
Abstract
Our group has shown that 1-year smoking cessation persisted or increased airway inflammation in chronic obstructive pulmonary disease (COPD). We compared adenosine and adenosine receptor (AR) expression in COPD and asymptomatic smokers (AS) before and after 1-year smoking cessation. Sputum cytospins and bronchial biopsies of (ex)smoking COPD patients and AS were studied for A1R, A2AR, A2BR, and A3R expression. Adenosine and inflammatory mediators were measured in sputum supernatants. At baseline, COPD patients had lower levels of adenosine and higher levels of vascular endothelial growth factor in sputum than AS. Smoking cessation induced significantly different effects in COPD than in AS, i.e. an increase in percentages of A3R expressing neutrophils and A1R expressing macrophages in COPD as increase in adenosine and monocyte chemoattractant protein-1 levels in sputum. Adenosine-related effector mechanisms may contribute to the persistence and progression of airway inflammation in COPD following 1-year smoking cessation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Chronic obstructive pulmonary disease (COPD) is a disease caused mainly by smoking and it is characterized by chronic airflow limitation and a range of pathological changes in the lung [1]. It is known that smoking cessation stops the accelerated lung function decline present in COPD [2, 3]. We found that smoking cessation improves both direct (methacholine) and indirect (adenosine 5′-monophosphate, AMP) airway hyperresponsiveness (AHR) after 1-year smoking cessation [4]. Interestingly, improvement of AHR was not associated with changes in lung function or sputum inflammation. Our previous paper showed that COPD patients who successfully ceased smoking for 1-year, overall airway inflammation persisted in bronchial biopsies, while the number of neutrophils, lymphocytes, interleukin (IL)-8, and eosinophil-cationic-protein levels significantly increased in sputum. In addition, no significant changes in vascular adhesion molecules (i.e. P-selectin, E-selectin, intercellular antigen-1 (ICAM-1) or vascular cell adhesion molecule (VCAM)) were found with smoking cessation in COPD patients also suggesting an ongoing inflammatory process in the airways. In asymptomatic smokers who successfully quitted smoking, some inflammatory markers (e.g. sputum macrophages, eosinophils, and IL-8) significantly decreased while others did not change [5]. This indicates that factors other than inflammatory cells or measured mediators likely induced the observed improvement in AHR.
Adenosine has been suggested to play a role in COPD [6]. Patients with COPD are significantly more responsive to AMP than healthy smoking volunteers [7, 8] and smokers have significantly increased concentrations of adenosine in the airway lining fluid [9]. Recently, a first report on the presence of adenosine receptors in lung parenchyma showed an enhanced density and decreased affinity of the adenosine receptors in subjects with COPD compared with control smokers [10]. Adenosine is a purine nucleoside that is released during tissue hypoxia and inflammation. It has both pro- and anti-inflammatory features, which are mediated by four different G-protein-coupled receptors, A1 (Gi coupled), A2A (Gs coupled), A2B (Gs and Gq11 coupled), and A3 (Gs and Gq11 coupled). Activation of adenosine receptors (AR) can have different effects, depending on the type of G protein that is involved and the cell type [6]. Activation of Gi-protein-coupled receptors decreases adenylyl cyclase activity and cAMP levels whereas activation of Gs coupled receptors increases cAMP levels [11]. With respect to the cell types involved, activation of A1R on neutrophils promotes adherence to endothelial cells and chemotaxis, indicating a pro-inflammatory response [12], whereas activation of A1R on cells from the monocytes/macrophage lineage inhibits the production of several pro-inflammatory cytokines (TNF, IL-8, and IL-6) and enhances the release of the anti-inflammatory cytokine IL-10, displaying an anti-inflammatory response [13, 14]. Therefore, each receptor can be either beneficially and/or detrimentally implicated in the inflammatory process of COPD.
In this study, our primary aim was to investigate whether adenosine-related effector mechanisms could be involved in the persistence and/or progression of the inflammatory response in COPD following 1-year smoking cessation. Therefore, adenosine content and receptor expression in sputum (macrophages and neutrophils) and bronchial biopsies of smokers with COPD and asymptomatic smokers were measured, both before and after 1-year smoking cessation. Since activation of adenosine receptors can influence the secretion of mediators from inflammatory cells, we related changes in adenosine and its receptors with changes in growth factors (VEGF) and chemokines (monocyte chemoattractant protein (MCP)-1) related to remodeling and inflammation that may underlie lung function loss in COPD.
Materials and methods
Subjects
Bronchial biopsy and sputum material of 11 COPD and 15 asymptomatic smoking subjects who successfully stopped smoking for at least 1 year from the study of Willemse et al. were studied (patient characteristics; Table 1) [5, 4, 15]. In short, the main important inclusion and exclusion criteria for both groups were: COPD (according to the American Thoracic Society (ATS)/ European Respiratory Society (ERS) guidelines [16]): forced expiratory volume (FEV)1/forced vital capacity post-bronchodilator <0.7, and chronic respiratory symptoms for at least 3 months for two successive years. Asymptomatic smokers: FEV1 ≥ 85%, predicted, and no chronic respiratory symptoms or airway obstruction. Participants in both groups were between 45 and 65 years of age, had >10 packyears smoking, smoked ≥10 cigarettes/day, had reversibility to salbutamol <9% of the predicted FEV1, did not use inhaled or oral corticosteroids in the previous 6 months, had no signs of atopy, and no respiratory tract infections 1 month prior to the study. During the study, COPD patients only used beta2-agonists or ipratropium on a regular basis; inhaled corticosteroids were not used. Only in case of an exacerbation, a short course of oral corticosteroids was allowed. Before each measurement, subjects were asked not to use long- or short-acting β2-agonists and/or ipratropium at least 12 h before the test. They did not suffer from a respiratory tract infection nor used oral corticosteroids in the month prior to any of the measurements.
The local medical ethics committee of our University Medical Center Groningen approved the study protocol and all subjects gave their written informed consent.
Study design and smoking cessation program
The study design and smoking cessation program have been described previously [5]. Sputum cells and supernatant as well as bronchial biopsies were only used if present both before and after 1-year smoking cessation.
Bronchoscopy and biopsy processing
Bronchial biopsies were taken from the subcarinae of the right, middle, or lower lobe and processed as described previously [17]. For immunostaining, sections (one per patient) were dried and fixed in acetone for 10 min and washed with phosphate-buffered saline (PBS). The following polyclonal antibodies were used: rabbit anti-rat adenosine A1 receptor, rabbit anti-canine adenosine A2A receptor, rabbit anti-human adenosine A2B receptor (all from Alpha Diagnostic International, San Antonio, TX, USA), and rabbit anti-human adenosine A3 receptor (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Antibodies directed against adenosine A1 and A2A receptors cross-reacted with human A1R and A2aR. The secondary antibody used was goat anti-rabbit biotin conjugated (SBA, Birmingham, AL, USA) and the tertiary antibody StreptABComplex peroxidase conjugated (DAKO, Glostrup, Denmark). As chromogen, 3-amino-9-ethyl-carbazol substrate was used; tissue was counterstained with Mayers’ Hematoxylin. Intensity of adenosine receptor expression in biopsies were scored in a blinded way (all by the same observer) with a semiquantitative three-point scale, with sections scored as negative, positive, or strongly positive. In addition, the total number of positive single cells in the biopsy was counted. Since not all biopsies presented all tissue structures scored, numbers presented can vary between graphs.
Sputum induction and processing
Sputum was induced by inhalation of hypertonic saline and processed as described previously [18]. Sputum samples consisting for >80% of squamous cells were excluded from analysis. Cytospins were dried and fixed in acetone for 10 min and washed with PBS for immunostaining. The same antibodies and chromogen substrate were used as described in the biopsy staining. Three hundred neutrophilic granulocytes and 300 macrophages were counted in every cytospin and scored as negative or positive; the percentage of cells expressing adenosine receptors was calculated. Due to the small numbers of lymphocytes present in sputum, we could not reliably quantify adenosine receptor expression on this cell type.
Measurement of adenosine in sputum supernatant using HPLC
Adenosine was measured using a Waters 2690 Alliance Separation Module HPLC system (Waters, Milford, MA, USA) coupled on-line to a Finnigan TSQ Quantum Triple Quadruple Mass Spectrometer (Finnigan Thermo Fisher Scientific, Waltham, MA, USA). Chromatographic separation was performed with a C18, 3 μm, Atlantis column (Waters) 2.1 mm I.D. × 100 mm length using a gradient of mobile phase A (ammonium acetate 20 mM, pH 7.0, in water) and B (ammonium acetate 20 mM, pH 7.0, in acetonitrile) at 0.2 mL/min flow rate. Before measurements samples were pre-treated as follows: 100 μl of sputum supernatant was acidified with 20 μl of HCl 120 mM, then 200 μl of chloroform was added to precipitate proteins and eliminate lipids. After vortexing and subsequent centrifugation, 80 μl of the aqueous supernatant was transferred to a glass injection vial and 5 μl of this solution was injected into the high-performance liquid chromatography (HPLC)-mass spectrometry (MS) system. MS detection was performed using ESI ionization in positive ion mode. Multiple-reaction monitoring mode was used to increase the sensitivity. Adenosine was quantified using the transition m/z 260 > m/z 136 corresponding to the loss of the ribofuranose ring. Samples were measured in a randomized way, twice independently at two different days. Results were in good agreement within a maximum of 10% variation. The calibration line consisted of eight points in the range of 5 to 1,000 ng/ml adenosine in a mixture of PBS and water (pretreated in the same way as the study samples). Adenosine levels are presented as concentrations.
Chemokine and growth factor measurement in sputum supernatant using multiplex ELISA
Sputum supernatant contents (sensitivity level) of monocyte chemoattractant protein (MCP)-1 (10 pg/ml), “regulated upon activation of normal T cell expressed and secreted” (RANTES, 15 pg/ml), tumor necrosis factor (TNF, 10 pg/ml), epidermal growth factor (EGF, 15 pg/ml), fibroblast growth factor (FGF)-basic (also known as FGF2, 15 pg/ml), vascular endothelial growth factor (VEGF, 15 pg/ml), and platelet-derived growth factor (PDGF)-BB (<10 pg/ml) were measured using a human custom multiplex antibody bead kit from Biosource International (Camarillo, CA, USA). Standard solutions were diluted with sputolysin reagent (Calbiochem®, Darmstadt, Germany) to copy the effect of it on sputum content during processing.
Statistical analysis
Statistics were performed using a Wilcoxon signed-rank test to assess differences between both measurements within the groups and a non-parametric Mann–Whitney U test to assess differences between both groups at baseline. A value of p < 0.05 was considered significant. Correlations between parameters were investigated using a Pearson’s Rho test. For some patients material at one time point was missing due to insufficient quality of the sample. At baseline the single measurements were used for baseline differences between both groups, but they were excluded from the paired test to obtain differences within the groups.
Results
Adenosine receptor expression in bronchial biopsies
The adenosine receptors A2A, A2B, and A3 were expressed by airway epithelial, endothelial, and smooth muscle cells in airway wall biopsies and by cells (morphologically macrophages) in the submucosa in both COPD and asymptomatic smokers (Fig. 1). In contrast, the A1 receptor was only expressed on a small number of cells (morphologically macrophages) in the tissue surrounding the airways. The pattern of expression of the four ARs was not different between COPD patients and their controls.

Adenosine receptor expression on bronchial biopsies from smoking asymptomatic (left panel) and COPD (right panel) subjects
The intensity of AR expression (scored semiquantitatively with immunohistochemistry) is presented in Fig. 2. There was no significant difference in AR expression between both groups at baseline. The A2A receptor expression on airway smooth muscle decreased significantly after 1-year smoking cessation (p = 0.025) in asymptomatic smokers, but not in COPD. A1R, A2BR, and A3R expression did not change with smoking cessation in both groups. In addition, no differences were found in the number of cells staining positive for all four ARs between the different groups. Total biopsy areas were similar for high and low numbers of positive cells.

Semi-quantitative scoring of the immunohistochemical staining of adenosine receptor expression on bronchial biopsies from smoking asymptomatic (AS) and COPD subjects, before and 1-year after smoking cessation. Structures present in bronchial biopsies were scored negative, positive, or strongly positive. Significant differences are depicted in the graphs. Paired data for 11 asymptomatic smokers and ten COPD patients
Adenosine receptor expression by sputum cells
In sputum, all four ARs were expressed by neutrophils and macrophages of both COPD and asymptomatic smokers. There was no difference in AR expression between both groups at baseline. The percentage of A3R positive neutrophils increased significantly in COPD patients after 1-year smoking cessation when compared to baseline (p = 0.038). This change in A3R expression was significantly different from the change occurring in asymptomatic smokers (p = 0.050, Fig. 3a). No changes were detected in percentages of neutrophils expressing A1R, A2AR, and A2BR. In COPD patients, the percentage of A1R positive macrophages increased after 1-year smoking cessation when compared to baseline (p = 0.017, Fig. 3b), an effect that was not observed in asymptomatic smokers. Smoking cessation had no effect on expression of the other ARs on macrophages. No significant differences were found in percentage expression of all ARs on macrophages between COPD and asymptomatic smokers.

Adenosine receptor expression on neutrophils (a) and macrophages (b) in sputum of COPD patients and asymptomatic smokers (AS), before and after 1-year smoking cessation. Significant differences are depicted in the graphs. Paired data for 11 asymptomatic smokers and ten COPD patients
Adenosine, growth factors, and cytokine levels in sputum supernatant
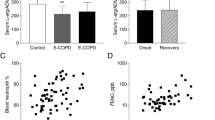
Adenosine levels in sputum supernatant were significantly lower in COPD at baseline (p = 0.017, Fig. 4a) than in asymptomatic smokers, whereas VEGF levels were higher (p = 0.020, Fig. 4b). In addition, the change in adenosine levels with smoking cessation was significantly different in COPD and asymptomatic smokers (p = 0.023), as was the change in MCP-1 (p = 0.013) (Fig. 4). No differences were detected in RANTES and other growth factors involved in tissue repair (EGF and PDGF-BB) (Fig. 4). Levels of FGF-b and TNF were below the sensitivity level and therefore not analyzed statistically.

Sputum adenosine (a), VEGF (b), EGF (c), PDGF-BB (d), MCP-1 (e), and RANTES (f) levels of CODP patients and asymptomatic smokers (AS), before and after 1-year smoking cessation. * Significant difference at baseline (p < 0.05) between COPD and AS, other significant differences are depicted in the graphs. Paired data for nine asymptomatic smokers and seven COPD patients
Discussion
We investigated the effect of smoking cessation on adenosine content and adenosine receptor expression in sputum and biopsies taken from COPD patients and asymptomatic smokers. Our most important observations were that COPD patients responded significantly different to smoking cessation than asymptomatic smokers with respect to sputum adenosine content, MCP-1 levels, and percentage of neutrophils expressing A3R and macrophages expressing A1R. This resulted in an increased adenosine content (trend) and increased percentage of A3R positive neutrophils and A1R positive macrophages in sputum of COPD patients. In addition, smokers with COPD have a lower level of adenosine and a higher level of VEGF in sputum than asymptomatic smokers.
Previous observations in these patients have elucidated that smoking cessation improved AHR, whereas inflammation persisted or even increased in sputum of COPD patients [4, 5]. Since adenosine is implicated in the development and persistence of inflammation, we investigated in the current study whether the observed increase in (aspects of) inflammation in COPD could be explained by the presence of adenosine and the expression of adenosine receptors as measured in two different compartments of the airways, i.e. sputum and bronchial biopsies.
We confirmed the pattern of ARs expression that was recently described by Varani et al. [10]. Thus, the A2AR and A3R were expressed on bronchial epithelium, smooth muscle cells, endothelial cells, and infiltrating cells (mostly macrophages), whereas A1R was only expressed by a few cells (morphologically macrophages). For A2BR, Varani and co-workers detected expression on mast cells and macrophages only, whereas we additionally detected expression on epithelium, smooth muscle and endothelial cells, using the same antibody. With respect to the intensity of AR expression, no differences were found between COPD and asymptomatic smokers in our study. Interestingly, Varani et al. detected an increased expression of A2AR and A3R at the mRNA level, a decreased expression of A2BR and no differences for A1R mRNA in airway wall tissue of COPD patients. We did not study mRNA expression of AR. Other differences between both studies may be explained by differences in the clinical characteristics of the investigated participants. At baseline, we investigated current smokers, whereas both the COPD and healthy smoking groups of Varani et al. consisted of a mixture of ex- and current smokers. Our data on the effects of 1-year smoking cessation show that this may have affected the results of the latter study. Also, some of the participants of Varani et al. were older and had higher packyears of smoking.
We report for the first time the expression of all four adenosine receptors on neutrophils and macrophages in sputum of COPD and asymptomatic smokers. At baseline, no differences were found between asymptomatic smokers and COPD patients. With respect to neutrophils, smoking cessation resulted in a significantly higher percentage of sputum neutrophils expressing A3R on their membrane in COPD, an effect not observed in asymptomatic smokers. The role of the human adenosine A3 receptor is poorly understood. mRNA for the adenosine A3 receptor is highest in the central nervous system, liver, and lung [19, 20]. A recent study showed that a ligand which combines adenosine A2A receptor agonist and A3 receptor antagonist activity inhibits the release of preformed granule proteins from neutrophils [21]. Another recent study showed that the A3R on neutrophils is additionally involved in neutrophilic chemotaxis [22]. This is of interest since we previously showed that the number of sputum neutrophils significantly increased in COPD patients who successfully quitted smoking for 1 year. The role of these neutrophils is still not clear but theoretically they could contribute to the enhanced adenosine levels after smoking cessation in COPD. The exact contribution of adenosine in the persistence and progression of the inflammatory response is further dictated by the expression pattern of the four different adenosine receptors.
The percentage of macrophages expressing the A1R was increased in COPD patients after 1-year of smoking cessation. Recently, a role for the A1R on monocytes was described in modulation of angiogenesis [23]. The A1R-selective agonist N6-cyclopenthyladenosine increased VEGF production by human monocytes in vitro and increased angiogenesis in a chicken chorioallantoic membrane model. In the present study, levels of chemokines and various growth factors were measured in sputum supernatant, to explain the increase in inflammation as a repair mechanism in the airways. Smoking cessation had a differential effect on MCP-1 production in sputum supernatant when comparing COPD with asymptomatic smokers. Since this chemokine is involved in recruitment of macrophages, the observed relative increase in MCP-1 in COPD with smoking cessation compared to asymptomatic individuals is in line with the previously published observation that the number of macrophages decreases upon smoking cessation in asymptomatic smokers, whereas it persists in COPD [5]. RANTES levels were similar in both groups and smoking cessation did not alter this. RANTES increases during COPD exacerbations [24] and is involved in chemotaxis of eosinophils. The percentage of eosinophils decreased in both COPD and asymptomatic patients after smoking cessation [5]. This thus could not be explained by RANTES. We found that COPD patients had higher levels of VEGF in sputum than asymptomatic smokers, compatible with findings described by Rovina et al. [25] and other research groups describing higher VEGF levels in sputum of patients suffering from chronic bronchitis and lower levels in patients with emphysema [25–27]. Cigarette smoking may upregulate VEGF, as suggested by an acute increase of VEGF plasma levels during smoking [28], yet we here show that smoking cessation did not reduce VEGF levels in both our groups.
Finally, we observed that smoking cessation decreased levels of adenosine in asymptomatic smokers, whereas it increased (trend) adenosine levels in COPD patients. This is of interest and in line with the observation that a number of inflammation markers significantly decreased in asymptomatic smokers who successfully quitted smoking while the number of sputum neutrophils, lymphocytes, interleukin-8, and eosinophil-cationic-protein levels significantly increased in COPD patients who successfully ceased smoking for 1 year [5].
Conclusions
We have shown that COPD patients respond significantly different to smoking cessation than asymptomatic smokers with respect to adenosine production and adenosine receptor expression. Our results may indicate that adenosine-related effector mechanisms are involved in the persistence and progression of the inflammatory response in COPD following 1-year smoking cessation.
References
Rabe KF, Hurd S, Anzueto A et al (2007) Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med 176:532–555
Anthonisen NR, Connett JE, Kiley JP et al (1994) Effects of smoking intervention and the use of an inhaled anticholinergic bronchodilator on the rate of decline of FEV1. The Lung Health Study. JAMA 272:1497–1505
Kanner RE, Connett JE, Williams DE et al (1999) Effects of randomized assignment to a smoking cessation intervention and changes in smoking habits on respiratory symptoms in smokers with early chronic obstructive pulmonary disease: the Lung Health Study. Am J Med 106:410–416
Willemse BW, ten Hacken NH, Rutgers B et al (2004) Smoking cessation improves both direct and indirect airway hyperresponsiveness in COPD. Eur Respir J 24:391–396
Willemse BW, ten Hacken NH, Rutgers B et al (2005) Effect of 1-year smoking cessation on airway inflammation in COPD and asymptomatic smokers. Eur Respir J 26:835–845
Polosa R (2002) Adenosine-receptor subtypes: their relevance to adenosine-mediated responses in asthma and chronic obstructive pulmonary disease. Eur Respir J 20:488–496
Oosterhoff Y, de Jong JW, Jansen MA et al (1993) Airway responsiveness to adenosine 5′-monophosphate in chronic obstructive pulmonary disease is determined by smoking. Am Rev Respir Dis 147:553–558
Oosterhoff Y, Jansen MA, Postma DS et al (1993) Airway responsiveness to adenosine 5′-monophosphate in smokers and nonsmokers with atopic asthma. J Allergy Clin Immunol 92:773–776
Driver AG, Kukoly CA, Ali S et al (1993) Adenosine in bronchoalveolar lavage fluid in asthma. Am Rev Respir Dis 148:91–97
Varani K, Caramori G, Vincenzi F et al (2006) Alteration of adenosine receptors in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 173:398–406
Cronstein BN (1994) Adenosine, an endogenous anti-inflammatory agent. 76:5–13
Cronstein BN, Daguma L, Nichols D et al (1990) The adenosine/neutrophil paradox resolved: human neutrophils possess both A1 and A2 receptors that promote chemotaxis and inhibit O2 generation, respectively. J Clin Invest 85:1150–1157
Hasko G, Szabo C, Nemeth ZH et al (1996) Adenosine receptor agonists differentially regulate IL-10, TNF-alpha, and nitric oxide production in RAW 264.7 macrophages and in endotoxemic mice. J Immunol 157:4634–4640
Le Moine O, Stordeur P, Schandene L et al (1996) Adenosine enhances IL-10 secretion by human monocytes. J Immunol 156:4408–4414
Willemse BW, ten Hacken NH, Rutgers B et al (2005) Association of current smoking with airway inflammation in chronic obstructive pulmonary disease and asymptomatic smokers. Respir Res 6:38
Celli BR, MacNee W (2004) Standards for the diagnosis and treatment of patients with COPD: a summary of the ATS/ERS position paper. Eur Respir J 23:932–946
Aleva RM, Kraan J, Smith M et al (1998) Techniques in human airway inflammation: quantity and morphology of bronchial biopsy specimens taken by forceps of three sizes. Chest 113:182–185
Rutgers SR, Timens W, Kaufmann HF et al (2000) Comparison of induced sputum with bronchial wash, bronchoalveolar lavage and bronchial biopsies in COPD. Eur Respir J 15:109–115
Ralevic V, Burnstock G (1998) Receptors for purines and pyrimidines. Pharmacol Rev 50:413–492
Fredholm BB, Arslan G, Halldner L et al (2000) Structure and function of adenosine receptors and their genes. Naunyn Schmiedebergs Arch Pharmacol 362:364–374
Bevan N, Butchers PR, Cousins R et al (2007) Pharmacological characterisation and inhibitory effects of (2R,3R,4S,5R)-2-(6-amino-2-{[(1S)-2-hydroxy-1-(phenylmethyl)ethyl]amino}-9 H-purin-9-yl)-5-(2-ethyl-2H-tetrazol-5-yl)tetrahydro-3,4-furandiol, a novel ligand that demonstrates both adenosine A(2A) receptor agonist and adenosine A(3) receptor antagonist activity. Eur J Pharmacol 564:219–225
Chen Y, Corriden R, Inoue Y et al (2006) ATP release guides neutrophil chemotaxis via P2Y2 and A3 receptors. Science 314:1792–1795
Clark AN, Youkey R, Liu X et al (2007) A1 adenosine receptor activation promotes angiogenesis and release of VEGF from monocytes. Circ Res 101:1075–7077
Zhu J, Qiu YS, Majumdar S et al (2001) Exacerbations of Bronchitis: bronchial eosinophilia and gene expression for interleukin-4, interleukin-5, and eosinophil chemoattractants. Am J Respir Crit Care Med 164:109–116
Rovina N, Papapetropoulos A, Kollintza A et al (2007) Vascular endothelial growth factor: an angiogenic factor reflecting airway inflammation in healthy smokers and in patients with bronchitis type of chronic obstructive pulmonary disease? Respir Res 8:53
Kanazawa H, Asai K, Hirata K et al (2003) Possible effects of vascular endothelial growth factor in the pathogenesis of chronic obstructive pulmonary disease. Am J Med 114:354–358
Kanazawa H, Asai K, Nomura S (2007) Vascular endothelial growth factor as a non-invasive marker of pulmonary vascular remodeling in patients with bronchitis-type of COPD. Respir Res 8:22
Wasada T, Kawahara R, Katsumori K et al (1998) Plasma concentration of immunoreactive vascular endothelial growth factor and its relation to smoking. Metabolism 47:27–30
Acknowledgement
This study was financially supported by the Groningen University Graduate School for Drug Exploration (GUIDE).
Conflicts of interest statement
We declare that we have no conflict of interest.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Versluis, M., ten Hacken, N., Postma, D. et al. Adenosine receptors in COPD and asymptomatic smokers: effects of smoking cessation. Virchows Arch 454, 273–281 (2009). https://doi.org/10.1007/s00428-009-0727-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00428-009-0727-9