
Abstract
Three new dinuclear triple helicates were synthesised using a ditopic semi-rigid pyridylylimine ligand L, separated by a diphenoxy-biphenol spacer providing considerable length to the backbone. L and the new large dinuclear triple helicate complexes [Fe2L3](BF4)4 (1), [Ni2L3](BF4)4 (2) and [Zn2L3](BF4)4 (3) have been characterised in solution and solid state. Single crystal X-ray diffraction was used to investigate overall complex ion shape as the coordination sphere was modulated by metal ion selection. Small differences in complex shape were seen to arise due to subtle distortions in coordination sphere environments. This study sheds light on how the length and twist of dinuclear triple helicates can be tuned by selection of coordinating metal ion.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Metal-directed self-assembly of complex supramolecular architectures remains an area of insatiable research interest [1,2,3,4,5]. Of particular interest is the dinuclear triple helicate, whose synthesis is made possible by design of suitable bis-bidentate ligand backbones [1,2,3,4,5,6,7,8,9,10,11,12,13,14]. These ligands must be tailored to provide suitable donor atoms to allow stable coordination environments with metal ions, while containing the delicate balance of required rigidity to undergo the conformational change necessary to achieve a helical assembly [1, 8,9,10,11, 15, 16]. The intrinsic nature in which these complexes assemble makes them good candidates to bind with DNA, so they are therefore interesting targets for molecular design [17,18,19]. Designable molecular assemblies that possess the ability to bind to DNA with recognition could give rise to a new class of therapeutic drugs [18, 20, 21].
As an extension to our previous work [6,7,8,9,10,11, 14] on dinuclear triple helicates we have designed three new dinuclear triple helicate assemblies. The ligand L (scheme 1) was achieved by incorporating the same long semi-rigid ligand backbone as our previous helicate [8], this time incorporating a pyridyl functional moiety that gives rise to a stronger ligand field. This study investigates the impact that coordination sphere size has on the shape and size of these assemblies, brought about by the changing of metal ions. The logic was to study the effect of three d-block metals with increasing coordination sphere size and distortion. For this, FeII, NiII and ZnII were selected, a d6, d8 and d10 electron configuration. With a strong-field ligand, these metals bear t2g6 occupancy with eg0, eg2 and eg4 configurations, respectively. By this method, we investigate the effect that d-orbital occupancy and coordination sphere size has on the shape and size of these assemblies. By investigating the effects that small changes have on the overall shape and size of these architectures, it may be possible to modulate the supramolecular contacts which they can form. This could result in interesting properties such as tuneable DNA binding affinity and varied crystal packing arrangements.

Synthesis of L from Schiff-base condensation reaction of 4,4′-(1,1′-Biphenyl-4,4′-diyldioxy)dianiline and 2-pyridinecarboxaldehyde in excess to ensure reaction completion
Here we describe three new [M2L3](BF4)4 helicates; [Fe2L3](BF4)4 (1), [Ni2L3](BF4)4 (2) and [Zn2L3](BF4)4 (3), have been characterised by SCXRD, HR ESI-MS, TGA-DSC, FT-IR and SEM-EDS. We also report structural characteristics for these new assemblies and relate them to the differences in their coordination sphere size. These structural characteristics were compared to those of our previously reported fully low spin (LS) FeII dinuclear triple helicate which employed 4-thiazolylimine chelating moieties. This work represents an important progression towards achieving new long dinuclear triple helicates, presenting a similar ligand bearing a stronger ligand field.
Experimental
Materials and instrumentation
All chemicals and reagents were purchased from commercial sources and used without further purification.
High-resolution electrospray ionisation–mass spectrometry (HR ESI-MS) data was obtained using a Waters Xevo QToF mass spectrometer, operating in positive ion mode. Samples were dissolved with acetonitrile (MeCN) and directly injected via syringe into the ESI source. Calibration of high-resolution masses was achieved using a Waters lock-spray system.
Fourier transform infrared (FT-IR) measurements were undertaken on a Bruker Vertex 70 with a diamond ATR crystal. Spectra were recorded over a 400–4000 cm− 1 range.
Thermogravimetric Analysis (TGA) and Differential Scanning Calorimetry (DSC) experiments were conducted using a NETZSCH STA449 Jupiter Simultaneous Thermal Analysis (STA) instrument. TGA measurements were conducted using Nitrogen for both the purge and protective gases; an aluminium crucible and a temperature range of 300–550 K at a rate of 10 K min− 1.
Scanning Electron Microscopy and Energy Dispersive X-ray Spectroscopy (SEM-EDS) analysis were carried out using an SEM JEOL JSM 6510LV with an attached silicon drift EDS detector and operated in low-vac with a chamber pressure of 30 Pa. The accelerating voltage was set to 25.0 kV. All samples were mounted to an aluminium stub with double-sided conductive carbon tape. Images were taken without surface coating.
Nuclear Magnetic Resonance (NMR) experiments were carried out on a Bruker Advance 400 MHz NMR Spectrometer.
Crystallographic data collection and refinement
Single Crystal X-ray Diffraction (SCXRD) experiments for complexes1–3 were carried out on a Bruker D8 Quest Single Crystal diffractometer with Photon II detector using a IµS 3.0 Microfocus source with Mo-Kα radiation (λ = 0.710723 Å) at the Mark Wainwright Analytical Centre at the University of New South Wales (UNSW). Data integration and reduction was undertaken using Bruker APEX 3 software. For all data, absorption corrections were applied using the multi scan method in SADABS [22]. The structures were solved by intrinsic phasing and the full-matrix least-squares refinements were carried out using a suite of SHELX programs [23, 24] via the OLEX2 graphical interface [25]. Non-hydrogen atoms were refined anisotropically except for some disordered anions and solvent molecules. Carbon-bound hydrogen atoms were included in idealised positions and refined using a riding model. In all structures, a solvent mask was applied to account for residual low intensity peaks, arising due to disordered solvent.
All crystallographic data in CIF format has been deposited at the Cambridge Crystallographic Data Centre with CCDC No: 2,329,598–2,329,600. It is available free of charge from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1 EZ, UK; fax: (+ 44) 1223-336-033; or e-mail: deposit@ccdc.cam.ac.uk. Specific refinement details and crystallographic data for each structure are present below in the crystallographic section.
Synthesis
Preparation of ligand (L)
To a stirring white suspension of 4,4′-(1,1′-Biphenyl-4,4′-diyldioxy)dianiline (1,000 mg, 2.71 mmol) in 25 mL of methanol (MeOH), 2-pyridinecarboxaldehyde (1,453 mg, 13.56 mmol) diluted in 10 mL of MeOH was added dropwise. Upon addition the suspension turned yellow. The reaction was allowed to reflux under nitrogen atmosphere overnight. The resultant creamy yellow precipitate was filtered while hot, the product was then washed with cold methanol and allowed to air dry, yielding the final product as a pale-yellow powder. Yield = 88%. HR ESI-MS, (positive ion detection, MeCN, m/z): calc. for [L + H]+, 547.2134; found 547.2081; FT-IR (ATR, νmax/cm− 1): 1580, 1567, 1496, 1433, 1268, 1252, 850, 826; 1H NMR (400 MHz, DMSO-d6) δ 8.72 (dd, J = 7.5, 1.6 Hz, 2 H, Ar-H), 8.65 (s, 2 H, N = C-H of Imine), 8.16 (dd, J = 7.5, 1.7 Hz, 2 H, Ar-H), 7.96 (td, J = 7.4, 1.5 Hz, 2 H, Ar-H), 7.70 (d, J = 8.5 Hz, 4 H, Ar-H), 7.53 (td, J = 7.4, 1.6 Hz, 2 H, Ar-H), 7.46 (d, J = 8.7 Hz, 4 H, Ar-H), 7.20–7.09 (m, 8 H, Ar-H).
Preparation of complexes 1–3
To a stirring suspension of L (200 mg, 0.36 mmol) in 20 mL of acetonitrile (MeCN), a solution of iron(II) tetrafluoroborate (82 mg, 0.24 mmol) in 5 mL of MeCN was added dropwise. The resulting deep purple solution was heated and reacted under a nitrogen atmosphere for 1–2 h. The resulting reaction mixture was filtered while hot, X-ray quality crystals were obtained by vapour diffusion of diethyl ether into the reaction mixture. Diffusion yielded large deep purple rod crystals, which were obtained by filtration, washed with diethyl ether, and dried in air. Yield = 44%. HR ESI-MS, (positive ion detection, MeCN, m/z): calc. for [Fe2L3]4+, 437.6229; found 437.5878. FT-IR (ATR, νmax/cm− 1): 1591, 1489, 1230, 1201, 1167, 1061.
To a stirring suspension of L (100 mg, 0.18 mmol) in 15 mL of MeCN, a solution of nickel(II) tetrafluoroborate (42 mg, 0.12 mmol) in 5 mL of MeCN was added dropwise. The resulting clear orange solution was heated and reacted under a nitrogen atmosphere for 1–2 h. The resulting reaction mixture was filtered while hot, X-ray quality crystals were obtained by vapour diffusion of diethyl ether into the reaction mixture. Diffusion yielded dark orange plate crystals, which were obtained by filtration, washed with diethyl ether, and dried in air. Crystals were not air stable. Yield = 29%. HR ESI-MS, (positive ion detection, MeCN, m/z): calc. for [Ni2L3]4+, 439.0992; found 439.1219; FT-IR (ATR, νmax/cm− 1): 1595, 1489, 1237, 1200, 1060.
To a stirring suspension of [L] (200 mg, 0.36 mmol) in 15 mL of MeCN, a solution of zinc(II) tetrafluoroborate (58 mg, 0.24 mmol) in 5 mL of MeCN was added dropwise. The resulting pale-yellow solution was heated and reacted under a nitrogen atmosphere for 1–2 h. The solution was filtered while hot, the remaining solvent was evaporated off under vacuum, to obtain a bright yellow precipitate. Yield = 64%. HR ESI-MS, (positive ion detection, MeCN, m/z): cald. for [Zn2L3]4+, 442.1187; found 442.0901; FT-IR (ATR, νmax/cm− 1): 1596, 1489, 1242, 1061. X-ray quality crystals were obtained by redissolving the crude product in MeCN, followed by vapour diffusion of diethyl ether into the solution, yielding clear yellow plate crystals. Crystals were not air stable.
Results and discussion
Synthesis and characterisation
The synthesis of L (Scheme 1) involves a Schiff-base condensation reaction using a 1:5 stoichiometric ratio of 4,4′-(1,1′-Biphenyl-4,4′-diyldioxy)dianiline and 2-pyridine carboxaldehyde in a methanol solution performed under inert conditions while the reaction mixture is refluxed overnight, adapted from previously reported methods [S9 for full descriptions and calculated values
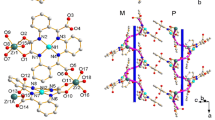
By this analysis we demonstrate that the distortion of the octahedral coordination sphere has a direct impact on the shape of the dinuclear triple helicate architecture. Subtle angular differences between the intermetallic axis and chelate vectors are correlated with slight changes to the shape of the overall complex ion (Fig. 4). Of course, larger coordination spheres contribute to the helicate length and twist, but these are not the only factor, as demonstrated by the analysis of the [LS-LS] FeII thiazolylimine helicate. There would be some contribution by the crystal packing on the helicate shape, which would act to balance the values of the chelate group angles. This is demonstrated by the relatively stable shape parameters in the presently reported series 1–3, as compared to the values established with the [LS-LS] FeII thiazolylimine dinuclear triple helicate.

Overlays of helicate units from SCXRD experiments. (a) The three structurally similar helicates of the presently reported series 1–3, coloured red (1), blue (2) and green (3), (b) The two structurally dissimilar Fe(II) helicates discussed in this study. 1 is shown in darker colours, while the 4-thaizolyl helicate is shown in lighter colours. Colour scheme for 1: Ligand A (red), symmetry equivalent ligand A (blue), ligand B (green). Colour scheme for 4-thiazolylimine analogue: Ligand C (pink), ligand B (light blue), ligand A (light green). Note that the disordered second position of ligand C has been omitted for clarity
