
Abstract
Peripheral T cell lymphomas are an aggressive group of non-Hodgkin lymphomas with poor outcomes for most subtypes and no accepted standard of care for relapsed patients. This study evaluated the efficacy and safety of forodesine, a novel purine nucleoside phosphorylase inhibitor, in patients with relapsed peripheral T cell lymphomas. Patients with histologically confirmed disease, progression after ≥ 1 prior treatment, and an objective response to last treatment received oral forodesine 300 mg twice-daily. The primary endpoint was objective response rate (ORR). Secondary endpoints included duration of response, progression-free survival (PFS), overall survival (OS), and safety. Forty-eight patients (median age, 69.5 years; median of 2 prior treatments) received forodesine. In phase 1 (n = 3 evaluable), no dose-limiting toxicity was observed during the first 28 days of forodesine treatment. In phase 2 (n = 41 evaluable), the ORR for the primary and final analyses was 22% (90% CI 12–35%) and 25% (90% CI 14–38%), respectively, including four complete responses (10%). Median PFS and OS were 1.9 and 15.6 months, respectively. The most common grade 3/4 adverse events were lymphopenia (96%), leukopenia (42%), and neutropenia (35%). Dose reduction and discontinuation due to adverse events were uncommon. Secondary B cell lymphoma developed in five patients, of whom four were positive for Epstein-Barr virus. In conclusion, forodesine has single-agent activity within the range of approved therapies in relapsed peripheral T cell lymphomas, with a manageable safety profile, and may represent a viable treatment option for this difficult-to-treat population.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Peripheral T cell lymphomas (PTCLs) are an aggressive group of non-Hodgkin lymphomas (NHLs), accounting for 5–10% of all NHLs in Western countries and approximately 20% of NHLs in Japan [1, 2]. The most common PTCL subtypes are PTCL, not otherwise specified (PTCL-NOS); anaplastic large cell lymphoma (ALCL); and angioimmunoblastic T cell lymphoma (AITL) [3, 4]. Newly diagnosed PTCL is most frequently treated with cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) or CHOP-like chemotherapies [5,6,7,8]. Although most patients respond initially, outcomes remain poor except in patients with anaplastic lymphoma kinase (ALK)-positive ALCL [5]. Median survival in a cohort of 153 North American patients with first-relapsed/refractory PTCLs was 5.5 months, underscoring the need for new treatments in this setting [9].
Prior to forodesine, several agents were approved for treatment of relapsed/refractory PTCL, including the folate antagonist pralatrexate [10] and the histone deacetylase inhibitors romidepsin [11] and belinostat [12] in the USA, and chidamide in China [13]. Additionally, the anti-CC chemokine receptor 4 monoclonal antibody mogamulizumab was approved in Japan for treatment of anti-CC chemokine receptor 4-positive relapsed/refractory PTCL [14], and the anti-CD30 antibody-drug conjugate brentuximab vedotin was approved for treatment of relapsed/refractory ALCL [15, 16]. Objective response rates (ORRs) for these agents in relapsed/refractory PTCL generally ranged from 25 to 35% [10,11,12,13,14], with brentuximab vedotin producing an ORR of 86% in ALCL [15] but 21% in non-ALCL PTCL [16].
Children born deficient in purine nucleoside phosphorylase (PNP) have reduced T cell counts, suggesting that PNP may be a target for treatment of T cell-mediated diseases [17]. Forodesine (BCX1777, Immucillin-H) is a novel PNP inhibitor, 100 to 1000 times more potent than other agents of this class [18, 19]. By inhibiting PNP, forodesine augments 2′-deoxyguanosine (dGuo) levels in plasma and T cells. The enzyme 2′-deoxycytidine kinase, which is highly upregulated in malignant T cells, phosphorylates dGuo to form deoxyguanosine monophosphate and then 2′-deoxyguanosine triphosphate. Accumulation of 2′-deoxyguanosine triphosphate causes an imbalance of the deoxyribonucleotide pool leading to T cell apoptosis. Forodesine was tolerated at doses of 200–300 mg once daily and exhibited preliminary evidence of anti-tumor efficacy in patients with PTCLs or cutaneous TCLs [19, 20]. Pharmacokinetic/pharmacodynamic data in healthy volunteers (data on file) suggested that a regimen of 300 mg twice-daily would be tolerable and provide forodesine exposure necessary for improving efficacy. Therefore, we conducted a phase 1/2 study of forodesine 300 mg twice-daily in Japanese patients with relapsed PTCL.
Patients and methods
Study design
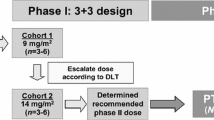
This multicenter, open-label study was conducted at 21 sites in Japan from January 2013 to February 2017. The study consisted of two parts: a phase 1 component designed to confirm the safety and tolerability of forodesine 300 mg twice-daily for 28 days in patients with relapsed/refractory PTCL and a phase 2 component designed to evaluate the efficacy and safety of this forodesine regimen in relapsed PTCL. The study was conducted in accordance with ethical principles of the Declaration of Helsinki and in compliance with International Council for Harmonization guidelines for Good Clinical Practice. The Institutional Review Boards of all participating institutions approved the study protocol, and all patients provided written informed consent. The study was registered at ClinicalTrials.gov (NCT01776411).
In phase 1, forodesine 300 mg (three 100-mg capsules) was given twice-daily after meals in a 28-day cycle. Phase 2 was initiated because none of the first three patients completing the 28-day cycle had dose-limiting toxicity (DLT; defined as treatment-related grade 3/4 non-hematologic toxicity excluding nausea, vomiting, or diarrhea or grade 4 neutropenia or thrombocytopenia lasting ≥ 7 days). During phase 2, patients received forodesine until disease progression (PD), unacceptable toxicity, or withdrawal of consent. Forodesine was stopped temporarily for ≤ 2 weeks in the event of DLT or if needed for management of adverse events (AEs). After recovery, a single-dose reduction to 200 mg twice-daily was allowed.
Patients had study visits on days 1 and 15 of cycles 1–4 and day 1 of subsequent cycles. After 22 patients completed 2 cycles, an interim efficacy analysis was conducted using a Simon minimax 2-step design to assess for futility (≤ 2 patients with objective responses); the study was to be terminated if futility was demonstrated. If futility was not demonstrated, the study was to be continued. Data cutoff was to be conducted when all the patients for efficacy evaluation completed the clinical study procedure by week 24. Based on data obtained until that time point, the primary analysis was to be conducted.
Patients
Japanese patients aged ≥ 20 years with histopathologically confirmed PTCL were eligible if they had PD after ≥ 1 prior treatment and had achieved an objective response on their last treatment. PTCL was defined according to the 2008 WHO Classification [3] and included PTCL-NOS, AITL, ALCL (ALK-positive or ALK-negative), extranodal natural killer cell/TCL (nasal type), enteropathy-associated TCL, hepatosplenic TCL, subcutaneous panniculitis-like TCL, and transformed mycosis fungoides. The PTCL subtype was diagnosed in each institution from lesion biopsy specimens and confirmed by an Independent Pathology Review Committee. Eligible patients had an enlarged lymph node or extranodal mass that was measurable in two perpendicular directions by computed tomography, with the greatest diameter > 1.5 cm; Eastern Cooperative Oncology Group performance status 0 or 1; and adequate hematopoietic, liver, and kidney function.
Patients who had received chemotherapy, radiation therapy, or high-dose corticosteroids (prednisolone ≥ 10 mg/day or equivalent) ≤ 21 days before the first dose of study drug were excluded, as were patients with a history of central nervous system involvement, allogeneic hematopoietic stem cell transplantation, autologous hematopoietic stem cell transplantation ≤ 100 days before study drug, severe cardiovascular or pulmonary disease, uncontrolled diabetes, or positivity for hepatitis B virus surface antigen, anti-hepatitis C virus antibody, or anti-human immunodeficiency virus antibody. Pregnant and lactating women and patients of child-bearing potential unwilling to use adequate contraception were also excluded.
Assessments
Tumor assessments were conducted after every 2 cycles for the first 24 weeks and then every 4 cycles. Efficacy was evaluated by an Independent Efficacy Assessment Committee (IEAC) according to revised International Working Group criteria [21], and classified as complete response (CR), partial response (PR), stable disease, or PD. The primary efficacy endpoint was IEAC-assessed ORR, consisting of the proportion of evaluable patients with CR or PR. Secondary efficacy endpoints included investigator-assessed ORR, duration of response (DoR), time to treatment failure, progression-free survival (PFS), and overall survival (OS). Primary analyses were conducted on data obtained by the time of data cutoff. In addition, final analyses on data from the entire period (including after the data cutoff) were also described for information purposes. For analyses other than the primary endpoint, results of final analyses were described.
Safety assessment
Safety was evaluated throughout the study and was comprised of AE monitoring, laboratory testing, physical examinations, vital-sign measurements, and 12-lead electrocardiograms. Severity of AEs was graded using the National Cancer Institute Common Terminology Criteria for Adverse Events, version 4.0.
Pharmacokinetics
The first seven patients comprised the full pharmacokinetics set; they received in-patient treatment for the first 4 days and had an additional visit on day 8. Blood samples were collected predose and at 1, 2, 4, 6, 8, and 12 h after the first dose on both days 1 and 15 and also predose on days 2, 3, 4, 8, 29, and 57. In all other patients, blood samples were collected predose for the first dose on days 1, 15, 29, and 57. Plasma forodesine concentrations were measured using a validated liquid chromatography-tandem mass spectrometry (LC/MS/MS) method, with a lower limit of quantitation of 2.5 ng/mL. Key pharmacokinetic parameters, including the observed maximum concentration (Cmax) and the area under the concentration-time curve to the last measurable drug concentration (AUClast), were determined by non-compartmental analysis using WinNonlin. Additional blood samples were collected at the above times for measurement of plasma dGuo concentrations by a validated LC/MS/MS with a lower limit of quantitation of 5.0 ng/mL.
Statistical analysis
Efficacy was evaluated in the full analysis set consisting of patients who received ≥ 1 dose of forodesine and had a postbaseline efficacy assessment. Using the Simon minimax two-step design [22], a sample size of 40 evaluable patients in phase 2 would have 80% statistical power at a one-sided α of 0.05 for showing a threshold ORR of 10%, assuming an expected ORR of 25%. A one-sided binomial test was used to determine if the observed ORR was above the predefined 10% threshold rate. To account for potential non-evaluable patients, the target sample size in phase 2 was set at 43. Time-to-event parameters were evaluated using Kaplan-Meier methods [23]. Safety was assessed in all patients who received ≥ 1 dose of study drug. AEs were coded to the Medical Dictionary for Regulatory Activities–Japanese, version 18.1. Safety parameters were summarized using descriptive statistics.
Results
Patients
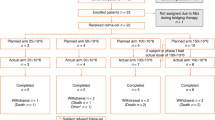
Forty-eight patients received forodesine (4 in phase 1, 44 in phase 2) (Fig. 1). Overall, the most common reasons for discontinuation were PD (n = 35) and AEs (n = 8). The study cohort had a median age of 69.5 years (range, 32–79 years) and had received a median of two prior treatment regimens (range, 1–9); most had PTCL-NOS (46%) or AITL (40%) (Table 1).

Disposition of patients in phase 1 and phase 2
Patients received forodesine for a median of 2.1 months (range, 0.2–36.0 months). Seventeen patients (35%) had a delay in forodesine dosing because of AEs, but only one patient (2%) had a dose reduction to 200 mg twice-daily (because of pneumonia). The mean daily dose of forodesine was 586.7 mg (standard deviation ± 37.2 mg).
Safety
Lymphopenia occurred in all patients (grade 3/4 in 46 patients [96%]), with all evaluated lymphocyte subsets (CD3+, CD4+, CD8+, CD16+, CD20+, CD56+) showing reductions from baseline (Fig. 2). Other common grade 3/4 hematologic toxicities included leukopenia (42%), neutropenia (35%), and thrombocytopenia (25%; Table 2). Febrile neutropenia occurred in six patients (13%). Grade 3/4 non-hematologic toxicities were uncommon. Adverse events that resulted in discontinuation occurred in 11 patients (23%; only Epstein-Barr virus [EBV]-associated lymphoma (n = 2) led to discontinuation in > 1 patient). One patient in phase 2 died from disseminated intravascular coagulation and multiorgan failure, which was attributed to underlying disease and considered not related to forodesine.

Scatterplots of lymphocyte subsets at baseline and on day 15
Twenty-two patients (46%) experienced serious AEs, most commonly infections (n = 8). Pneumonia (n = 4) was the only serious infection reported in > 1 patient. Secondary B cell lymphoma was reported in five patients; all were women aged 65 to 75 years, and three entered the study with AITL and two with PTCL-NOS. In four of five patients, lymphoma cells were positive for EBV encoded RNA-1 on in situ hybridization. Based on an exploratory analysis, the patients who developed secondary B cell lymphoma had received forodesine for a median of 11.6 months (range, 2.2–16.6 months); the median duration from forodesine initiation to development of secondary B cell lymphoma was 14.3 months (range, 6.7–16.6 months). Median trough lymphocyte counts in patients who did (n = 5) or did not (n = 43) develop secondary B cell lymphoma were 87 per mm3 (range, 51–120) and 71 per mm3 (range, 0–731), respectively. Median trough CD4+ lymphocyte counts in patients who did (n = 5) or did not (n = 42) develop secondary B cell lymphoma were 44 per mm3 (range, 18–162) and 51/mm3 (range, 5–3274), respectively. Outcome for patients who developed secondary B cell lymphoma are shown in Table 3. One patient achieved CR to treatment for secondary B cell lymphoma and survived with PTCL at the time of the final analysis. Three patients died of lymphoma (PTCL and/or secondary B cell lymphoma), and the outcome of one patient is unknown.
Efficacy
Among the 41 evaluable patients in phase 2, the ORR (IEAC assessment) for the primary analysis was 22% (90% CI 12–35%), and included four with CR (10%) and five with PR (12%) (Table 4), which was significantly greater than the predefined 10% threshold rate (P = 0.018). The investigator-assessed ORR was 22%, and included three patients with CR and six with PR. One of four patients in phase 1 also achieved a PR. Furthermore, the ORR at the final analysis was 25% (90% CI 14–38%), and included four patients with CR (10%) and six with PR (15%) (Table 4). The median time to response was 2.8 months (range, 1.8–12.8 months), and the median time to treatment failure was 2.2 months (95% CI 1.8–5.0 months) (data not shown). Median DoR was 10.4 months (95% CI 5.9–16.0 months) (Fig. 3a), median PFS was 1.9 months (95% CI 1.8–4.6 months) (Fig. 3b), and median OS was 15.6 months (95% CI 10.7–NE months) (Fig. 3c) among evaluable patients. Two-year OS was 39%. Compared with non-responders, the hazard ratios for PFS and OS among responders were 0.21 (95% CI 0.09–0.48) and 0.19 (95% CI 0.04–0.80), respectively (Fig. 4).

Duration of response (n = 10 responding patients) (a), progression-free survival (b), and overall survival (OS) (c) among evaluable phase 2 patients (n = 41)

Progression-free survival (a) and overall survival (b) among responders and non-responders in the phase 2 populations. CI confidence interval, NE not estimable
For the major PTCL subtypes in the phase 1 and 2 cohorts combined, the ORR was 33% (95% CI 13–59%) among 18 evaluable patients with AITL and 23% (95% CI 8–45%) among 22 evaluable patients with PTCL-NOS. ORRs > 30% were observed in several predefined subgroups, including age < 65 years (6/16; 38%), two prior treatment regimens (3/9; 33%), stage III disease (7/19; 37%), and low/normal lactate dehydrogenase (9/25; 36%) (Table 5). In general, patients with CR and PR showed a progressive reduction in target tumor size over time after starting forodesine (Fig. 5).

Reduction of target lesions measured by the sum of the products of the greatest diameters in the phase 2 populations: waterfall plot of maximum reduction (a) and target lesion reduction rate (b). CR complete response, PD progressive disease, PR partial response, SD stable disease
Pharmacokinetics
Plasma forodesine concentrations increased over 4 h after the first dose (mean [± standard deviation] Cmax, 435.7 [± 152.9] ng/mL) and then decreased gradually (Fig. 6). On day 15, the mean pretreatment concentration was 509.7 ng/mL (± 180.4) and after dosing, again increased over 4 h to a mean of 683.1 ng/mL (± 162.9) before gradually decreasing. Throughout the treatment period, mean trough plasma forodesine concentrations at each time point remained within a range of 460 to 540 ng/mL. The AUClast values after dosing on days 1 and 15 were 3540 and 6520 ng h/mL, indicating an accumulation ratio of 1.8. Plasma dGuo levels increased over 8 h after forodesine dosing, with mean trough concentrations at each time point within a range of 551 to 840 ng/mL. Plasma forodesine and dGuo concentrations on days 1 and 15 showed a positive relationship according to a maximum drug effect model.

Plasma forodesine and 2′-deoxyguanosine (dGuo) concentrations. Concentration-time profile of plasma forodesine on days 1 and 15 (a), trough plasma forodesine concentrations from days 1 to 57 (b), concentration-time profile of plasma dGuo on days 1 and 15 (c), and trough plasma dGuo concentrations from days 1 to 57 (d)
Discussion
This study demonstrated that forodesine has promising single-agent activity and led to its approval in Japan for treatment of relapsed/refractory PTCL. The ORR (22–25%) is comparable with ORRs reported in phase 2 studies of several recently approved agents for PTCL, including pralatrexate, romidepsin, and belinostat [10,11,12]. Differences in patient populations, histopathologic subtype distributions, disease status, and pretreatment characteristics make comparisons across studies difficult. Our study cohort mostly consisted of patients with PTCL-NOS and AITL; the ORR was numerically higher for those with AITL (33%) than for those with PTCL-NOS (23%). In a recent phase 2 study of patients with relapsed/refractory PTCL, lenalidomide demonstrated an ORR of 22%, and also showed a higher ORR in the AITL subset (31%) [24]. Given etiologic differences underlying the various PTCL histologies, it is plausible that specific agents may be used preferentially for specific PTCL subtypes. Indeed, brentuximab vedotin is highly active in patients with relapsed/refractory CD30+ ALCL but has less activity against other PTCL subtypes [15, 16]. Gemcitabine also showed promising activity in a small study of patients with PTCL-NOS and mycosis fungoides [25].
The median time to objective response was 2.8 months, and responses to forodesine appeared durable. In the final data analysis, the ORR was 25% (one patient reached PR after 13 months of administration) and median DoR was 10.4 months (range, 5.9–16.0 months).
The safety profile of forodesine 300 mg twice-daily was acceptable. Although dose delays because of AEs occurred in 35% of patients, dose reduction was only needed in 1 patient (2%), and discontinuation due to AEs occurred in 11 patients (23%). Toxicity consisted mostly of lymphopenia and other hematologic AEs; non-hematologic toxicities were generally mild/moderate in severity. The high rate of lymphopenia is thought to reflect the mechanism of action of forodesine. By inhibiting PNP, forodesine induces lymphocyte apoptosis (mainly T cells), leading to a reduction in lymphocyte counts and causing an immunosuppressive effect that may result in an increased risk of infection and secondary B cell lymphoma.
In this study, five patients developed secondary B cell lymphoma, of whom three had AITL and two had PTCL-NOS, consistent with the general distribution in our study cohort. EBV-driven B cell lymphoproliferation and EBV-related B cell lymphoma secondary to immunosuppression have been reported in patients with AITL and PTCL-NOS [26, 27]. Clonal expansion of EBV-negative B cells has also been described in patients with PTCLs [28, 29]. EBV status was not assessed at enrollment in our study; patients were not treated with anti-viral agents as prophylaxis, and all patients had received prior immunosuppressive chemotherapy. Thus, it cannot be excluded that the secondary lymphomas were already evolving before forodesine initiation. No clear difference was observed between total lymphocyte and CD4+ lymphocyte counts for patients who did or did not develop secondary B cell lymphoma, and risk factors for development of secondary B cell lymphoma were not identified in this study. Future studies should investigate whether EBV status at baseline or during forodesine treatment influences risk of secondary B cell lymphoma. In addition, sufficient attention must be placed on risk of opportunistic infection given that forodesine’s mechanism of action leads to T cell reductions.
In conclusion, forodesine has clinically meaningful single-agent activity, with durable responses, and a manageable safety profile in patients with relapsed PTCL. Compared with PTCL options that require intravenous infusion with frequent or prolonged clinic visits, the oral formulation makes forodesine easier to administer and, in turn, may be more convenient and less burdensome to patients. New therapeutic strategies with forodesine, including combination therapy, are being considered.
References
Anderson JR, Armitage JO, Weisenburger DD, for the Non-Hodgkin’s Lymphoma Classification Project (1998) Epidemiology of the non-Hodgkin’s lymphomas: distributions of the major subtypes differ by geographic locations. Ann Oncol 9:717–720
Chihara D, Ito H, Matsuda T, Shibata A, Katsumi A, Nakamura S, Tomotaka S, Morton LM, Weisenburger DD, Matsuo K (2014) Differences in incidence and trends of haematological malignancies in Japan and the United States. Br J Haematol 164:536–545
Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW (2008) WHO Classification of Tumours of Haematopoietic and Lymphoid Tissue. International Agency for Research on Cancer (IARC), Lyon
Campo E, Swerdlow SH, Harris NL, Pileri S, Stein H, Jaffe ES (2011) The 2008 WHO classification of lymphoid neoplasms and beyond: evolving concepts and practical applications. Blood 117:5019–5032
NCCN Clinical practice guidelines in oncology. T-cell lymphomas Version 3.2018. http://www.nccn.org. Accessed 13 March 2018
d’Amore F, Gaulard P, Trümper L, Corradini P, Kim WS, Specht L, Bjerregaard Pedersen M, Ladeto M, on behalf of the ESMO Guidelines Committee (2015) Peripheral T-cell lymphomas: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol 26(suppl 5):v108–v115
Van Obbergh F, Van Hoof A, Verhoef G, Dierickx D, De Wilde V, Offner F, Bron D, Sonet A, André M, Janssens A, Bonnet C, Deprijck B, Zachée P, Kentos A, Schroyens W, Van Den Neste E (2013) Treatment of peripheral T-cell lymphomas: recommendations of the Belgian hematological society (BHS). Belg J Hematol 4:90–101
Kitahara H, Maruyama D, Maeshima AM, Makita S, Miyamoto KI, Fukuhara S, Munakata W, Suzuki T, Kobayashi Y, Tajima K, Terauchi T, Kurihara H, Taiguchi H, Komatsu N, Tobinai K (2017) Prognosis of patients with peripheral T cell lymphoma who achieve complete response after CHOP/CHOP-like chemotherapy without autologous stem cell transplantation as an initial treatment. Ann Hematol 96:411–420
Mak V, Hamm J, Chhanabhai M, Shenkier T, Klasa R, Sehn LH, Villa D, Gascoyne RD, Connors JM, Savage KJ (2013) Survival of patients with peripheral T-cell lymphoma after first relapse or progression: spectrum of disease and rare long-term survivors. J Clin Oncol 31:1970–1976
O’Connor OA, Pro B, Pinter-Brown L, Bartlett N, Popplewell L, Coiffier B, Lechowicz MJ, Savage KJ, Shustov AR, Gisselbrecht C, Jacobsen E, Zinzani PL, Furman R, Goy A, Haioun C, Crump M, Zain JM, His E, Boyd A, Horwitz S (2011) Pralatrexate in patients with relapsed or refractory peripheral T-cell lymphoma: results from the pivotal PROPEL study. J Clin Oncol 29:1182–1189
Coiffier B, Pro B, Prince HM, Foss F, Sokol L, Greenwood M, Caballero D, Borchmann P, Morschhauser F, Wilhelm M, Pinter-Brown L, Padmanabhan S, Shustov A, Nichols J, Carroll S, Balser J, Balser B, Horwitz S (2012) Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy. J Clin Oncol 30:631–636
O’Connor OA, Horwitz S, Masszi T, Van Hoof A, Brown P, Doorduijn J, Hess G, Jurczak W, Knoblauch P, Chawla S, Bhat G, Choi MR, Walewski J, Savage K, Foss F, Allen LF, Shustov A (2015) Belinostat in patients with relapsed or refractory peripheral T-cell lymphoma: results of the pivotal phase II BELIEF (CLN-19) study. J Clin Oncol 33:2492–2499
Shi Y, Dong M, Hong X, Zhang W, Feng J, Zhu J, Yu L, Ke X, Huang H, Shen Z, Fan Y, Li W, Zhao X, Qi J, Huang H, Zhou D, Ning Z, Lu X (2015) Results from a multicenter, open-label, pivotal phase II study of chidamide in relapsed or refractory peripheral T-cell lymphoma. Ann Oncol 26:1766–1771
Ogura M, Ishida T, Hatake K, Taniwaki M, Ando K, Tobinai K, Fujimoto K, Yamamoto K, Miyamoto T, Uike N, Tanimoto M, Tsukasaki K, Ishizawa K, Suzumiya J, Inagaki H, Tamura K, Akinaga S, Tomonaga M, Ueda R (2014) Multicenter phase II study of mogamulizumab (KW-0761), a defucosylated anti-CC chemokine receptor 4 antibody, in patients with relapsed peripheral T-cell lymphoma and cutaneous T-cell lymphoma. J Clin Oncol 32:1157–1163
Pro B, Advani R, Brice P, Bartlett NL, Rosenblatt JD, Illidge T, Matous J, Ramchandren R, Fanale M, Connors JM, Yang Y, Sievers EL, Kennedy DA, Sustov A (2012) Brentuximab vedotin (SGN-35) in patients with relapsed or refractory systemic anaplastic large-cell lymphoma: results of a phase II study. J Clin Oncol 30:2190–2196
Lamarque M, Bossard C, Contejean A, Brice P, Parrens M, Le Gouill S, Brière J, Bouabdallah R, Canioni D, Tilly H, Bouchindhomme B, Bachy E, Delarue R, Haioun C (2016) Gaulard P (2016) Brentuximab vedotin in refractory or relapsed peripheral T-cell lymphomas: the French named patient program experience in 56 patients. Haematologica 101:e103–e106
Markert ML (1991) Purine nucleoside phosphorylase deficiency. Immunodefic Rev 3:45–81
Kicska GA, Long L, Hörig H, Fairchild C, Tyler PC, Furneaux RH, Schramm VL, Kaufman HL (2001) Immucillin H, a powerful transition-state analog inhibitor of purine nucleoside phosphorylase, selectively inhibits human T lymphocytes. Proc Natl Acad Sci U S A 98:4593–4598
Ogura M, Tsukasaki K, Nagai H, Uchida T, Oyama T, Suzuki T, Taguchi J, Maruyama D, Hotta T, Tobinai K (2012) Phase I study of BCX1777 (forodesine) in patients with relapsed or refractory peripheral T/natural killer-cell malignancies. Cancer Sci 103:1290–1295
Dummer R, Duvic M, Scarisbrick J, Olsen EA, Rozati S, Eggmann N, Goldinger SM, Hutchinson K, Geskin L, Illidge TM, Giuliano E, Kim YH (2014) Final results of a multicenter phase II study of the purine nucleoside phosphorylase (PNP) inhibitor forodesine in patients with advanced cutaneous T-cell lymphomas (CTCL) (mycosis fungoides and Sézary syndrome). Ann Oncol 25:1807–1812
Cheson BD, Pfistner B, Juweid ME, Gascoyne RD, Specht L, Horning SJ, Coiffier B, Fisher RI, Hagenbeek A, Zucca E, Rosen ST, Stroobants S, Lister TA, Hoppe RT, Dreyling M, Tobinai K, Vose JM, Connors JM, Federico M, Diehl V (2007) Revised response criteria for malignant lymphoma. J Clin Oncol 25:579–586
Simon R (1989) Optimal two-stage designs for phase II clinical trials. Control Clin Trials 10:1–10
Kaplan EL, Meier P (1958) Nonparametric estimation from incomplete observations. J Am Stat Assoc 53:457–481
Morschhauser F, Fitoussi O, Haioun C, Thieblemont C, Quach H, Delarue R, Glaisner J, Bosly A, Lister J, Li J, Coiffier B (2013) A phase 2, multicentre, single-arm, open-label study to evaluate the safety and efficacy of single-agent lenalidomide (Revlimid) in subjects with relapsed or refractory peripheral T-cell non-Hodgkin lymphoma: the EXPECT trial. Eur J Cancer 49:2869–2876
Zinzani PL, Venturini F, Stefoni V, Fina M, Pellegrini C, Derenzini E, Gandolfi L, Broccoli A, Argnani L, Quirini F, Pileri S, Baccarani M (2010) Gemcitabine as a single agent in pretreated T-cell lymphoma patients: evaluation of the long-term outcomes. Ann Oncol 21:860–863
Zettl A, Lee SS, Rüdger T, Starostik P, Marino M, Kirchner T, Ott M, Müller-Hermelink HK, Ott G (2002) Epstein-Barr virus-associated B-cell lymphoproliferative disorders in angioimmunoblastic T-cell lymphoma and peripheral T-cell lymphoma, unspecified. Am J Clin Pathol 117:368–379
Takahashi T, Maruyama R, Mishima S, Inoue M, Kawakami K, Onishi C, Miyake T, Tanaka J, Nabika T, Ishikura H (2010) Small bowl perforation caused by Epstein-Barr virus-associated B cell lymphoma in a patient with angioimmunoblastic T-cell lymphoma. J Clin Exp Hematopathol 50:59–63
Balagué O, Martinez A, Colomo L, Roselló E, Garcia A, Martinez-Bernal M, Palacin A, Fu K, Weisenburger D, Colomer D, Burke JS, Warnke RA, Campo E (2007) Epstein-Barr virus negative clonal plasma cell proliferaitons and lymphomas in peripheral T-cell lymphomas: a phenomenon with distinctive clincopathologic features. Am J Surg Pathol 31:1310–1322
Willenbrock K, Bräuninger A, Hansmann ML (2007) Frequent occurrence of B-cell lymphomas in angioimmunoblastic T-cell lymphoma and proliferation of Epstein-Barr virus-infected cells in early cases. Br J Haematol 138:733–739
Acknowledgements
The authors thank the patients, their families, doctors, nurses, and staff members who participated in this multicenter trial. We thank trial investigator Dr. Yasunobu Abe (Kyushu Cancer Center) who passed away prior to submission of this publication. We also thank Dr. Takashi Terauchi (National Cancer Center Hospital), Dr. Ukihide Tateishi (Yokohama City University Hospital), and Mitsuaki Tatsumi (Osaka University Hospital) who served as members of Image Evaluation Committee; Dr. Yoshihiro Matsuno (Hokkaido University Hospital), Shigeo Nakamura (Nagoya University Hospital), and Koichi Oshima (Kurume University Hospital) who served as members of central pathology review; and Dr. Noboru Horikoshi (Juntendo University) and Dr. Noriko Usui (Jikei University Daisan Hospital) who served as members of the Independent Data Monitoring Committee. Medical writing and editorial assistance was provided by Team 9 Science.
Funding
This work was supported by Mundipharma K.K., Tokyo, Japan.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Dai Maruyama reports research funding from Mundipharma K.K., Chugai Pharma, Kyowa Hakko Kirin, Ono Pharmaceutical, Celgene, Janssen, GlaxoSmithKline, Eisai, Takeda, SERVIER, and Abbvie, MSD, and receiving honoraria from Mundipharma K.K., Takeda, Janssen, Eisai, Biomedis International, Celgene, Sanofi, Kyowa Hakko Kirin, Fujifilm, Mochida Pharmaceutical Co., Ltd., Ono Pharmaceutical, and Chugai Pharma. Toshiki Uchida reports receiving an honorarium from Janssen. Yoshinobu Maeda reports research funding from Chugai Pharma, Kyowa Hakko Kirin, and Ono Pharmaceutical, and an honorarium from Mundipharma K.K. Hirokazu Nagai reports research funding from Chugai Pharma, Mundipharma K.K., Eisai, Sanofi K.K., Janssen, Takeda, Ono Pharmaceutical, and Kyowa Hakko Kirin. Hirohiko Shibayama reports research funding from Takeda, Janssen, Ono Pharmaceutical, Astellas, Tei** Pharma, Shionogi, Taiho, and Mundipharma KK, and honoraria from Takeda, Celgene, Novartis, Janssen, Kyowa Hakko Kirin, and Mundipharma KK. Kiyoshi Ando reports research funding from Mundipharma K.K. Isao Yoshida reports research funding from Kyowa Hakko Kirin and Chugai Pharma, and receiving honoraria from Mundipharma K.K., Celgene, and Kyowa Hakko Kirin. Michihiro Hidaka reports research funding from Mundipharma K.K. and Chugai Pharma. Tohru Murayama reports research funding from Celltrion, and honoraria from Nippon Shinyaku, Taiho Pharma, Janssen, Siemens, Kyowa Hakko Kirin, Novartis, Celgene, Ono Pharmaceutical, Pfizer, and Bristol-Myers Squibb. Junji Suzumiya reports research funding from Kyowa Hakko Kirin, Chugai-Roche, Shinnipponkagaku-PPD, Astellas, Toyama Chemical, Eisai, Takeda, Sumitomo Dainippon, Shionogi, Taiho, Yakult, and SymBio, and receiving honoraria from Kyowa Hakko Kirin, Chugai-Roche, Janssen, Eisai, Takeda, Sumitomo Dainippon, Otsuka, Celgene, Alexion, Novartis, Pfizer, AstraZeneca, Bristol-Myers Squibb, Merck, Ono Pharmaceutical, Eli Lilly, Shire, Gilead, and Zenyaku. Kazuo Tamura reports receiving honoraria from Ono Pharmaceutical, Kyowa Hakko Kirin, and Eli Lilly. Ryuzo Ueda reports research funding and an honorarium from Kyowa Hakko Kirin, serving as a consultant for Mundipharma K.K. and Terumo, and receiving an honorarium from Chugai Pharma. Kensei Tobinai reports receiving research funding from Mundipharma K.K., Chugai Pharma, Kyowa Hakko Kirin, Ono Pharmaceutical, Celgene, Janssen, GlaxoSmithKline, Eisai, Takeda, Servier, and Abbvie, and honoraria from Zenyaku Kogyo, Eisai, Takeda, Mundipharma K.K., Janssen, HUYA Bioscience International, Kyowa Hakko Kirin, Celgene, Chugai Pharma, and Ono Pharmaceutical. All remaining authors declared that they have no conflicts of interest.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declarations and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in this study.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Maruyama, D., Tsukasaki, K., Uchida, T. et al. Multicenter phase 1/2 study of forodesine in patients with relapsed peripheral T cell lymphoma. Ann Hematol 98, 131–142 (2019). https://doi.org/10.1007/s00277-018-3418-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00277-018-3418-2