2018) Redox system and phospholipid metabolism in the kidney of hypertensive rats after FAAH inhibitor URB597 administration. Redox Biol 15:41–50
Farber G, Parks MM, Lustgarten Guahmich N, Zhang Y, Monette S, Blanchard SC, Di Lorenzo A, Blobel CP (2019) ADAM10 controls the differentiation of the coronary arterial endothelium. Angiogenesis 22:237–250
Bøtker HE, Hausenloy D, Andreadou I, Antonucci S, Boengler K, Davidson SM, Deshwal S, Devaux Y, Di Lisa F, Di Sante M, Efentakis P, Femmino S, Garcia-Dorado D, Giricz Z, Ibanez B, Iliodromitis E, Kaludercic N, Kleinbongard P, Neuhauser M, Ovize M, Pagliaro P, Rahbek-Schmidt M, Ruiz-Meana M, Schluter KD, Schulz R, Skyschally A, Wilder C, Yellon DM, Ferdinandy P, Heusch G (2018) Practical guidelines for rigor and reproducibility in preclinical and clinical studies on cardioprotection. Basic Res Cardiol 113:39. https://doi.org/10.1007/s00395-018-0696-8
Zhou H, Wang S, Hu S, Chen Y, Ren J (2018) ER-mitochondria microdomains in cardiac ischemia-reperfusion injury: a fresh perspective. Front Physiol 9:755
Zhou H, Wang S, Zhu P, Hu S, Chen Y, Ren J (2018) Empagliflozin rescues diabetic myocardial microvascular injury via AMPK-mediated inhibition of mitochondrial fission. Redox Biol 15:335–346
Souza LEB, Beckenkamp LR, Sobral LM, Fantacini DMC, Melo FUF, Borges JS, Leopoldino AM, Kashima S, Covas DT (2018) Pre-culture in endothelial growth medium enhances the angiogenic properties of adipose-derived stem/stromal cells. Angiogenesis 21:15–22
Deleon-Pennell KY, Mouton AJ, Ero OK, Ma Y, Padmanabhan Iyer R, Flynn ER, Espinoza I, Musani SK, Vasan RS, Hall ME, Fox ER, Lindsey ML (2018) LXR/RXR signaling and neutrophil phenotype following myocardial infarction classify sex differences in remodeling. Basic Res Cardiol 113:40. https://doi.org/10.1007/s00395-018-0699-5
Ding M, Ning J, Feng N, Li Z, Liu Z, Wang Y, Wang Y, Li X, Huo C, Jia X, Xu R, Fu F, Wang X, Pei J (2018) Dynamin-related protein 1-mediated mitochondrial fission contributes to post-traumatic cardiac dysfunction in rats and the protective effect of melatonin. J Pineal Res 64(1). https://doi.org/10.1111/jpi.12447
Zhou H, Wang J, Hu S, Zhu H, Toanc S, Ren J (2019) BI1 alleviates cardiac microvascular ischemia-reperfusion injury via modifying mitochondrial fission and inhibiting XO/ROS/F-actin pathways. J Cell Physiol 234:5056–5069
Zhu P, Hu S, ** Q, Li D, Tian F, Toan S, Li Y, Zhou H, Chen Y (2018) Ripk3 promotes ER stress-induced necroptosis in cardiac IR injury: a mechanism involving calcium overload/XO/ROS/mPTP pathway. Redox Biol 16:157–168
Deussen A (2018) Mechanisms underlying coronary autoregulation continue to await clarification. Basic Res Cardiol 113:34
Illarioshkin SN, Klyushnikov SA, Vigont VA, Seliverstov YA, Kaznacheyeva EV (2018) Molecular pathogenesis in Huntington’s disease. Biochemistry (Mosc) 83:1030–1039
LaE Erland, Shukla MR, Singh AS, Murch SJ, Saxena PK (2018) Melatonin and serotonin: mediators in the symphony of plant morphogenesis. J Pineal Res 64:e12452
Geng C, Wei J, Wu C (2018) Yap-Hippo pathway regulates cerebral hypoxia-reoxygenation injury in neuroblastoma N2a cells via inhibiting ROCK1/F-actin/mitochondrial fission pathways. Acta Neurol Belg 23:1–4
Angelova PR, Barilani M, Lovejoy C, Dossena M, Vigano M, Seresini A, Piga D, Gandhi S, Pezzoli G, Abramov AY, Lazzari L (2018) Mitochondrial dysfunction in Parkinsonian mesenchymal stem cells impairs differentiation. Redox Biol 14:474–484
Zhu H, ** Q, Li Y, Ma Q, Wang J, Li D, Zhou H, Chen Y (2018) Melatonin protected cardiac microvascular endothelial cells against oxidative stress injury via suppression of IP3R-[Ca(2+)]c/VDAC-[Ca(2+)]m axis by activation of MAPK/ERK signaling pathway. Cell Stress Chaperones 23:101–113
Zhou H, Zhu P, Wang J, Zhu H, Ren J, Chen Y (2018) Pathogenesis of cardiac ischemia reperfusion injury is associated with CK2alpha-disturbed mitochondrial homeostasis via suppression of FUNDC1-related mitophagy. Cell Death Differ 25:1080–1093
Darden J, Payne LB, Zhao H, Chappell JC (2019) Excess vascular endothelial growth factor-A disrupts pericyte recruitment during blood vessel formation. Angiogenesis 22:167–183
Dickinson JD, Sweeter JM, Warren KJ, Ahmad IM, De Deken X, Zimmerman MC, Brody SL (2018) Autophagy regulates DUOX1 localization and superoxide production in airway epithelial cells during chronic IL-13 stimulation. Redox Biol 14:272–284
LaE Erland, Yasunaga A, Li ITS, Murch SJ, Saxena PK (2018) Direct visualization of location and uptake of applied melatonin and serotonin in living tissues and their redistribution in plants in response to thermal stress. J Pineal Res 66:e12527
Gonzalez NR, Liou R, Kurth F, Jiang H, Saver J (2018) Antiangiogenesis and medical therapy failure in intracranial atherosclerosis. Angiogenesis 21:23–35
Hassanshahi M, Hassanshahi A, Khabbazi S, Su YW, **an CJ (2017) Bone marrow sinusoidal endothelium: damage and potential regeneration following cancer radiotherapy or chemotherapy. Angiogenesis 20:427–442
Hooshdaran B, Kolpakov MA, Guo X, Miller SA, Wang T, Tilley DG, Rafiq K, Sabri A (2017) Dual inhibition of cathepsin G and chymase reduces myocyte death and improves cardiac remodeling after myocardial ischemia reperfusion injury. Basic Res Cardiol 112:62
Das N, Mandala A, Naaz S, Giri S, Jain M, Bandyopadhyay D, Reiter RJ, Roy SS (2017) Melatonin protects against lipid-induced mitochondrial dysfunction in hepatocytes and inhibits stellate cell activation during hepatic fibrosis in mice. J Pineal Res 62:e12404
Hatori Y, Inouye S, Akagi R, Seyama T (2018) Local redox environment beneath biological membranes probed by palmitoylated-roGFP. Redox Biol 14:679–685
Dominguez-Rodriguez A, Abreu-Gonzalez P, De La Torre-Hernandez JM, Gonzalez-Gonzalez J, Garcia-Camarero T, Consuegra-Sanchez L, Garcia-Saiz MD, Aldea-Perona A, Virgos-Aller T, Azpeitia A, Reiter RJ (2017) Effect of intravenous and intracoronary melatonin as an adjunct to primary percutaneous coronary intervention for acute ST-elevation myocardial infarction: results of the Melatonin Adjunct in the acute myocaRdial Infarction treated with Angioplasty trial. J Pineal Res 62:e12374
Yuan F, **e Q, Wu J, Bai Y, Mao B, Dong Y, Bi W, Ji G, Tao W, Wang Y, Yuan Z (2011) MST1 promotes apoptosis through regulating Sirt1-dependent p53 deacetylation. J Biol Chem 286:6940–6945
** Q, Li R, Hu N, **n T, Zhu P, Hu S, Ma S, Zhu H, Ren J, Zhou H (2018) DUSP1 alleviates cardiac ischemia/reperfusion injury by suppressing the Mff-required mitochondrial fission and Bnip3-related mitophagy via the JNK pathways. Redox Biol 14:576–587
Liu Z, Liu Y, Xu Q, Peng H, Tang Y, Yang T, Yu Z, Cheng G, Zhang G, Shi R (2017) Critical role of vascular peroxidase 1 in regulating endothelial nitric oxide synthase. Redox Biol 12:226–232
Camare C, Pucelle M, Negre-Salvayre A, Salvayre R (2017) Angiogenesis in the atherosclerotic plaque. Redox Biol 12:18–34
Gao L, Zhao YC, Liang Y, Lin XH, Tan YJ, Wu DD, Li XZ, Ye BZ, Kong FQ, Sheng JZ, Huang HF (2016) The impaired myocardial ischemic tolerance in adult offspring of diabetic pregnancy is restored by maternal melatonin treatment. J Pineal Res 61:340–352
Zhou H, Wang J, Zhu P, Zhu H, Toan S, Hu S, Ren J, Chen Y (2018) NR4A1 aggravates the cardiac microvascular ischemia reperfusion injury through suppressing FUNDC1-mediated mitophagy and promoting Mff-required mitochondrial fission by CK2alpha. Basic Res Cardiol 113:23
Galley HF, Mccormick B, Wilson KL, Lowes DA, Colvin L, Torsney C (2017) Melatonin limits paclitaxel-induced mitochondrial dysfunction in vitro and protects against paclitaxel-induced neuropathic pain in the rat. J Pineal Res 63:e12444
Cuadrado A, Kugler S, Lastres-Becker I (2018) Pharmacological targeting of GSK-3 and NRF2 provides neuroprotection in a preclinical model of tauopathy. Redox Biol 14:522–534
Sajib S, Zahra FT, Lionakis MS, German NA, Mikelis CM (2018) Mechanisms of angiogenesis in microbe-regulated inflammatory and neoplastic conditions. Angiogenesis 21:1–14
Meyer IS, Leuschner F (2018) The role of Wnt signaling in the healing myocardium: a focus on cell specificity. Basic Res Cardiol 113:44
Zhou H, Zhu P, Guo J, Hu N, Wang S, Li D, Hu S, Ren J, Cao F, Chen Y (2017) Ripk3 induces mitochondrial apoptosis via inhibition of FUNDC1 mitophagy in cardiac IR injury. Redox Biol 13:498–507
Rusnati M, Borsotti P, Moroni E, Foglieni C, Chiodelli P, Carminati L, Pinessi D, Annis DS, Paiardi G, Bugatti A, Gori A, Longhi R, Belotti D, Mosher DF, Colombo G, Taraboletti G (2019) The calcium-binding type III repeats domain of thrombospondin-2 binds to fibroblast growth factor 2 (FGF2). Angiogenesis 22:133–144
Ter Horst EN, Krijnen PaJ, Hakimzadeh N, Robbers L, Hirsch A, Nijveldt R, Lommerse I, Fontijn RD, Meinster E, Delewi R, Van Royen N, Zijlstra F, Van Rossum AC, Van Der Schoot CE, Van Der Pouw Kraan T, Horrevoets AJ, Van Der Laan AM, Niessen HWM, Piek JJ (2018) Elevated monocyte-specific type I interferon signalling correlates positively with cardiac healing in myocardial infarct patients but interferon alpha application deteriorates myocardial healing in rats. Basic Res Cardiol 114:1
Zhou H, Li D, Zhu P, Hu S, Hu N, Ma S, Zhang Y, Han T, Ren J, Cao F, Chen Y (2017) Melatonin suppresses platelet activation and function against cardiac ischemia/reperfusion injury via PPARgamma/FUNDC1/mitophagy pathways. J Pineal Res 63:e12438
Zhou H, Hu S, ** Q, Shi C, Zhang Y, Zhu P, Ma Q, Tian F, Chen Y (2017) Mff-Dependent mitochondrial fission contributes to the pathogenesis of cardiac microvasculature ischemia/reperfusion injury via induction of mROS-mediated cardiolipin oxidation and HK2/VDAC1 disassociation-involved mPTP opening. J Am Heart Assoc 6:e005328
Zhang Y, Zhou H, Wu W, Shi C, Hu S, Yin T, Ma Q, Han T, Zhang Y, Tian F, Chen Y (2016) Liraglutide protects cardiac microvascular endothelial cells against hypoxia/reoxygenation injury through the suppression of the SR-Ca(2 +)-XO-ROS axis via activation of the GLP-1R/PI3 K/Akt/survivin pathways. Free Radic Biol Med 95:278–292
Zhou H, Zhang Y, Hu S, Shi C, Zhu P, Ma Q, ** Q, Cao F, Tian F, Chen Y (2017) Melatonin protects cardiac microvasculature against ischemia/reperfusion injury via suppression of mitochondrial fission-VDAC1-HK2-mPTP-mitophagy axis. J Pineal Res 63:e12413
Kim N, Do J, Bae JS, ** HK, Kim JH, Inn KS, Oh MS, Lee JK (2018) Piperlongumine inhibits neuroinflammation via regulating NF-kappaB signaling pathways in lipopolysaccharide-stimulated BV2 microglia cells. J Pharmacol Sci 137:195–201
Zhou H, Li D, Zhu P, Ma Q, Toan S, Wang J, Hu S, Chen Y, Zhang Y (2018) Inhibitory effect of melatonin on necroptosis via repressing the Ripk3-PGAM5-CypD-mPTP pathway attenuates cardiac microvascular ischemia-reperfusion injury. J Pineal Res 65:e12503
Riehle C, Bauersachs J (2018) Of mice and men: models and mechanisms of diabetic cardiomyopathy. Basic Res Cardiol 114:2
Gundamaraju R, Vemuri R, Chong WC, Bulmer AC, Eri R (2019) Bilirubin attenuates ER stress-mediated inflammation, escalates apoptosis and reduces proliferation in the LS174T colonic epithelial cell line. Int J Med Sci 16:135–144
Tyka K, Jorns A, Turatsinze JV, Eizirik DL, Lenzen S, Gurgul-Convey E (2019) MCPIP1 regulates the sensitivity of pancreatic beta-cells to cytokine toxicity. Cell Death Dis 10:29
Zhang YH, Wang DW, Xu SF, Zhang S, Fan YG, Yang YY, Guo SQ, Wang S, Guo T, Wang ZY, Guo C (2018) Alpha-lipoic acid improves abnormal behavior by mitigation of oxidative stress, inflammation, ferroptosis, and tauopathy in P301S Tau transgenic mice. Redox Biol 14:535–548
Valentine MS, Link PA, Herbert JA, Gninzeko FK, Schneck MB, Shankar K, Nkwocha J, Reynolds AM, Heise RL (2018) Inflammation and monocyte recruitment due to aging and mechanical stretch in alveolar epithelium are inhibited by the molecular chaperone 4-phenylbutyrate. Cell Mol Bioeng 11:495–508
Zhou H, Ma Q, Zhu P, Ren J, Reiter RJ, Chen Y (2018) Protective role of melatonin in cardiac ischemia-reperfusion injury: from pathogenesis to targeted therapy. J Pineal Res 64:e12471
Shi C, Cai Y, Li Y, Li Y, Hu N, Ma S, Hu S, Zhu P, Wang W, Zhou H (2018) Yap promotes hepatocellular carcinoma metastasis and mobilization via governing cofilin/F-actin/lamellipodium axis by regulation of JNK/Bnip3/SERCA/CaMKII pathways. Redox Biol 14:59–71
Yang Y, Tang X, Hao F, Ma Z, Wang Y, Wang L, Gao Y (2018) Bavachin induces apoptosis through mitochondrial regulated ER stress pathway in HepG2 cells. Biol Pharm Bull 41:198–207
Celardo I, Costa AC, Lehmann S, Jones C, Wood N, Mencacci NE, Mallucci GR, Loh SH, Martins LM (2016) Mitofusin-mediated ER stress triggers neurodegeneration in pink1/parkin models of Parkinson’s disease. Cell Death Dis 7(6):e2271. https://doi.org/10.1038/cddis.2016.173
Zhang J, Qiu J, Zhou Y, Wang Y, Li H, Zhang T, Jiang Y, Gou K, Cui S (2018) LIM homeobox transcription factor Isl1 is required for melatonin synthesis in the pig pineal gland. J Pineal Res 65:e12481
Olson KR, Gao Y, Arif F, Arora K, Patel S, Deleon ER, Sutton TR, Feelisch M, Cortese-Krott MM, Straub KD (2018) Metabolism of hydrogen sulfide (H2S) and production of reactive sulfur ppecies (RSS) by superoxide dismutase. Redox Biol 15:74–85
Tyagi A, Nguyen CU, Chong T, Michel CR, Fritz KS, Reisdorph N, Knaub L, Reusch JEB, Pugazhenthi S (2018) SIRT3 deficiency-induced mitochondrial dysfunction and inflammasome formation in the brain. Sci Rep 8:17547
Czapski GA, Cieslik M, Wencel PL, Wojtowicz S, Strosznajder RP, Strosznajder JB (2018) Inhibition of poly(ADP-ribose) polymerase-1 alters expression of mitochondria-related genes in PC12 cells: relevance to mitochondrial homeostasis in neurodegenerative disorders. Biochim Biophys Acta Mol Cell Res 1865:281–288
Hsu PC, Yang CT, Jablons DM, You L (2018) The role of Yes-Associated Protein (YAP) in regulating programmed death-ligand 1 (PD-L1) in thoracic cancer. Biomedicines 6:114
Li R, **n T, Li D, Wang C, Zhu H, Zhou H (2018) Therapeutic effect of Sirtuin 3 on ameliorating nonalcoholic fatty liver disease: the role of the ERK-CREB pathway and Bnip3-mediated mitophagy. Redox Biol 18:229–243
Wang J, Hong Y, Shao S, Zhang K, Hong W (2018) FFAR1-and FFAR4-dependent activation of Hippo pathway mediates DHA-induced apoptosis of androgen-independent prostate cancer cells. Biochem Biophys Res Commun 506:590–596
Funding
None.
Author information
Authors and Affiliations
Contributions
SH and LLW conceived the research; GPZ and SH performed the experiments; all authors participated in discussing and revising the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Data availability
All data generated or analyzed during this study are included in this published article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
About this article

Cite this article
Hou, S., Wang, L. & Zhang, G. Mitofusin-2 regulates inflammation-mediated mouse neuroblastoma N2a cells dysfunction and endoplasmic reticulum stress via the Yap-Hippo pathway. J Physiol Sci 69, 697–709 (2019). https://doi.org/10.1007/s12576-019-00685-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12576-019-00685-6