
Abstract
Mesenchymal stem cells (MSCs) have immune-modulatory and tissue-regenerative properties that make them a suitable and promising tool for cell-based therapy application. Since the bio-chemo-mechanical environment influences MSC fate and behavior, the understanding of the mechanosensors involved in the transduction of mechanical inputs into chemical signals could be pivotal. In this context, the nuclear pore complex is a molecular machinery that is believed to have a key role in force transmission and in nucleocytoplasmic shuttling regulation. To fully understand the nuclear pore complex role and the nucleocytoplasmic transport dynamics, recent advancements in fluorescence microscopy provided the possibility to study passive and facilitated nuclear transports also in mechanically stimulated cell culture conditions. Here, we review the current available methods for the investigation of nucleocytoplasmic shuttling, including photo-perturbation-based approaches, fluorescence correlation spectroscopy, and single-particle tracking techniques. For each method, we analyze the advantages, disadvantages, and technical limitations. Finally, we summarize the recent knowledge on mechanical regulation of nucleocytoplasmic translocation in MSC, the relevant progresses made so far, and the future perspectives in the field.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Over 10 years, thanks to their unique properties and multiple clinical benefits, mesenchymal stem cells (MSCs) have been studied and used as suitable and promising tools for cell-based therapy applications. MSCs are adult multipotent cells with the great potential to self-renew and to differentiate into multiple cell lineages mainly derived from the mesodermal layer but also from the ectoderma and endoderma under specific conditions (Fig. 1) (Moon et al. 2018; Le and Yao 2017).

Graphical representation of mesenchymal stem cells differentiation pathways
MSCs can be easily harvested from different mesenchymal tissues that are bone marrow, adipose tissue, umbilical cord, and dermis. Cell isolation is a simple procedure based on the collection of a heterogeneous population of plastic-adherent cells screened by the expression of specific surface antigens (Barry and Murphy 2004). The use of this type of adult stem cells arises low ethical issues (Volarevic et al. 2018), shows a low risk of tumorigenicity (Meier et al. 2013), and possesses a broad spectrum of immune regulatory and tissue organ repairing ability. Thanks to MSC immune-modulatory properties, these cells were used for some preclinical cell-based treatment of different autoimmune diseases, such as systemic lupus erythematosus (Cras et al. 2015), inflammatory bowel’s disease (He et al. 2012), and rheumatoid arthritis (Abd Elhalem et al. 2018). These first studies showed great benefits on clinical and biochemical markers but appeared highly dependent on the host inflammatory state. Exploiting their tissue-regenerative properties, MSCs have been also applied for cell-based therapy in case of several other pathologies, such as liver disease (Zhao et al. 2018), myocardial infarction (Madigan and Atoui 2018), pancreatitis (Ahmed et al. 2018), and stroke (Bang et al. 2016).
Focusing on improving the clinical cell application and the control of MSC fate and behavior, researchers have been able to modulate MSC differentiation by regulating the chemical or mechanical environment. Until some decades ago, they guided MSC differentiation by adding exogenous chemical molecules to cell culture. However, these techniques do not allow translation to the clinic because they could induce immune reactions or even cause tumors onset. The main challenge today is being able to effectively control the differentiation in vitro without using chemical factors. Therefore, with the aim of mimicking the entire bio-chemo-mechanical environment, recent promising approaches are focused on the mechanical influence of the MSC environment (Raimondi et al. 2012).
Mechanical regulation of MSC differentiation
In the last decades, researchers evaluated how mechanical environment (matrix and external inputs) is transduced into a cell biochemical signal leading to gene transcription. This mechanism is defined as mechanotranscription. Among all the transcription factors involved in MSC differentiation pathways, few factors have been studied in the context of mechanotranscription. In Table 1, we listed the main transcription factors (TFs) involved in the promotion of the earlier stages of MSC differentiation. As this table clearly shows, so far, the pathways investigated in connection to the mechanotranscription mechanism are those characterizing the three mesodermal cell lineages defined by the International Society of Cellular Therapy for the determination of MSC population: adipogenesis, osteogenesis, and chondrogenesis.
Matrix microenvironment (architecture, stiffness, composition) and external mechanical stimuli clearly influence both in vitro and in vivo cell growth. Substrate and matrix stiffness regulate cell properties, such as differentiation, proliferation, and cell shape (Nava et al. 2012, 2014; Sun et al. 2016), its skeleton destabilization is transduced in chromatin rearrangement and epigenetic modifications, leading to the exposure of specific binding sites to the transcription machinery (Killaars et al. 2018; Heo et al. 2015; Arnsdorf et al. 2010). Thus, besides the chromatin remodeling, understanding the nucleocytoplasmic shuttling of proteins—such as transcription factors—could be determined to fully characterize and control the mechanotransduction events.
In a famous study, Sirio Dupont highlighted an interesting phenomenon related to the migration of transcription factors involved in key cellular mechanisms. He showed how two mechanotransduction mediators YAP (yes-associated protein)/TAZ (transcriptional coactivator with PDZ-binding motif) localization is strictly related to the rigidity and cell shape (Dupont et al. 2011). They plated MSCs on micropillars arrays of different rigidity and observed a higher percentage of nuclear YAP/TAZ when MSCs were plated on rigid micropillars with rather than elastic ones.
Within this scenario, the aim of this review was to summarize the state of the art of the nucleocytoplasmic transport through the nuclear pore complexes in mesenchymal stem cells subjected to mechanically stressed conditions.
The nuclear pore complex
The NPC is a complex molecular machine (∼ 125 MDa) composed by approximately 30 nucleoporins (Nup), giving to the NPC a cylindrical structure that spans from the cytoplasmic to the nucleoplasmic side of the cell (Fig. 4) (Garcia et al. 2016). In the cytoplasm side, the gate is the cytoplasmic ring: it is a diaphragm with a diameter around 100–150 nm equipped by eight filaments (50–70 nm in length) catching cargoes and macromolecules to facilitate their transport to the nucleus.

Scheme of the nuclear pore complex. (a) Cytoplasmic and (b) nucleoplasmic view of the NPC. (c) Illustration of FG-nucleoporin filament in the NPC central channel. (d) Illustration of nucleoporins (Nups) composing a section of the NPC. (e) Vertical section of the NPC: the cytoplasmic ring (CR) is marked in blue, the central ring/spoke ring (SR) is shown in purple, the nucleoplasmic ring (NR) and the nuclear basket filaments are labelled in green. The translocation paths are represented with light blue arrows and the red ones are representative of the secondary channels. Image taken from (Garcia et al. 2016) with permission
The spoke ring is the NPC structure localized in the nuclear envelope (NE) lipid bilayer. It is a cylinder of 50-nm length and width between 20 and 80 nm, filled with protein filaments, and the phenylalanine–glycine repeats nucleoporins (FG-Nups). FG-Nups have the role to block big inert molecules (> 70 kDa) and facilitate cargo passage to the nucleus. The exchange of ion and small molecule is also allowed from a group of secondary channels (around 4 nm in diameter), placed around the spoke ring. The transmembrane ring located between the two NE lipid membranes confers stability to the NPC cylinder.
Finally, the nuclear and the distal rings are in the nucleoplasmic side and are connected by Nup153 and Tpr proteins forming the nuclear basket (50–75-nm length) (Gu 2018; Garcia et al. 2016; Wente and Rout 2010).
Transports through the NPC could be passive or facilitated. They are bidirectional and share the central diffusion channel located within the central pore: molecules smaller than ~ 70 kDa in size (corresponding to a maximum diameter ~ 10 nm) can passively diffuse across the central part of the pore and their translocation capability is function of size (Gerace and Burke 1998; Keminer and Peters 1999; Paine et al. 1975).
Molecules bigger than 70 kDa pass through the NPC by facilitated diffusion only in the presence of specific motifs (nuclear localization and nuclear export signals (NLS/NES)). The carrier is aided to translocate principally by FG-Nup filaments with cell energy expenditure. In fact, the release of molecules into the nucleus or into the cytoplasm is driven by the state of the Ran nucleotide that cycles between the GDP and the GTP bound states.
NPC molecular machinery is so efficient that in a single pore could translocate up to 1000 molecules/s corresponding to a mass flow nearly 100 MDa/s (Ribbeck and Gorlich 2001; Stewart 2007). In the last decades, the development of genetically encoded fluorescent proteins and fluorescent synthetic dyes has opened the door to study protein localization and trafficking at the level of single cell and single pore (Chalfie 1995; Los et al. 2008; Keppler et al. 2002; Giepmans et al. 2006). Different translocation models have been proposed and well described (Fahrenkrog and Aebi et al. 2003). Nevertheless, since the mechanism of nucleocytoplasmic translocation remains poorly understood, it is currently intensely investigated with the techniques described in the following section.
Fluorescence microscopy techniques applied to characterize nucleocytoplasmic transport
Fluorescence microscopy provides an efficient approach to study diffusion and transport in and out subcellular compartments. In fact, in the last decade, significant advances have been made not only in the field of fluorescent dye\protein engineering but also in microscope set-up development and quantitative fluorescence microscopy techniques. Today, molecular events can be studied both at the level of the single live cell (microns) and single pore (nanometers), allowing visualization and analysis of molecular dynamics through a single NPC. The main techniques used to evaluate fluorescent molecule and protein diffusion or fluxes between different cell compartments are photo-perturbation, correlation spectroscopy, and single-molecule tracking (Fig. 5 and Table 2).

Schematic representation of three approaches to measure molecule nucleocytoplasmic translocation. a FRAP is the most used photo-perturbation technique. It is based on the nuclear photo-bleaching and the subsequent measure of the fluorescence recovery as intensity average of the cell nucleus. Fast recovery is indicative of diffusion and a strong molecule binding leads to an immobile fraction of non-fluorescent proteins. b FCS is a fluctuation-based method. It measures fluorescence fluctuations arising from fluorescent proteins movement across the excitation volume. The autocorrelation function G(τ) represents the probability that the protein of interest remains within the excitation volume for a time longer than τ. c SMT is based on the detection of individual fluorescent proteins. This is the reason why it requires a very low concentration (pM) of fluorescent molecules. To maximize the signal to noise ratio, the excitation occurs by tilting the laser beam (HILO illumination)
Photo-perturbation approaches are based on changing the fluorescent dye photo-physical properties and studying the redistribution of fluorescence. Among these techniques, Fluorescence Recovery After Photo-bleaching (FRAP) is a powerful method used to investigate protein mobility into the cells (Reits and Neefjes 2001; Kang et al. 2009). It consists of irreversibly photo-bleach fluorescent protein in a selected region of interest (ROI). Thanks to the protein diffusion, it is possible to analyze the fluorescence recovery into the ROI. Two main parameters can be extracted from the FRAP measurement: the mobile fraction (Mf), representing the protein fraction diffusing in the selected ROI, and the characteristic time of the diffusion t1/2. Small and highly diffusing proteins show fast recovery, while no fluorescence recovery is observed with an immobile molecule. The speed of the recovery is dependent on the molecular size, the environment viscosity, or the interaction degree between the protein of interest and other molecules (Lippincott-Schwartz et al. 2001).
In photo-perturbation experiments, the protein of interest is linked to a fluorescent protein or dye chosen relying on their physico-chemical features such as quantum yield, its low tendency to photo-bleach, and its photo-stability during post-bleach image acquisition (Lippincott-Schwartz and Patterson 2003). FRAP measurement is widely used and is relatively easy to perform. Furthermore, it can be accomplished on any standard confocal microscope or on a wide-fields microscope equipped with a laser able to bleach a limited area. In any case, it is necessary to keep in account that this technique has some disadvantages (Bancaud et al. 2010; Mueller et al. 2012, 2013; Mazza et al. 2007; Braeckmans et al. 2003; Blumenthal et al. 2015):
-
The high expression level of fluorescence is required, precluding the study of proteins that need to be over-expressed.
-
Photo-bleaching is generated by a strong laser pulse that could induce cell photo-toxicity.
-
Photo-bleaching is not completely irreversible for several fluorescent molecules based on GFP technology, leaving some uncertainty on the measure of goodness.
-
Results are dependent on the size of the bleached ROI and the profile of the bleached volume making it difficult to measure diffusion on large areas and volumes.
-
A quantitative interpretation of FRAP measurements is not trivial and data need to be fitted with the correct kinetic model.
Another photo-perturbation method more recently used is the fluorescence loss in photo-bleaching (FLIP). It could be used in place or paired with FRAP to investigate proteins diffusion. This technique is based on the cell image acquisition between bleaching pulses on a fixed ROI. In this case, if the protein is free to move and able to enter in the bleaching region, a fluorescent signal decay is measured over the entire cell (Ishikawa-Ankerhold et al. 2012). Among photo-perturbation approaches, there are also emerging techniques based on photo-switchable and photo-activatable proteins. After an UV laser pulse, these molecules can shift their emission spectrum or switch from a dark state to a bright state (or vice versa) allowing to study protein mobility. Since the optical properties modulation can be induced with relatively low laser intensities, with these tools, it is possible to perform diffusion measurement, limiting the sample photo-damage (Bancaud et al. 2010).
First investigations to study passive diffusion process through the NPC in single live cells started using inert fluorescent proteins microinjected in Xenopus ovocyte or transfected in eukaryotic cells. In 2003, Wei et al. (2003) used FRAP to prove that EGFP (an inert fluorescent protein of 27 kDa) diffuses bi-directionally through the pore with a rate reduction up to ∼ 100-fold with respect to the diffusion within the nucleus or the cytoplasm, due to the reduced size of the NPC channel available. Moreover, any significant variation in EGFP diffusion through the NPC was observed by Ca2+ depletion demonstrating that EGFP nucleocytoplasmic translocation is a passive diffusion event.
After the pioneering studies on inert tracers, photo-perturbation techniques were successfully used to examine the nucleocytoplasmic-facilitated transport of other molecules (Ando et al. 2004; Köster et al. 2005; Sunn et al. 2005; Chudakov et al. 2007; Davies et al. 2010; Cardarelli et al. 2011a), such as the proteins with nuclear localization/export signals (NLS/NES) and importins. Measures confirm the hypothesis that facilitated nuclear import regulation is mediated by the binding with the β-domain of importinα and that both passive and facilitated transports occur through the central pore channel without interfering each other. Otherwise, different molecules transported by the same pathway hamper each other (Naim et al. 2007; Cardarelli et al. 2009; Bizzarri et al. 2012).
In fluorescence correlation spectroscopy (FCS), a small volume of the sample is illuminated and specimen fluorescent fluctuations are acquired over time. Using the autocorrelation function G, fluorescence fluctuations provide information about molecules concentration and diffusion. In fact, G amplitude is inversely proportional to the average number of fluorescent molecules and the decay time represents the diffusion capability (Elson 2011). FCS is a relatively non-invasive technique and it works best at low molecule concentration (nM concentration with respect to the μM concentration for FRAP), which hinders the use of simple transient transfections to make cells fluorescent. In contrast to FRAP, FCS is well suited for fast diffusion systems (in the order of sub-millisecond) and it is able also to determine the density and aggregation state of the protein of interest.
Using photo-perturbation approaches or FCS, it is possible to measure the mobility properties corresponding to the average behavior of the observed molecules, but the data need to be fitted with the right diffusion model to avoid inaccurate interpretation (Mueller et al. 2012, 2013). The main limitation of FCS is to provide information on a single point of the sample that is often not useful in a non-homogeneous system–like cells. For these kinds of samples, new techniques were developed, including both spatial and temporal correlation of fluorescence that reveals the direction and velocity of systematic motion. For example, the pair correlation fluorescence (pCF) technique has been widely used to study nucleocytoplasmic protein translocation. Its basic principle is to measure the time it takes from a molecule to migrate between two points, analyzing fluorescence fluctuation on a linear ROI. The spatial and temporal correlation among two arbitrary points of the line provides a map of protein transport and shows the presence of barriers or obstacles to diffusion with a millisecond time resolution (Cardarelli and Gratton 2010). Thanks to these technical advancements, in the last decade, correlation spectroscopy techniques appeared as ideal alternative strategies to investigate nucleocytoplasmic shuttling (Cardarelli et al. 2011b). In particular, the pair correlation function (pCF) method guarantees single-molecule sensitivity also in samples with high concentration of fluorescent molecules. pCF is therefore suitable to investigate the nucleocytoplasmic translocation of fluorescent proteins and the role of the NLS in nuclear-facilitated transport. Cardarelli et al., for example, calculated the NLS-GFP transit time through the nuclear pore in the 1–40-ms range and demonstrated that fastest cytoplasm-to-nucleus transit happens very close to the NE barrier, where endogenous importin carriers are accumulated (Cardarelli and Gratton 2010).
The single-molecule tracking (SMT) technique allows to detect and track in time and space individual fluorescent particles. It provides rich data sets that describe diffusion and binding kinetics of the protein of interest (Liu et al. 2016). During an image acquisition, a fluorescent single molecule produces a spot limited by the diffraction law. If molecules are at a very low concentration, they can be resolved and localized with a precision up to 20 nm by using post-processing algorithms (Mortensen et al. 2010). The particle localization precision depends, besides the molecule concentration, on the signal to noise ratio (SNR) of the images. Therefore, to maximize localization and SNR, it is primary to minimize the contribution of out-of-focus molecules and to express protein at very low concentration (pM), for example by using microinjection techniques or new labelling technology (like SNAP-TAG or HALO-TAG approaches). To avoid imaging photo-bleaching it is suitable to use very stable and bright fluorophores (Chow et al. 2016; Los et al. 2008; Keppler et al. 2002). Therefore, to maximize the SNR, a highly illuminated and laminated optical light sheet (HILO) set-up is used: the highly inclined and thin laser beam creates an excitation volume in a limited depth range (microns) and in the center of the object field. Since only a thin layer of the sample is illuminated, the SNR increases about eight times with respect to epi-illumination (Tokunaga et al. 2008). Using this technique, the frame rate acquisition of the particle tracking spans from 100 to 10,000 frames/s, depending on the speed of the camera readout and the illumination time necessary to detect the optical probe (Liu et al. 2016). It allows a wide range of investigations like particle tracking, molecular interaction site, and molecule association/dissociation kinetics (Loffreda et al. 2017; Cui et al. 2018). Using the single-molecule tracking to analyze the nucleocytoplasmic transport allowed to show that the movement along the pore axis is bidirectional and it has the characteristics of a random walk. Furthermore, this technique provided a measurement of the interaction time (or residence time) between the fluorescent molecule and the pore, which is the spending time the protein takes to interact with the nuclear pore central channel proteins (Yang et al. 2004; Kubitscheck et al. 2005; Dange et al. 2008). The range spans from 1 to around 33 ms, as a function of the protein feature, but most of the proteins take 5–10 ms to cross the NPC (Tu and Musser 2011). Interestingly, not all signal-dependent nuclear import events complete the translocation through the NPC. In fact, molecules in proximity to the cytoplasmic periphery and those partially penetrated the central channel can more easily abort the transport, spending the majority of its interaction time moving within the NPC central pore. Moreover, cargo signal-dependent transport efficiency is a function of importin concentration. In fact, for example, NLS-2XGFP flux is reduced up to 50% in case of low importinβ level (Yang 2004; Yang and Musser 2006). Recently, by introducing the single-point edge-excitation sub-diffraction microscopy method (SPEED) again, Yang and colleague obtained a three-dimensional density map of the transient interactions with a spatiotemporal resolution of 9 nm and 400 μs (Ma and Yang 2010; Goryaynov et al. 2012; Goryaynov and Yang 2014). They demonstrated the following:
-
1.
Electrostatic interaction between transiting molecules and FG-Nups does not play a dominant role in determining nuclear transport;
-
2.
The spatial density of interaction sites between importinβ1 and FG-Nups increases as a function of the space and reaches its maximum in the central pore region;
-
3.
Cargo rarely occupies the central NPC channel to pass from the cytosol to the nucleus;
-
4.
The facilitated translocation pathway strictly depends on the FG-Nups interaction.
Mechanoregulation of nucleocytoplasmic translocation
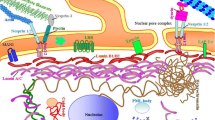
While it has been extensively proven that mechanotransduction regulates cellular mechanisms, such as cytoskeleton organization and gene regulation (Buxboim et al. 2010; McMurray et al. 2015; Bao et al. 2018; Keeling et al. 2017; Tajik et al. 2016; Miroshnikova et al. 2017), only in recent years a new idea is emerging that mechanical signals are transduced also by the nuclear membrane and, in particular, by the NPC. The connection between focal adhesions and the nucleus via the actin cytoskeleton likely allows to transmit internal forces that stretch the nuclear membrane and the NPCs, reducing the resistance to molecular transport through the nuclear membrane, thus increasing the molecules fluxes (Garcia et al. 2016).
The current understanding of mechanosensing at the nuclear envelope by NPC stretch activation and its possible effect in physiology and pathology is still poor (Donnaloja et al. 2019).
Currently, there are two theories concerning the mechanical opening of the pore. The first suggests that the tensions inside the cell stretch the nuclear envelope, increasing the pore size (Elosegui-Artola 2018). However, nowadays, the measurements supporting this theory are not completely reliable because they are carried out with a standard transmission electron microscopy procedure. This means that the sample must be fixed and included in resin, then cut using the microtome to obtain the slices imaged by the TEM. The use of a sliced plane does not allow to know the effective direction and depth of microstructures and, therefore, involves a systematic error in the measure. To overcome this limitation, it would be necessary to use a scanning EM tomography (STEM) or a focused ion beam combined with scanning electron microscopy (FIB-SEM) that allows a three-dimensional imaging of the sample and the measure of the effective dimension of the NPC without parallax errors. Using the STEM microscopy, we measured the nuclear ring area in non-adherent MSCs (which take roundish shape) and spread MSCs finding no significant differences (Garcia et al. 2016).
The second theory, which is yet to be proven, is that the cellular internal forces act on the nuclear part of the nuclear pore and precisely on the basket. The hypothesis seems to be reasonable, since it would give an explanation to the presence of the basket in the nuclear pore complex and above all plausible since the basket is formed by eight nucleoplasmic filaments resulting in a rotational symmetry (Lezon et al. 2009; Knockenhauer and Schwartz 2016). An external force, coming from the cytoskeleton and acting on the basket, could unroll the net facilitating the passage of TF collected in the basket (Donnaloja et al. 2019).
Apart from the opening mechanism of the pore that still must be investigated, what is known today is that in a cell subjected to mechanical stimulation, the transcription factors flow towards the nucleus increases. Elosegui-Artola and colleagues studied the NPC mechanotransduction evaluating the YAP nucleocytoplasmic translocation in fibroblasts (Elosegui-Artola 2018). YAP is a mechanosensitive transcription factor, notoriously involved in cancer, regeneration, and organ size control. They analyzed several elements related to the NPC mechanotransduction by using the FRAP technique. They demonstrated that applying forces to the cell nucleus, the YAP nuclear translocation increased by decreasing the restriction of NPC to protein transport. Moreover, modulating the stiffness of the substrate or using drugs to depolymerize the cell cytoskeleton, they have proven that YAP translocation was mediated by forces transmitted to the nucleus and that the import is related to the force transmission via actin cytoskeleton and not microtubules organization.
In the last years, we are working on the TF nucleocytoplasmic transport in MSCs. We evaluated the inert green-fluorescent protein (GFP) nucleocytoplasmic passive diffusion in MSCs grown on flat substrates or in three-dimensional substrates able to modify cell morphology (García-González et al. 2018). We cultured MSCs in a three-dimensional (3D) substrate, the “Nichoid,” able to condition cell adhesion at the single-cell scale, in order to maintain a roundish nuclear configuration, and on a flat glass substrate where the spread cell configuration induces a disk-like shape to the nucleus. We set up a numerical model of diffusive molecules transport through the NE, based on NPC deformation, and we compared results with those obtained measuring the GFP diffusion through the nuclear envelope by fluorescence recovery after photo-bleaching (FRAP). Our results show that cell stretching modulates the characteristic time needed for passive nuclear import of diffusive molecules, correlating a faster import with the nuclear spreading (Fig.6).

GFP fluorescence recovery after photo-bleaching curves measured on spread cells adhered on a flat substrate or roundish cells adhered in a Nichoid. (b) SEM image of a Nichoid. (c) Examples of GFP-expressing MSCs (green) grown into the Nichoid. In red, the Nichoid fluorescence and the cell DNA are visible. (d) Examples of GFP-expressing MSCs (green) grown on flat glass substrate. In red, the cell nuclei are highlighted
The question arisen is whether the flow of transcription factors in the nucleus is really due to a mechanical stimulus or if it was triggered by a molecular process. In fact, since scaffolds generate gradual and constant mechanical conditioning, it is reasonable to think that the mechanoregulation events could involve also not-immediate mechanical phenomena, such as chemical regulation given by importins and RAN-GTP concentration. However, the nuclear protein import results obtained from Elosegui-Artola by coupling the fluorescence microscopy atomic force microscopy (AFM) techniques, removed any doubt. They used AFM to apply a constant force to the cell nucleus and observed that force application increased the nuclear/cytosolic YAP ratio and that the YAP localization returned to the cytosol upon force release (Fig. 7). Therefore, they demonstrated that force application to the nucleus is enough to drive an immediate YAP nuclear translocation independently of RAN-GTP concentration and scaffold stiffness.

Top: Nuclear/cytosolic YAP ratio (red) and Hoechst nuclear average intensity (blue) for fibroblast cells seeded on 5 kPa gels (n = 9 cells) and transfected with EGFP-YAP during AFM indentation. Force was applied with an AFM cantilever with a 20-mm-diameter spherical tip. Sequentially: No force (1 min), 1.5 nN force (5 min), and no force (4 min). Bottom: Examples of color maps showing YAP fluorescence intensity in the conditions measured. Image taken from (Elosegui-Artola 2018) with permission
Despite MSCs being extremely sensitive to mechanical stimuli and therefore an optimal candidate to investigate mechanotransduction mechanisms, to the best of our knowledge, in literature, there are no other articles investigating the TF nucleocytoplasmic transport in MSCs. This is probably due to the difficulty of achieving MSCs expressing fluorescent molecules. In fact, standard DNA transfection procedures result less efficient in the case of stem cells (Maurisse 2010; Hamann 2019). On the other hand, other techniques useful to directly insert fluorescent probes into cells—like microinjection—are very time consuming and often unsuitable for application on non-standard substrates, such as truly 3D scaffolds like the Nichoid.
Nowadays, as well as understanding the effective mechanisms of the nuclear pore opening, the other important questions still open in the field of nucleocytoplasmic translocation are related to the characterization of TF-facilitated transport in relation to the cell environment and the nuclear shape.
Currently, we are studying the facilitated transport of a transcription factor involved in MSC differentiation towards the myocardial phenotype: MyoD (Vandromme et al. 1995). The main difference compared with the work conducted by Elosegui-Artola et al. is that the scaffold we use for cell growth (the Nichoid) is truly 3D and more representative of the physiological stem cell niche than a two-dimensional system.
We are considering two methods of investigation, FRAP and SMT, and facing their technical challenges. Apart from the difficulty of achieving MSCs expressing fluorescent transcription factors, each of these two methods has specific complexities. In the case of FRAP, for example, the extensive cellular three-dimensionality, induced by the cell growth in the Nichoid, complicates the measurement. In fact, what occurs during the bleaching phase of a nuclear ROI is that also many fluorescent proteins in the cytosol are bleached out. Moreover, as shown in Figs. 6 and 8, the nuclear section area on which the bleaching is carried out is about 3–4 times greater for cells grown on glass flat substrate than in the Nichoid. Although it is very difficult to carry out these measures, we have obtained the first results and we have developed a computational model of nuclear diffusion/deformation to better interpret the results from FRAP measurements.

Images showing MSCs expressing MyoD-GFP grown on glass flat substrate (a) and in the Nichoid (b). In yellow, the cell nuclei are highlighted
Regarding SMT, the major problems with this technique concern the three-dimensionality of the investigated systems and the sample fluorescence of the Nichoid. In fact, using a HILO microscope, the highly inclined beam does not allow the observation of the entire sample. Acquiring only a small part of the sample, the three-dimensionality is partially lost. This limitation could theoretically be overcome using a light sheet microscope. Instead, the Nichoid fluorescence problem is more complicated to solve. In fact, as explained in the previous paragraph, to make good SMT measurements, it is very important to maximize the SNR to contrast the brightness of the single molecules with respect to the background and to be able to make an accurate tracking. Since our sample fluoresces in the same wavelengths as the fluorophore, the application of this technique on cells grown in the Nichoid is still challenging (Fig. 9). We are trying to overcome this concerning aspect by varying the composition of the material in which it is produced and diminishing the Nichoid fluorescence.

Examples of a SMT acquisition with HILO microscope on glass flat substrate (a) and in the Nichoid (b). Cells are MCF7 expressing fluorescent p53 protein. Images acquired at Istituto Scientifico Ospedale San Raffaele, Centro di Imaging Sperimentale, Milano
Conclusion
Mechanosensors and mechanotransduction mechanisms have a key role in modulating MSC fate by controlling the master switch between stemness maintenance and differentiation in these cells. As it has clearly emerged in some recent studies, the NPC plays a determinant role as a mechanosensor by regulating the nuclear import of transcription factors likely based on a stretch-activation mechanism. Despite the huge advancements in fluorescence microscopy to measure nucleocytoplasmic shuttling and to deepen the involvement of NPC in mechanotransduction, the optimal acquisition method is still to be defined, mainly due to the difficulty of transferring the existing techniques to 3D cell models.
The understanding of these mechanisms will allow the development and design of more performing substrates to mechanoguide and control cellular fate. These innovative systems could be used to improve cell-based therapy in regenerative medicine and in the field of personalized medicine.
References
Abd Elhalem SS, Haggag NZ, El-Shinnawy NA (2018) Bone marrow mesenchymal stem cells suppress IL-9 in adjuvant-induced arthritis. Autoimmunity 51(1):25–34. https://doi.org/10.1080/08916934.2018.1428956
Ahmed SM, Morsi M, Ghoneim NI, Abdel-Daim MM, El-Badri N (2018) Mesenchymal stromal cell therapy for pancreatitis: a systematic review. Oxidative Med Cell Longev 18:3250864. https://doi.org/10.1155/2018/3250864
Ando R, Mizuno H, Miyawaki A (2004) Regulated fast nucleocytoplasmic shuttling observed by reversible protein highlighting. Science 306(5700):1370–1373. https://doi.org/10.1126/science.1102506
Araújo JAM, Hilscher MM, Marques-Coelho D, Golbert DCF, Cornelio DA, Batistuzzo de Medeiros SR, Leão RN, Costa MR (2018) Direct reprogramming of adult human somatic stem cells into functional neurons using Sox2, Ascl1, and Neurog2. Front Cell Neurosci 12:155. https://doi.org/10.3389/fncel.2018.00155
Arnsdorf EJ, Tummala P, Jacobs CR (2009) Non-canonical Wnt signaling and N-cadherin related beta-catenin signaling play a role in mechanically induced osteogenic cell fate. PLoS One 4(4):e5388. https://doi.org/10.1371/journal.pone.0005388
Arnsdorf EJ, Tummala P, Castillo AB, Zhang F, Jacobs CR (2010) The epigenetic mechanism of mechanically induced osteogenic differentiation. J Biomech 43(15):2881–6.88. https://doi.org/10.1016/j.jbiomech.2010.07.033
Bancaud A, Huet S, Rabut G, Ellenberg J (2010) Fluorescence perturbation techniques to study mobility and molecular dynamics of proteins in live cells: FRAP, photoactivation, photoconversion, and FLIP. Cold Spring Harb Protoc (12):pdb.top90. https://doi.org/10.1101/pdb.top90
Bang OY, Kim EH, Cha JM, Moon GJ (2016) Adult stem cell therapy for stroke: challenges and progress. J Stroke 18(3):256–266. https://doi.org/10.5853/jos.2016.01263
Bao M, **e J, Huck WTS (2018) Recent advances in engineering the stem cell microniche in 3D. Adv Sci (Weinh) 5(8):1800448. https://doi.org/10.1002/advs.201800448
Barry FP, Murphy JM (2004) Mesenchymal stem cells: clinical applications and biological characterization. Int J Biochem Cell Biol 36(4):568–584. https://doi.org/10.1016/j.biocel.2003.11.001
Bizzarri R, Cardarelli F, Serresi M, Beltram F (2012) Fluorescence recovery after photobleaching reveals the biochemistry of nucleocytoplasmic exchange. Anal Bioanal Chem 403(8):2339–2351. https://doi.org/10.1007/s00216-012-6025-4
Blumenthal D, Goldstien L, Edidin M, Gheber LA (2015) Universal approach to FRAP analysis of arbitrary bleaching patterns. Sci Rep 5:11655. https://doi.org/10.1038/srep11655
Bonnet N, Ferrari SL (2010) Exercise and the skeleton: how it works and what it really does. IBMS BoneKEy 7(7):235–248. https://doi.org/10.1138/20100454
Braeckmans K, Peeters L, Sanders NN, De Smedt SC, Demeester J (2003) Three-dimensional fluorescence recovery after photobleaching with the confocal scanning laser microscope. Biophys J 85(4):2240–2252. https://doi.org/10.1016/S0006-3495(03)74649-9
Browning JA, Walker RE, Hall AC, Wilkins RJ (1999) Modulation of Na+ x H+ exchange by hydrostatic pressure in isolated bovine articular chondrocytes. Acta Physiol Scand 166(1):39–45. https://doi.org/10.1046/j.1365-201x.1999.00534.x
Buxboim A, Ivanovska IL, Discher DE (2010) Matrix elasticity, cytoskeletal forces and physics of the nucleus: how deeply do cells ‘feel’ outside and in? J Cell Sci 123(3):297–308. https://doi.org/10.1242/jcs.041186
Cardarelli F, Gratton E (2010) In vivo imaging of single-molecule translocation through nuclear pore complexes by pair correlation functions. PLoS One 5(5):e10475. https://doi.org/10.1371/journal.pone.0010475
Cardarelli F, Bizzarri R, Serresi M, Albertazzi L, Beltram F (2009) Probing nuclear localization signal-importin alpha binding equilibria in living cells. J Biol Chem 284:36638–36646. https://doi.org/10.1074/jbc.M109.036699
Cardarelli F, Serresi M, Albanese A, Bizzarri R, Beltram F (2011a) Quantitative analysis of Tat peptide binding to import carriers reveals unconventional nuclear transport properties. J Biol Chem 286:12292–12299. https://doi.org/10.1074/jbc.M110.203083
Cardarelli F, Lanzano L, Gratton E (2011b) Fluorescence correlation spectroscopy of intact nuclear pore complexes. Biophys J 101(4):L27–L29. https://doi.org/10.1016/j.bpj.2011.04.057
Case N, Thomas J, **e Z, Sen B, Styner M, Rowe D, Rubin J (2013) Mechanical input restrains PPARγ2 expression and action to preserve mesenchymal stem cell multipotentiality. Bone 52(1):454–464. https://doi.org/10.1016/j.bone.2012.08.122
Chalfie M (1995) Green fluorescent protein. Photochem Photobiol 62(4):651–656. https://doi.org/10.1111/j.1751-1097.1995.tb08712.x
Chen YJ, Huang CH, Lee IC, Lee YT, Chen MH, Young TH (2008) Effects of cyclic mechanical stretching on the mRNA expression of tendon/ligament-related and osteoblast-specific genes in human mesenchymal stem cells. Connect Tissue Res 49(1):7–14. https://doi.org/10.1080/03008200701818561
Chow YT, Chen S, Wang R, Liu C, Kong CW, Li RA, Cheng SH, Sun D (2016) Single cell transfection through precise microinjection with quantitatively controlled injection volumes. Sci Rep 6:24127. https://doi.org/10.1038/srep24127
Chubinskiy-Nadezhdin VI, Vasileva VY, Pugovkina NA, Vassilieva IO, Morachevskaya EA, Nikolsky NN, Negulyaev YA (2017) Local calcium signalling is mediated by mechanosensitive ion channels in mesenchymal stem cells. Biochem Biophys Res Commun 482(4):563–568. https://doi.org/10.1016/j.bbrc.2016.11.074
Chudakov DM, Lukyanov S, Lukyanov KA (2007) Using photoactivatable fluorescent protein Dendra2 to track protein movement. Biotechniques 42(5):553, 555, 557, passim. https://doi.org/10.2144/000112470
Cras A, Farge D, Carmoi T, Lataillade JJ, Wang DD, Sun L (2015) Update on mesenchymal stem cell-based therapy in lupus and scleroderma. Arthritis Res Ther 17:301. https://doi.org/10.1186/s13075-015-0819-7
Cui Y, Yu M, Yao X, **ng J, Lin J, Li X (2018) Single-particle tracking for the quantification of membrane protein dynamics in living plant cells. Mol Plant 11(11):1315–1327. https://doi.org/10.1016/j.molp.2018.09.008
Dange T, Grunwald D, Grunwald A, Peters R, Kubitscheck U (2008) Autonomy and robustness of translocation through the nuclear pore complex: a single-molecule study. J Cell Biol 183:77–86. https://doi.org/10.1083/jcb.200806173
Davies RG, Jans DA, Wagstaff KM (2010) Use of fluorescence photobleaching techniques to measure the kinetics of intracellular transport. Microscopy: Science, Technology, Applications and Education. ISBN-13: 978-84-614-6189-9
Donnaloja F, Jacchetti E, Soncini M, Raimondi MT (2019) Mechanosensing at the nuclear envelope by nuclear pore complex stretch activation and its effect in physiology and pathology. Front Physiol. https://doi.org/10.3389/fphys.2019.00896
Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, Elvassore N, Piccolo S (2011) Role of YAP/TAZ in mechanotransduction. Nature 474(7350):179–183. https://doi.org/10.1038/nature10137
Elosegui-Artola A, Andreu I, Beedle AEM, Lezamiz A, Uroz M, Kosmalska AJ, Oria R, Kechagia JZ, Rico-Lastres P, Le Roux AL, Shanahan CM, Trepat X, Navajas D, Garcia-Manyes S, Roca-Cusachs P (2017) Force triggers YAP nuclear entry by regulating transport across nuclear pores. Cell 171(6):1397–1410.e14. https://doi.org/10.1016/j.cell.2017.10.008
Elson EL (2011) Fluorescence correlation spectroscopy: past, present, future. Biophys J 101(12):2855–2870. https://doi.org/10.1016/j.bpj.2011.11.012
Engler AJ, Sen S, Sweeney HL, Discher DE (2006) Matrix elasticity directs stem cell lineage specification. Cell. 126(4):677–689
Fahrenkrog B, Aebi U (2003) The nuclear pore complex: nucleocytoplasmic transport and beyond. Nat Rev Mol Cell Biol 4(10):757–766. https://doi.org/10.1038/nrm1230
Garcia A, Rodriguez Matas JF, Raimondi MT (2016) Modeling of the mechano-chemical behaviour of the nuclear pore complex: current research and perspectives. Integr Biol (Camb) 8(10):1011–1021. https://doi.org/10.1039/c6ib00153j
García-González A, Jacchetti E, Marotta R, Tunesi M, Rodríguez Matas JF, Raimondi MT (2018) The effect of cell morphology on the permeability of the nuclear envelope to diffusive factors. Front Physiol 9:925. https://doi.org/10.3389/fphys.2018.00925
Gerace L, Burke B (1998) Functional organization of the nuclear envelope. Annu Rev Cell Biol 4:335–374. https://doi.org/10.1146/annurev.cb.04.110188.002003
Giepmans BN, Adams SR, Ellisman MH, Tsien RY (2006) The fluorescent toolbox for assessing protein location and function. Science 312(5771):217–224. https://doi.org/10.1126/science.1124618
Goryaynov A, Yang W (2014) Role of molecular charge in nucleocytoplasmic transport. PLoS One 9(2):e88792. https://doi.org/10.1371/journal.pone.0088792
Goryaynov A, Ma J, Yang W (2012) Single-molecule studies of nucleocytoplasmic transport: from one dimension to three dimensions. Integr Biol (Camb) 4(1):10–21. https://doi.org/10.1039/c1ib00041a
Gu Y (2018) The nuclear pore complex: a strategic platform for regulating cell signaling. New Phytol 219(1):25–30. https://doi.org/10.1111/nph.14756
Guerquin MJ, Charvet B, Nourissat G, Havis E, Ronsin O, Bonnin MA, Ruggiu M, Olivera-Martinez I, Robert N, Lu Y, Kadler KE, Baumberger T, Doursounian L, Berenbaum F, Duprez D (2013) Transcription factor EGR1 directs tendon differentiation and promotes tendon repair. J Clin Invest 123(8):3564–3576. https://doi.org/10.1172/JCI67521
Guilak F, Cohen DM, Estes BT, Gimble JM, Liedtke W, Chen CS (2009) Control of stem cell fate by physical interactions with the extracellular matrix. Cell Stem Cell 5(1):17–26. https://doi.org/10.1016/j.stem.2009.06.016
Hamann A, Nguyen A, Pannier AK (2019) Nucleic acid delivery to mesenchymal stem cells: a review of nonviral methods and applications. J Biol Eng 13:7. https://doi.org/10.1186/s13036-019-0140-0
Haws HJ, McNeil MA, Hansen MD (2016) Control of cell mechanics by RhoA and calcium fluxes during epithelial scattering. Tissue Barriers 4(3):e1187326. https://doi.org/10.1080/21688370.2016.1187326
He XW, He XS, Lian L, Wu XJ, Lan P (2012) Systemic infusion of bone marrow-derived mesenchymal stem cells for treatment of experimental colitis in mice. Dig Dis Sci 57(12):3136–3144. https://doi.org/10.1007/s10620-012-2290-5
Heo SJ, Thorpe SD, Driscoll TP, Duncan RL, Lee DA, Mauck RL (2015) Biophysical regulation of chromatin architecture instills a mechanical memory in mesenchymal stem cells. Sci Rep 5:16895. https://doi.org/10.1038/srep16895
Hime G, Abud H (2013) Transcriptional and translational regulation of stem cells. Adv Exp Med Biol 786:213–219. ISBN 978-94-007-6621-1 (EDS). https://doi.org/10.1007/978-94-007-6621-1
Huang CY, Hagar KL, Frost LE, Sun Y, Cheung HS (2004) Effects of cyclic compressive loading on chondrogenesis of rabbit bone-marrow derived mesenchymal stem cells. Stem Cells 22(3):313–323. https://doi.org/10.1634/stemcells.22-3-313
Huang Y, Jia X, Bai K, Gong X, Fan Y (2010) Effect of fluid shear stress on cardiomyogenic differentiation of rat bone marrow mesenchymal stem cells. Arch Med Res 41(7):497–505. https://doi.org/10.1016/j.arcmed.2010.10.002
Ishikawa-Ankerhold HC, Ankerhold R, Drummen GP (2012) Advanced fluorescence microscopy techniques--FRAP, FLIP, FLAP, FRET and FLIM. Molecules 17(4):4047–4132. https://doi.org/10.3390/molecules17044047
Jortikka MO, Parkkinen JJ, Inkinen RI, Kärner J, Järveläinen HT, Nelimarkka LO, Tammi MI, Lammi MJ (2000) The role of microtubules in the regulation of proteoglycan synthesis in chondrocytes under hydrostatic pressure. Arch Biochem Biophys 374:172–180. https://doi.org/10.1006/abbi.1999.1543
Kang M, Day CA, Drake K, Kenworthy AK, Di Benedetto E (2009) A generalization of theory for two-dimensional fluorescence recovery after photobleaching applicable to confocal laser scanning microscopes. Biophys J 97(5):1501–1511. https://doi.org/10.1016/j.bpj.2009.06.017
Keeling MC, Flores LR, Dodhy AH, Murray ER, Gavara N (2017) Actomyosin and vimentin cytoskeletal networks regulate nuclear shape, mechanics and chromatin organization. Sci Rep 7(1):5219. https://doi.org/10.1038/s41598-017-05467-x
Keminer O, Peters R (1999) Permeability of single nuclear pores. Biophys J 77(1):217–228. https://doi.org/10.1016/S0006-3495(99)76883-9
Keppler A, Gendreizig S, Gronemeyer T, Pick H, Vogel H, Johnsson K (2002) A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat Biotechnol 21:86–89. https://doi.org/10.1038/nbt765
Killaars AR, Grim JC, Walker CJ, Hushka EA, Brown TE, Anseth KS (2018) Extended exposure to stiff microenvironments leads to persistent chromatin remodeling in human mesenchymal stem cells. Adv Sci (Weinh) 6(3):1801483. https://doi.org/10.1002/advs.201801483
Knockenhauer KE, Schwartz TU (2016) The nuclear pore complex as a flexible and dynamic gate. Cell 164:1162–1171. https://doi.org/10.1016/j.cell.2016.01.034
Köster M, Frahm T, Hauser H (2005) Nucleocytoplasmic shuttling revealed by FRAP and FLIP technologies. Curr Opin Biotechnol 16(1):28–34. https://doi.org/10.1016/j.copbio.2004.11.002
Kubitscheck U, Grünwald D, Hoekstra A, Rohleder D, Kues T, Siebrasse JP, Peters R (2005) Nuclear transport of single molecules: dwell times at the nuclear pore complex. J Cell Biol 168:233–243. https://doi.org/10.1083/jcb.200411005
Le W, Yao J (2017) The effect of myostatin (GDF-8) on proliferation and tenocyte differentiation of rat bone marrow-derived mesenchymal stem cells. J Hand Surg Asian Pac 22(2):200–207. https://doi.org/10.1142/S0218810417500253
Lee HJ, Choi BH, Min BH, Son YS, Park SR (2006) Low-intensity ultrasound stimulation enhances chondrogenic differentiation in alginate culture of mesenchymal stem cells. Artif Organs 30(9):707–715. https://doi.org/10.1111/j.1525-1594.2006.00288.x
Lezon TR, Sali A, Bahar I (2009) Global motions of the nuclear pore complex: insights from elastic network models. PLoS Comput Biol 5:e1000496. https://doi.org/10.1371/journal.pcbi.1000496
Li R, Liang L, Dou Y, Huang Z, Mo H, Wang Y, Yu B (2015) Mechanical stretch inhibits mesenchymal stem cell adipogenic differentiation through TGFβ1/Smad2 signaling. J Biomech 48(13):3665–3671. https://doi.org/10.1016/j.jbiomech.2015.08.013
Lippincott-Schwartz J, Patterson GH (2003) Development and use of fluorescent protein markers in living cells. Science 300(5616):87–91. https://doi.org/10.1126/science.1082520
Lippincott-Schwartz J, Snapp E, Kenworthy A (2001) Studying protein dynamics in living cell. Nat Rev Mol Cell Biol 2(6):444–456. https://doi.org/10.1038/35073068
Liu H, Zhang C, Zhu S, Lu P, Zhu T, Gong X, Zhang Z, Hu J, Yin Z, Heng BC, Chen X, Ouyang HW (2015) Mohawk promotes the tenogenesis of mesenchymal stem cells through activation of the TGFβ signaling pathway. Stem Cells 33(2):443–455. https://doi.org/10.1002/stem.1866
Liu C, Liu YL, Perillo EP, Dunn AK, Yeh HC (2016) Single-molecule tracking and its application in biomolecular binding detection. IEEE J Sel Top Quantum Electron 22(4):6804013. https://doi.org/10.1109/JSTQE.2016.2568160
Loffreda A, Jacchetti E, Antunes S, Rainone P, Daniele T, Morisaki T, Bianchi ME, Tacchetti C, Mazza D (2017) Live-cell p53 single-molecule binding is modulated by C-terminal acetylation and correlates with transcriptional activity. Nat Commun 8(1):313. https://doi.org/10.1038/s41467-017-00398-7
Lombardi ML, Jaalouk DE, Shanahan CM, BurkeB RKJ, Lammerding J (2011) The interaction between nesprins and sun proteins at the nuclear envelope is critical for force transmission between the nucleus and cytoskeleton. J Biol Chem 286:26743–26753. https://doi.org/10.1074/jbc.M111.233700
Los GV, Encell LP, McDougall MG, Hartzell DD, Karassina N, Zimprich C, Wood MG, Learish R (2008) HaloTag: a novel protein labeling technology for cell imaging and protein analysis. ACS Chem Biol 3(6):373–382. https://doi.org/10.1021/cb800025k
Luo ZJ, Seedhom BB (2007) Light and low-frequency pulsatile hydrostatic pressure enhances extracellular matrix formation by bone marrow mesenchymal cells: an in-vitro study with special reference to cartilage repair. Proc Inst Mech Eng H 221(5):499–507. https://doi.org/10.1243/09544119JEIM199
Ma J, Yang W (2010) Three-dimensional distribution of transient interactions in the nuclear pore complex obtained from single-molecule snapshots. PNAS 107(16):7305–7310. https://doi.org/10.1073/pnas.0908269107
Madigan M, Atoui R (2018) Therapeutic use of stem cells for myocardial infarction. Bioengineering (Basel) 5(2):28. https://doi.org/10.3390/bioengineering5020028
Marrelli M, Paduano F, Tatullo M (2015) Human periapical cyst-mesenchymal stem cells differentiate into neuronal cells. J Dent Res 94(6):843–852. https://doi.org/10.1177/0022034515570316
Maurisse R, De Semir D, Emamekhoo H, Bedayat B, Abdolmohammadi A, Parsi H, Gruenert DC (2010) Comparative transfection of DNA into primary and transformed mammalian cells from different lineages. BMC Biotechnol 10:9. https://doi.org/10.1186/1472-6750-10-9
Mazza D, Cella F, Vicidomini G, Krol S, Diaspro A (2007) Role of three-dimensional bleach distribution in confocal and two-photon fluorescence recovery after photobleaching experiments. Appl Opt 46(30):7401–7411. https://doi.org/10.1364/AO.46.007401
McMurray RJ, Dalby MJ, Tsimbouri PM (2015) Using biomaterials to study stem cell mechanotransduction, growth and differentiation. J Tissue Eng Regen Med 9:528–539. https://doi.org/10.1002/term.1957
Meier RP, Müller YD, Morel P, Gonelle-Gispert C, Bühler LH (2013) Transplantation of mesenchymal stem cells for the treatment of liver diseases, is there enough evidence? Stem Cell Res 11(3):1348–1364. https://doi.org/10.1016/j.scr.2013.08.011
Miroshnikova YA, Nava MM, Wickström SA (2017) Emerging roles of mechanical forces in chromatin regulation. J Cell Sci 130(14):2243–2250. https://doi.org/10.1242/jcs.202192
Moon MY, Kim HJ, Choi BY, Sohn M, Chung TN, Suh SW (2018) Zinc promotes adipose-derived mesenchymal stem cell proliferation and differentiation towards a neuronal fate. Stem Cells Int 2018:5736535. https://doi.org/10.1155/2018/5736535
Mortensen KI, Churchman LS, Spudich JA, Flyvbjerg H (2010) Optimized localization analysis for single-molecule tracking and super-resolution microscopy. Nat Methods 7(5):377–381. https://doi.org/10.1038/nmeth.1447
Mueller F, Morisaki T, Mazza D, McNally JG (2012) Minimizing the impact of photoswitching of fluorescent proteins on FRAP analysis. Biophys J 102(7):1656–1665. https://doi.org/10.1016/j.bpj.2012.02.029
Mueller F, Stasevich TJ, Mazza D, McNally JG (2013) Quantifying transcription factor kinetics: at work or at play? Crit Rev Biochem Mol Biol 48(5):492–514. https://doi.org/10.3109/10409238.2013.833891
Murphy CM, Matsiko A, Haugh MG, Gleeson JP, O’Brien FJ (2012) Mesenchymal stem cell fate is regulated by the composition and mechanical properties of collagen-glycosaminoglycan scaffolds. J Mech Behav Biomed Mater 11:53–62. https://doi.org/10.1016/j.jmbbm.2011.11.009
Naim B, Brumfeld V, Kapon R, Kiss V, Nevo R, Reich Z (2007) Passive and facilitated transport in nuclear pore complexes is largely uncoupled. J Biol Chem 282(6):3881–3888. https://doi.org/10.1074/jbc.M608329200
Nava MM, Raimondi MT, Pietrabissa R (2012) Controlling self-renewal and differentiation of stem cells via mechanical cues. J Biomed Biotechnol 2012:797410. https://doi.org/10.1155/2012/797410
Nava MM, Raimondi MT, Pietrabissa R (2014) Bio-chemo-mechanical models for nuclear deformation in adherent eukaryotic cells. Biomech Model Mechanobiol 13(5):929–943. https://doi.org/10.1007/s10237-014-0558-8
Oldenburg AR, Collas P (2016) Map** nuclear lamin-genome interactions by chromatin immunoprecipitation of nuclear lamins. Methods Mol Biol 1411:315–324. https://doi.org/10.1007/978-1-4939-3530-7_20
Paine PL, Moore LC, Horowitz SB (1975) Nuclear envelope permeability. Nature 254(5496):109–114. https://doi.org/10.1038/254109a0
Park JS, Chu JS, Cheng C, Chen F, Chen D, Li S (2004) Differential effects of equiaxial and uniaxial strain on mesenchymal stem cells. Biotechnol Bioeng 88(3):359–368. https://doi.org/10.1002/bit.20250
Pownall ME, Gustafsson MK, Emerson CP Jr (2002) Myogenic regulatory factors and the specification of muscle progenitors in vertebrate embryos. Annu Rev Cell Dev Biol 18:747–783. https://doi.org/10.1146/annurev.cellbio.18.012502.105758
Raimondi MT, Eaton SM, Nava MM, Laganà M, Cerullo G, Osellame R (2012) Two-photon laser polymerization: from fundamentals to biomedical application in tissue engineering and regenerative medicine. J Appl Biomater Funct Mater 10(1):55–65. https://doi.org/10.5301/JABFM.2012.9278
Reits EA, Neefjes JJ (2001) From fixed to FRAP: measuring protein mobility and activity in living cells. Nat Cell Biol 3(6):E145–E147. https://doi.org/10.1038/35078615
Ribbeck K, Gorlich D (2001) Kinetic analysis of translocation through nuclear pore complexes. EMBO J 20(6):1320–1330. https://doi.org/10.1093/emboj/20.6.1320
Schäck L, Budde S, Lenarz T, Krettek C, Gross G, Windhagen H, Hoffmann A, Warnecke A (2016) Induction of neuronal-like phenotype in human mesenchymal stem cells by overexpression of neurogenin1 and treatment with neurotrophins. Tissue Cell 48(5):524–532. https://doi.org/10.1016/j.tice.2016.06.011
Sen B, **e Z, Case N, Styner M, Rubin CT, Rubin J (2011) Mechanical signal influence on mesenchymal stem cell fate is enhanced by incorporation of refractory periods into the loading regimen. J Biomech 44(4):593–599. https://doi.org/10.1016/j.jbiomech.2010.11.022
Sikavitsas VI, Bancroft GN, Holtorf HL, Jansen JA, Mikos AG (2003) Mineralized matrix deposition by marrow stromal osteoblasts in 3D perfusion culture increases with increasing fluid shear forces. Proc Natl Acad Sci U S A 100(25):14683–14688. https://doi.org/10.1073/pnas.2434367100
Sittichokechaiwut A, Edwards JH, Scutt AM, Reilly GC (2010) Short bouts of mechanical loading are as effective as dexamethasone at inducing matrix production by human bone marrow mesenchymal stem cell. Eur Cell Mater 20:45–57. https://doi.org/10.22203/eCM.v020a05
Steward AJ, Wagner DR, Kelly DJ (2013) The pericellular environment regulates cytoskeletal development and the differentiation of mesenchymal stem cells and determines their response to hydrostatic pressure. Eur Cell Mater 25:167–178. https://doi.org/10.22203/eCM.v025a12
Stewart M (2007) Molecular mechanism of the nuclear protein import cycle. Nat Rev Mol Cell Biol 8(3):195–208. https://doi.org/10.1038/nrm2114
Sun M, Chi G, Li P, Lv S, Xu J, Xu Z, **a Y, Tan Y, Xu J, Li L, Li Y (2018) Effects of matrix stiffness on the morphology, adhesion, proliferation and osteogenic differentiation of mesenchymal stem cells. Int J Med Sci 15(3):257–268. https://doi.org/10.7150/ijms.21620
Sunn KL, Eisman JA, Gardiner EM, Jans DA (2005) FRAP analysis of nucleocytoplasmic dynamics of the vitamin D receptor splice variant VDRB1: preferential targeting to nuclear speckles. Biochem J 388(Pt 2):509–514. https://doi.org/10.1042/BJ20042040
Swift J, Ivanovska IL, Buxboim A, Harada T, Dingal PC, Pinter J, Pajerowski JD, Spinler KR, Shin JW, Tewari M, Rehfeldt F, Speicher DW, Discher DE (2013) Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science 341(6149):1240104. https://doi.org/10.1126/science.1240104
Tajik A, Zhang Y, Wei F, Sun J, Jia Q, Zhou W, Singh R, Khanna N, Belmont AS, Wang N (2016) Transcription upregulation via force-induced direct stretching of chromatin. Nat Mater 15(12):1287–1296. https://doi.org/10.1038/nmat4729
Terry LJ, Wente SR (2009) Flexible gates: dynamic topologies and functions for FG nucleoporins in nucleocytoplasmic transport. Eukaryot Cell 8(12):1814–1827. https://doi.org/10.1128/EC.00225-09
Tokunaga M, Imamoto N, Sakata-Sogawa K (2008) Highly inclined thin illumination enables clear single-molecule imaging in cells. Nat Methods 5:159–161. https://doi.org/10.1038/nmeth1171
Tu LC, Musser SM (2011) Single molecule studies of nucleocytoplasmic transport. Biochim Biophys Acta 1813(9):1607–1618. https://doi.org/10.1016/j.bbamcr.2010.12.011
Vandromme M, Cavadore JC, Bonnieu A, Froeschlé A, Lamb N, Fernandez A (1995) Two nuclear localization signals present in the basic-helix 1 domains of MyoD promote its active nuclear translocation and can function independently. Proc Natl Acad Sci U S A 92(10):4646–4650. https://doi.org/10.1073/pnas.92.10.4646
Volarevic V, Markovic BS, Gazdic M, Volarevic A, Jovicic N, Arsenijevic N, Armstrong L, Djonov V, Lako M, Stojkovic M (2018) Ethical and safety issues of stem cell-based therapy. Int J Med Sci 15(1):36–45. https://doi.org/10.7150/ijms.21666
Wang D, Jiang X, Lu A, Tu M, Huang W, Huang P (2018) BMP14 induces tenogenic differentiation of bone marrow mesenchymal stem cells in vitro. Exp Ther Med 16(2):1165–1174. https://doi.org/10.3892/etm.2018.6293
Wei X, Henke VG, Strubing C, Brown EB, Clapham DE (2003) Real-time imaging of nuclear permeation by EGFP in single intact cells. Biophys J 84:1317–1327. https://doi.org/10.1016/S0006-3495(03)74947-9
Wente SR, Rout MP (2010) The nuclear pore complex and nuclear transport. Cold Spring Harb Perspect Biol 2(10):a000562. https://doi.org/10.1101/cshperspect.a000562
Wright MO, Stockwell RA, Nuki G (1992) Response of plasma membrane to applied hydrostatic pressure in chondrocytes and fibroblasts. Connect Tissue Res 28(1–2):49–70. https://doi.org/10.3109/03008209209014227
Wu J, Lewis AH, Grandl J (2017) Touch, tension, and transduction - the function and regulation of piezo ion channels. Trends Biochem Sci 42(1):57–71. https://doi.org/10.1016/j.tibs.2016.09.004
Yanagisawa M, Suzuki N, Mitsui N, Koyama Y, Otsuka K, Shimizu N (2007) Effects of compressive force on the differentiation of pluripotent mesenchymal cells. Life Sci 81(5):405–412. https://doi.org/10.1016/j.lfs.2007.06.004
Yang W, Musser SM (2006) Nuclear import time and transport efficiency depend on importin β concentration. J Cell Biol 174(7):951–961. https://doi.org/10.1083/jcb.200605053
Yang W, Gelles J, Musser SM (2004) Imaging of single-molecule translocation through nuclear pore complexes. Proc Natl Acad Sci U S A 101(35):12887–12892. https://doi.org/10.1073/pnas.0403675101
Yang C, Tibbitt MW, Basta L, Anseth KS (2014) Mechanical memory and dosing influence stem cell fate. Nat Mater 13(6):645–652. https://doi.org/10.1038/nmat3889
Yourek G, McCormick SM, Mao JJ, Reilly GC (2010) Shear stress induces osteogenic differentiation of human mesenchymal stem cells. Regen Med 5(5):713–724. https://doi.org/10.2217/rme.10.60
Zhao L, Chen S, Shi X, Cao H, Li L (2018) A pooled analysis of mesenchymal stem cell-based therapy for liver disease. Stem Cell Res Ther 9(1):72. https://doi.org/10.1186/s13287-018-0816-2
Acknowledgments
The authors wish to thank Dr. Davide Mazza and Emanuele Colombo, Istituto Scientifico Ospedale San Raffaele, Centro di Imaging Sperimentale in Milano, for the single-molecule images used in this paper.
Funding
This project received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (grant agreement no. 646990 - NICHOID).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Boeri, L., Albani, D., Raimondi, M.T. et al. Mechanical regulation of nucleocytoplasmic translocation in mesenchymal stem cells: characterization and methods for investigation. Biophys Rev 11, 817–831 (2019). https://doi.org/10.1007/s12551-019-00594-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12551-019-00594-3