
Abstract
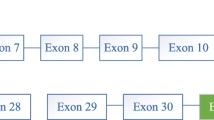
Pompe disease or glycogen storage disease type II is a glycogen storage disorder associated with malfunction of the acid α-glucosidase enzyme (GAA; EC.3.2.1.3) leading to intracellular aggregations of glycogenin muscles. The infantile-onset type is the most life-threatening form of this disease, in which most of patients suffer from cardiomyopathy and hypotonia in early infancy. In this study, a typical case of Pompe disease was reported in an Iranian patient using molecular analysis of the GAA gene. Our results revealed a new c.1824_1828dupATACG mutation in exon 13 of the GAA gene. In conclusion, with the finding of this novel mutation, the genotypic spectrum of Iranian patients with Pompe disease has been extended, facilitating the definition of disease-related mutations.

Similar content being viewed by others

References
Hirschhorn R, Reuser AJ (2001) Glycogen storage disease type II: acid alpha-glucosidase (acid maltase) deficiency. In: Scriver C, Beaudet A, Sly W (eds) The metabolic and molecular bases of inherited disease. McGraw Hill, New York, pp 3389–3420
Ausems M, Verbiest J, Hermans M, Kroos M, Beemer F, Wokke J, Sandkuijl L, Reuser A, Van der Ploeg A (1999) Frequency of glycogen storage disease type II in The Netherlands: implications for diagnosis and genetic counselling. Eur J Hum Genet EJHG 7(6):713
Kroos M, Pomponio RJ, van Vliet L, Palmer RE, Phipps M, Van der Helm R, Halley D, Reuser A (2008) Update of the Pompe disease mutation database with 107 sequence variants and a format for severity rating. Hum Mutat 29(6):E13–E26
Hers H (1963) α-Glucosidase deficiency in generalized glycogen-storage disease (Pompe’s disease). Biochem J 86(1):11
Salafsky I, Nadler H (1973) A fluorometric assay of alpha-glucosidase and its application in the study of Pompe’s disease. J Lab Clin Med 81(3):450
Kishnani PS, Hwu W-L, Mandel H, Nicolino M, Yong F, Corzo D (2006) A retrospective, multinational, multicenter study on the natural history of infantile-onset Pompe disease. J Pediatr 148(5):671–676
Brown BI, Brown DH, Jeffrey PL (1970) Simultaneous absence of α-1, 4-glucosidase and α-1, 6-glucosidase activities (pH 4) in tissues of children with type II glycogen storage disease. Biochemistry 9(6):1423–1428
Swallow DM, Corney G, Harris H, Hirschhorn R (1975) Acid α-glucosidase: a new polymorphism in man demonstrable by ‘affinity’electrophoresis. Ann Hum Genet 38(4):391–406
Martiniuk F, Honig J, Hirschhorn R (1984) Further studies of the structure of human placental acid α-glucosidase. Arch Biochem Biophys 231(2):454–460
Martiniuk F, Mehler M, Pellicer A, Tzall S, La Badie G, Hobart C, Ellenbogen A, Hirschhorn R (1986) Isolation of a cDNA for human acid alpha-glucosidase and detection of genetic heterogeneity for mRNA in three alpha-glucosidase-deficient patients. Proc Natl Acad Sci 83(24):9641–9644
Martiniuk F, Mehler M, Tzall S, Meredith G, Hirschhorn R (1990) Sequence of the cDNA and 5′-flanking region for human acid α-glucosidase, detection of an intron in the 5′ untranslated leader sequence, definition of 18-bp polymorphisms, and differences with previous cDNA and amino acid sequences. DNA Cell Biol 9(2):85–94
Hoefsloot L, Hoogeveen-Westerveld M, Kroos M, Van Beeumen J, Reuser A, Oostra B (1988) Primary structure and processing of lysosomal alpha-glucosidase; homology with the intestinal sucrase-isomaltase complex. EMBO J 7(6):1697
Kroos M, Waitfield A, Joosse M, Winchester B, Reuser A, MacDermot K (1997) A novel acid α-glucosidase mutation identified in a Pakistani family with glycogen storage disease type II. J Inherit Metab Dis 20(4):556–558
Pagon RA, Adam MP, Bird TD, Dolan CR, Fong C-T, Stephens K, Leslie N, Tinkle BT (2013) Glycogen Storage Disease Type II (Pompe Disease)
Hudgson P, Gardner-Medwin D, Worsfold M, Pennington R, Walton JN (1968) Adult myopathy from glycogen storage disease due to acid maltase deficiency. Brain 91(3):435–462
Hermans MM, Dv Leenen, Kroos MA, Beesley CE, Van der Ploeg AT, Sakuraba H, Wevers R, Kleijer W, Michelakakis H, Kirk EP (2004) Twenty-two novel mutations in the lysosomal α-glucosidase gene (GAA) underscore the genotype–phenotype correlation in glycogen storage disease type II. Hum Mutat 23(1):47–56
Müller-Felber W, Horvath R, Gempel K, Podskarbi T, Shin Y, Pongratz D, Walter MC, Baethmann M, Schlotter-Weigel B, Lochmüller H (2007) Late onset Pompe disease: clinical and neurophysiological spectrum of 38 patients including long-term follow-up in 18 patients. Neuromuscul Disord 17(9):698–706
Kroos M, Hoogeveen-Westerveld M, Michelakakis H, Pomponio R, Van der Ploeg A, Halley D, Reuser A, Augoustides-Savvopoulou P, Ausems M, Llona JB (2012) Update of the pompe disease mutation database with 60 novel GAA sequence variants and additional studies on the functional effect of 34 previously reported variants. Hum Mutat 33(8):1161–1165
Acknowledgments
We would like to thank Genzyme Company and their representative, Dr. Soheila Saadati, for their support in the diagnosis and treatment of Pompe patients. This work was supported by a grant from Dr. M. Houshmand, to whom we give our greatest appreciation.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Aryani, O., Manshadi, M.D., Tondar, M. et al. A newly identified c.1824_1828dupATACG mutation in exon 13 of the GAA gene in infantile-onset glycogen storage disease type II (Pompe disease). Mol Biol Rep 41, 6211–6214 (2014). https://doi.org/10.1007/s11033-014-3500-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-014-3500-3