
Abstract
Background
Co-occurrence of polycystic kidney disease and hyperinsulinemic hypoglycemia has been reported in children in a few families associated with a variant in the promotor of the PMM2 gene, at position -167 upstream of the coding sequence. PMM2 encodes phosphomannomutase 2, a key enzyme in N-glycosylation. While biallelic coding PMM2 mutations are involved in congenital disorder of glycosylation CDG1A, that particular variant in the promoter of the gene, either in the homozygous state or associated with a mutation in the coding exons of the gene, is thought to restrict the N-glycosylation defect to the kidney and the pancreas.
Methods
Targeted exome sequencing of a panel of genes involved in monogenic kidney diseases.
Results
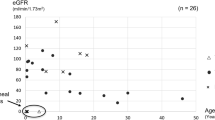
We identified a PMM2 variant at position -167 associated with a pathogenic PMM2 variant in the coding exons in 3 families, comprising 6 cases affected with a cystic kidney disease. The spectrum of phenotypes was very broad, from extremely enlarged fetal cystic kidneys in the context of a COACH-like syndrome, to isolated cystic kidney disease with small kidneys, slowly progressing toward kidney failure in adulthood. Hypoglycemia was reported only in one case.
Conclusion
These data show that the PMM2 promotor variation, in trans of a PMM2 coding mutation, is associated with a wide spectrum of kidney phenotypes, and is not always associated with extra-renal symptoms. When present, extra-renal defects may include COACH-like syndrome. These data prompt screening of PMM2 in unresolved cases of fetal hyperechogenic/cystic kidneys as well as in cystic kidney disease in children and adults.

Graphical Abstract


Similar content being viewed by others


References
Soares AR, Figueiredo CM, Quelhas D, Silva ES, Freitas J, Oliveira MJ, Faria S, Fortuna AM, Borges T (2020) Hyperinsulinaemic hypoglycaemia and polycystic kidney disease - a rare case concerning PMM2 gene pleiotropy. Eur Endocrinol 16:66–68. https://doi.org/10.17925/EE.2020.16.1.66
Moreno Macián F, De Mingo Alemany C, León Cariñena S, Ortega López P, Rausell Felix D, Aparisi Navarro M, Martinez Matilla M, Cardona Gay C, Martinez Castellano F, Albiach Mesado V (2020) Mutations in PMM2 gene in four unrelated Spanish families with polycystic kidney disease and hyperinsulinemic hypoglycemia. J Pediatr Endocrinol Metab 33:1283–1288. https://doi.org/10.1515/jpem-2020-0168
Cabezas OR, Flanagan SE, Stanescu H, García-Martínez E, Caswell R, Lango-Allen H, Antón-Gamero M, Argente J, Bussell A-M, Brandli A, Cheshire C, Crowne E, Dumitriu S, Drynda R, Hamilton-Shield JP, Hayes W, Hofherr A, Iancu D, Issler N, Jefferies C, Jones P, Johnson M, Kesselheim A, Klootwijk E, Koettgen M, Lewis W, Martos JM, Mozere M, Norman J, Patel V, Parrish A, Pérez-Cerdá C, Pozo J, Rahman SA, Sebire N, Tekman M, Turnpenny PD, Hoff WV, Viering DHHM, Weedon MN, Wilson P, Guay-Woodford L, Kleta R, Hussain K, Ellard S, Bockenhauer D (2017) Polycystic kidney disease with hyperinsulinemic hypoglycemia caused by a promoter mutation in phosphomannomutase 2. J Am Soc Nephrol 28:2529–2539. https://doi.org/10.1681/ASN.2016121312
Heidet L, Morinière V, Henry C, De Tomasi L, Reilly ML, Humbert C, Alibeu O, Fourrage C, Bole-Feysot C, Nitschké P, Tores F, Bras M, Jeanpierre M, Pietrement C, Gaillard D, Gonzales M, Novo R, Schaefer E, Roume J, Martinovic J, Malan V, Salomon R, Saunier S, Antignac C, Jeanpierre C (2017) Targeted exome sequencing identifies PBX1 as involved in monogenic congenital anomalies of the kidney and urinary tract. J Am Soc Nephrol 28:2901–2914. https://doi.org/10.1681/ASN.2017010043
Matthijs G, Schollen E, Bjursell C, Erlandson A, Freeze H, Imtiaz F, Kjaergaard S, Martinsson T, Schwartz M, Seta N, Vuillaumier-Barrot S, Westphal V, Winchester B (2000) Mutations in PMM2 that cause congenital disorders of glycosylation, type Ia (CDG-Ia). Hum Mutat 16:386–394. https://doi.org/10.1002/1098-1004(200011)16:5<386::AID-HUMU2>3.0.CO;2-Y
Barone R, Sturiale L, Sofia V, Ignoto A, Fiumara A, Sorge G, Garozzo D, Zappia M (2008) Clinical phenotype correlates to glycoprotein phenotype in a sib pair with CDG-Ia. Am J Med Genet A 146A:2103–2108. https://doi.org/10.1002/ajmg.a.32446
van Ommen CH, Peters M, Barth PG, Vreken P, Wanders RJ, Jaeken J (2000) Carbohydrate-deficient glycoprotein syndrome type 1a: a variant phenotype with borderline cognitive dysfunction, cerebellar hypoplasia, and coagulation disturbances. J Pediatr 136:400–403. https://doi.org/10.1067/mpd.2000.103503
Briones P, Vilaseca MA, García-Silva MT, Pineda M, Colomer J, Ferrer I, Artigas J, Jaeken J, Chabás A (2001) Congenital disorders of glycosylation (CDG) may be underdiagnosed when mimicking mitochondrial disease. Eur J Paediatr Neurol 5:127–131. https://doi.org/10.1053/ejpn.2001.0483
Freeze HH, Westphal V (2001) Balancing N-linked glycosylation to avoid disease. Biochimie 83:791–799. https://doi.org/10.1016/s0300-9084(01)01292-5
Kjaergaard S, Schwartz M, Skovby F (2001) Congenital disorder of glycosylation type Ia (CDG-Ia): phenotypic spectrum of the R141H/F119L genotype. Arch Dis Child 85:236–239. https://doi.org/10.1136/adc.85.3.236
Vuillaumier-Barrot S, Le Bizec C, De Lonlay P, Madinier-Chappat N, Barnier A, Dupré T, Durand G, Seta N (2006) PMM2 intronic branch-site mutations in CDG-Ia. Mol Genet Metab 87:337–340. https://doi.org/10.1016/j.ymgme.2005.10.015
Romano S, Bajolle F, Valayannopoulos V, Lyonnet S, Colomb V, de Baracé C, Vouhe P, Pouard P, Vuillaumier-Barrot S, Dupré T, de Keyzer Y, Sidi D, Seta N, Bonnet D, de Lonlay P (2009) Conotruncal heart defects in three patients with congenital disorder of glycosylation type Ia (CDG Ia). J Med Genet 46:287–288. https://doi.org/10.1136/jmg.2008.057620
Thong MK, Fietz M, Nicholls C, Lee MH, Asma O (2009) Congenital disorder of glycosylation type Ia in a Malaysian family: clinical outcome and description of a novel PMM2 mutation. J Inherit Metab Dis 32(Suppl 1):S41–S44. https://doi.org/10.1007/s10545-009-1031-1
Bell CJ, Dinwiddie DL, Miller NA, Hateley SL, Ganusova EE, Mudge J, Langley RJ, Zhang L, Lee CC, Schilkey FD, Sheth V, Woodward JE, Peckham HE, Schroth GP, Kim RW, Kingsmore SF (2011) Carrier testing for severe childhood recessive diseases by next-generation sequencing. Sci Transl Med 3:65ra4. https://doi.org/10.1126/scitranslmed.3001756
Vega AI, Pérez-Cerdá C, Abia D, Gámez A, Briones P, Artuch R, Desviat LR, Ugarte M, Pérez B (2011) Expression analysis revealing destabilizing mutations in phosphomannomutase 2 deficiency (PMM2-CDG): expression analysis of PMM2-CDG mutations. J Inherit Metab Dis 34:929–939. https://doi.org/10.1007/s10545-011-9328-2
Bortot B, Cosentini D, Faletra F, Biffi S, De Martino E, Carrozzi M, Severini GM (2013) PMM2-CDG: phenotype and genotype in four affected family members. Gene 531:506–509. https://doi.org/10.1016/j.gene.2013.07.083
Lazarin GA, Haque IS, Nazareth S, Iori K, Patterson AS, Jacobson JL, Marshall JR, Seltzer WK, Patrizio P, Evans EA, Srinivasan BS (2013) An empirical estimate of carrier frequencies for 400+ causal Mendelian variants: results from an ethnically diverse clinical sample of 23,453 individuals. Genet Med 15:178–186. https://doi.org/10.1038/gim.2012.114
Ream MA, Mikati MA (2014) Clinical utility of genetic testing in pediatric drug-resistant epilepsy: a pilot study. Epilepsy Behav 37:241–248. https://doi.org/10.1016/j.yebeh.2014.06.018
Stefanits H, Konstantopoulou V, Kuess M, Milenkovic I, Matula C (2014) Initial diagnosis of the congenital disorder of glycosylation PMM2-CDG (CDG1a) in a 4-year-old girl after neurosurgical intervention for cerebral hemorrhage. J Neurosurg Pediatr 14:546–549. https://doi.org/10.3171/2014.7.PEDS14102
Andreotti G, Monti MC, Citro V, Cubellis MV (2015) Heterodimerization of two pathological mutants enhances the activity of human phosphomannomutase2. PLoS One 10:e0139882. https://doi.org/10.1371/journal.pone.0139882
Yuste-Checa P, Gámez A, Brasil S, Desviat LR, Ugarte M, Pérez-Cerdá C, Pérez B (2015) The effects of PMM2-CDG-causing mutations on the folding, activity, and stability of the PMM2 protein. Hum Mutat 36:851–860. https://doi.org/10.1002/humu.22817
Chan B, Clasquin M, Smolen GA, Histen G, Powe J, Chen Y, Lin Z, Lu C, Liu Y, Cang Y, Yan Z, **a Y, Thompson R, Singleton C, Dorsch M, Silverman L, Su S-SM, Freeze HH, ** S (2016) A mouse model of a human congenital disorder of glycosylation caused by loss of PMM2. Hum Mol Genet 25:2182–2193. https://doi.org/10.1093/hmg/ddw085
Rossi M, Medina Escobar A, Ameghino L, Merello M (2017) Expanding the phenotype of phosphomannomutase-2 gene congenital disorder of glycosylation: cervical dystonia. J Neurol Sci 378:52–54. https://doi.org/10.1016/j.jns.2017.04.037
Vals M-A, Morava E, Teeäär K, Zordania R, Pajusalu S, Lefeber DJ, Õunap K (2017) Three families with mild PMM2-CDG and normal cognitive development. Am J Med Genet A 173:1620–1624. https://doi.org/10.1002/ajmg.a.38235
Bastaki F, Bizzari S, Hamici S, Nair P, Mohamed M, Saif F, Malik EM, Al-Ali MT, Hamzeh AR (2018) Single-center experience of N-linked congenital disorders of glycosylation with a summary of molecularly characterized cases in Arabs. Ann Hum Genet 82:35–47. https://doi.org/10.1111/ahg.12220
Rego S, Dagan-Rosenfeld O, Zhou W, Sailani MR, Limcaoco P, Colbert E, Avina M, Wheeler J, Craig C, Salins D, Röst HL, Dunn J, McLaughlin T, Steinmetz LM, Bernstein JA, Snyder MP (2018) High-frequency actionable pathogenic exome variants in an average-risk cohort. Cold Spring Harb Mol Case Stud 4:a003178. https://doi.org/10.1101/mcs.a003178
Aldinger KA, Timms AE, Thomson Z, Mirzaa GM, Bennett JT, Rosenberg AB, Roco CM, Hirano M, Abidi F, Haldipur P, Cheng CV, Collins S, Park K, Zeiger J, Overmann LM, Alkuraya FS, Biesecker LG, Braddock SR, Cathey S, Cho MT, Chung BHY, Everman DB, Zarate YA, Jones JR, Schwartz CE, Goldstein A, Hopkin RJ, Krantz ID, Ladda RL, Leppig KA, McGillivray BC, Sell S, Wusik K, Gleeson JG, Nickerson DA, Bamshad MJ, Gerrelli D, Lisgo SN, Seelig G, Ishak GE, Barkovich AJ, Curry CJ, Glass IA, Millen KJ, Doherty D, Dobyns WB (2019) Redefining the etiologic landscape of cerebellar malformations. Am J Hum Genet 105:606–615. https://doi.org/10.1016/j.ajhg.2019.07.019
Ceyhan-Birsoy O, Murry JB, Machini K, Lebo MS, Yu TW, Fayer S, Genetti CA, Schwartz TS, Agrawal PB, Parad RB, Holm IA, McGuire AL, Green RC, Rehm HL, Beggs AH, BabySeq Project Team (2019) Interpretation of genomic sequencing results in healthy and Ill newborns: results from the BabySeq Project. Am J Hum Genet 104:76–93. https://doi.org/10.1016/j.ajhg.2018.11.016
Lefrère B, Stepanian A, Charles P, Foulon-Pinto G, Béranger N, Alhenc-Gelas M, Drouet L, Siguret V (2019) Multifactorial hypercoagulable state associated with a thrombotic phenotype in phosphomannomutase-2 congenital disorder of glycosylation (PMM2-CDG): case report and brief review of the literature. Thromb Res 178:75–78. https://doi.org/10.1016/j.thromres.2019.04.010
Chong M, Yoon G, Susan-Resiga D, Chamberland A, Cheillan D, Paré G, Seidah NG (2020) Hypolipidaemia among patients with PMM2-CDG is associated with low circulating PCSK9 levels: a case report followed by observational and experimental studies. J Med Genet 57:11–17. https://doi.org/10.1136/jmedgenet-2019-106102
Peng T, Lv C, Tan H, Huang J, He H, Wang Y, Zeng M, Yi D, Li J, Deng H, Shi X, **ao H (2020) Novel PMM2 missense mutation in a Chinese family with non-syndromic premature ovarian insufficiency. J Assist Reprod Genet 37:443–450. https://doi.org/10.1007/s10815-019-01675-8
Powis Z, Towne MC, Hagman KDF, Blanco K, Palmaer E, Castro A, Sajan SA, Radtke K, Feyma TJ, Juliette K, Tang S, Sidiropoulos C (2020) Clinical diagnostic exome sequencing in dystonia: genetic testing challenges for complex conditions. Clin Genet 97:305–311. https://doi.org/10.1111/cge.13657
Yıldız Y, Arslan M, Çelik G, Kasapkara ÇS, Ceylaner S, Dursun A, Sivri HS, Coşkun T, Tokatlı A (2020) Genotypes and estimated prevalence of phosphomannomutase 2 deficiency in Turkey differ significantly from those in Europe. Am J Med Genet A 182:705–712. https://doi.org/10.1002/ajmg.a.61488
Noelle V, Knuepfer M, Pulzer F, Schuster V, Siekmeyer W, Matthijs G, Vogtmann C (2005) Unusual presentation of congenital disorder of glycosylation type 1a: congenital persistent thrombocytopenia, hypertrophic cardiomyopathy and hydrops-like aspect due to marked peripheral oedema. Eur J Pediatr 164:223–226. https://doi.org/10.1007/s00431-004-1611-x
Casado M, O’Callaghan MM, Montero R, Pérez-Cerda C, Pérez B, Briones P, Quintana E, Muchart J, Aracil A, Pineda M, Artuch R (2012) Mild clinical and biochemical phenotype in two patients with PMM2-CDG (congenital disorder of glycosylation Ia). Cerebellum 11:557–563. https://doi.org/10.1007/s12311-011-0313-y
Resende C, Carvalho C, Alegria A, Oliveira D, Quelhas D, Bandeira A, Proença E (2014) Congenital disorders of glycosylation with neonatal presentation. BMJ Case Rep 2014:bcr2013010037. https://doi.org/10.1136/bcr-2013-010037
Coutelier M, Hammer MB, Stevanin G, Monin M-L, Davoine C-S, Mochel F, Labauge P, Ewenczyk C, Ding J, Gibbs JR, Hannequin D, Melki J, Toutain A, Laugel V, Forlani S, Charles P, Broussolle E, Thobois S, Afenjar A, Anheim M, Calvas P, Castelnovo G, de Broucker T, Vidailhet M, Moulignier A, Ghnassia RT, Tallaksen C, Mignot C, Goizet C, Le Ber I, Ollagnon-Roman E, Pouget J, Brice A, Singleton A, Durr A, Paraplegia S, Network A (2018) Efficacy of exome-targeted capture sequencing to detect mutations in known cerebellar ataxia genes. JAMA Neurol 75:591–599. https://doi.org/10.1001/jamaneurol.2017.5121
Schiff M, Roda C, Monin M-L, Arion A, Barth M, Bednarek N, Bidet M, Bloch C, Boddaert N, Borgel D, Brassier A, Brice A, Bruneel A, Buissonnière R, Chabrol B, Chevalier M-C, Cormier-Daire V, De Barace C, De Maistre E, De Saint-Martin A, Dorison N, Drouin-Garraud V, Dupré T, Echenne B, Edery P, Feillet F, Fontan I, Francannet C, Labarthe F, Gitiaux C, Héron D, Hully M, Lamoureux S, Martin-Coignard D, Mignot C, Morin G, Pascreau T, Pincemaille O, Polak M, Roubertie A, Thauvin-Robinet C, Toutain A, Viot G, Vuillaumier-Barrot S, Seta N, De Lonlay P (2017) Clinical, laboratory and molecular findings and long-term follow-up data in 96 French patients with PMM2-CDG (phosphomannomutase 2-congenital disorder of glycosylation) and review of the literature. J Med Genet 54:843–851. https://doi.org/10.1136/jmedgenet-2017-104903
Parisi MA (2009) Clinical and molecular features of Joubert syndrome and related disorders. Am J Med Genet C: Semin Med Genet 151C:326–340. https://doi.org/10.1002/ajmg.c.30229
Iannicelli M, Brancati F, Mougou-Zerelli S, Mazzotta A, Thomas S, Elkhartoufi N, Travaglini L, Gomes C, Ardissino GL, Bertini E, Boltshauser E, Castorina P, D’Arrigo S, Fischetto R, Leroy B, Loget P, Bonnière M, Starck L, Tantau J, Gentilin B, Majore S, Swistun D, Flori E, Lalatta F, Pantaleoni C, Penzien J, Grammatico P, International JSRD Study Group, Dallapiccola B, Gleeson JG, Attie-Bitach T, Valente EM (2010) Novel TMEM67 mutations and genotype-phenotype correlates in meckelin-related ciliopathies. Hum Mutat 31:E1319–E1331. https://doi.org/10.1002/humu.21239
Doherty D, Parisi MA, Finn LS, Gunay-Aygun M, Al-Mateen M, Bates D, Clericuzio C, Demir H, Dorschner M, van Essen AJ, Gahl WA, Gentile M, Gorden NT, Hikida A, Knutzen D, Ozyurek H, Phelps I, Rosenthal P, Verloes A, Weigand H, Chance PF, Dobyns WB, Glass IA (2010) Mutations in 3 genes (MKS3, CC2D2A and RPGRIP1L) cause COACH syndrome (Joubert syndrome with congenital hepatic fibrosis). J Med Genet 47:8–21. https://doi.org/10.1136/jmg.2009.067249
Höck M, Wegleiter K, Ralser E, Kiechl-Kohlendorfer U, Scholl-Bürgi S, Fauth C, Steichen E, Pichler K, Lefeber DJ, Matthjis G, Keldermans L, Maurer K, Zschocke J, Karall D (2015) ALG8-CDG: novel patients and review of the literature. Orphanet J Rare Dis 10:73. https://doi.org/10.1186/s13023-015-0289-7
Tham E, Eklund EA, Hammarsjö A, Bengtson P, Geiberger S, Lagerstedt-Robinson K, Malmgren H, Nilsson D, Grigelionis G, Conner P, Lindgren P, Lindstrand A, Wedell A, Albåge M, Zielinska K, Nordgren A, Papadogiannakis N, Nishimura G, Grigelioniene G (2016) A novel phenotype in N-glycosylation disorders: Gillessen-Kaesbach-Nishimura skeletal dysplasia due to pathogenic variants in ALG9. Eur J Hum Genet 24:198–207. https://doi.org/10.1038/ejhg.2015.91
Marques-da-Silva D, Francisco R, Webster D, Dos Reis Ferreira V, Jaeken J, Pulinilkunnil T (2017) Cardiac complications of congenital disorders of glycosylation (CDG): a systematic review of the literature. J Inherit Metab Dis 40:657–672. https://doi.org/10.1007/s10545-017-0066-y
Boskovski MT, Yuan S, Pedersen NB, Goth CK, Makova S, Clausen H, Brueckner M, Khokha MK (2013) The heterotaxy gene GALNT11 glycosylates Notch to orchestrate cilia type and laterality. Nature 504:456–459. https://doi.org/10.1038/nature12723
Hofherr A, Köttgen M (2013) Induced pluripotent stem cells from polycystic kidney disease patients: a novel tool to model the pathogenesis of cystic kidney disease. J Am Soc Nephrol 24:1507–1509. https://doi.org/10.1681/ASN.2013070767
Hu J, Harris PC (2020) Regulation of polycystin expression, maturation and trafficking. Cell Signal 72:109630. https://doi.org/10.1016/j.cellsig.2020.109630
Inoue Y, Sohara E, Kobayashi K, Chiga M, Rai T, Ishibashi K, Horie S, Su X, Zhou J, Sasaki S, Uchida S (2014) Aberrant glycosylation and localization of polycystin-1 cause polycystic kidney in an AQP11 knockout model. J Am Soc Nephrol 25:2789–2799. https://doi.org/10.1681/ASN.2013060614
Besse W, Chang AR, Luo JZ, Triffo WJ, Moore BS, Gulati A, Hartzel DN, Mane S, Regeneron Genetics Center, Torres VE, Somlo S, Mirshahi T (2019) ALG9 mutation carriers develop kidney and liver cysts. J Am Soc Nephrol 30:2091–2102. https://doi.org/10.1681/ASN.2019030298
Kane MS, Davids M, Bond MR, Adams CJ, Grout ME, Phelps IG, O’Day DR, Dempsey JC, Li X, Golas G, Vezina G, Gunay-Aygun M, Hanover JA, Doherty D, He M, Malicdan MCV, Gahl WA, Boerkoel CF (2017) Abnormal glycosylation in Joubert syndrome type 10. Cilia 6:2. https://doi.org/10.1186/s13630-017-0048-6
Acknowledgements
We thank all patients and families. We thank all clinicians who recorded data in health records. We thank Patrick Nitschké from the Bioinformatic Plateform, Paris Descartes Sorbonne Paris Cité University, Imagine Institute, Paris, France, and Cecile Fourrage from the Genetic Department, APHP, Hôpital universitaire Necker-Enfants malades, Paris, France, for their help with NGS.
Funding
This work was supported by state funding from the gence Nationale de la Recherche (ANR) under “Investissements d’avenir” program (ANR-10-IAHU-01) and by a public grant overseen by the ANR as part of the second “Investissements d’Avenir” program (reference: ANR-17-RHUS-0002).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Dorval, G., Jeanpierre, C., Morinière, V. et al. Cystic kidney diseases associated with mutations in phosphomannomutase 2 promotor: a large spectrum of phenotypes. Pediatr Nephrol 36, 2361–2369 (2021). https://doi.org/10.1007/s00467-021-04953-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-021-04953-9