
Abstract
Veillonella spp. are Gram-negative opportunistic pathogens present in the respiratory, digestive, and reproductive tracts of mammals. An abnormal increase in Veillonella relative abundance in the body is closely associated with periodontitis, inflammatory bowel disease, urinary tract infections, and many other diseases. We designed a pair of primers and a probe based on the 16S rRNA gene sequences of Veillonella and conducted real-time quantitative PCR (qPCR) and droplet digital PCR (ddPCR) to quantify the abundance of Veillonella in fecal samples. These two methods were tested for specificity and sensitivity using simulated clinical samples. The sensitivity of qPCR was 100 copies/μL, allowing for the accurate detection of a wide range of Veillonella concentrations from 103 to 108 CFU/mL. The sensitivity of ddPCR was 11.3 copies/μL, only allowing for the accurate detection of Veillonella concentrations from 101 to 104 CFU/mL because of the limited number of droplets generated by ddPCR. ddPCR is therefore more suitable for the detection of low-abundance Veillonella samples. To characterize the validity of the assay system, clinical samples from children with inflammatory bowel disease were collected and analyzed, and the results were verified using isolation methods. We conclude that molecular assays targeting the 16S rRNA gene provides an important tool for the rapid diagnosis of chronic and infectious diseases caused by Veillonella and also supports the isolation and identification of Veillonella for research purposes.
Key points
• With suitable primer sets, the qPCR has a wider detection range than ddPCR.
• ddPCR is suitable for the detection of low-abundance samples.
• Methods successfully guided the isolation of Veillonella in clinical sample.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Veillonella spp. are Gram-negative, obligate anaerobes that are present in the oral cavity and form part of the normal flora of mammalian organisms (Yuan et al. 2021). To date, 15 species of Veillonella (V. atypica, V. caviae, V. criceti, V. denticariosi, V. dispar, V. infantium, V. magna, V. montpellierensis, V. nakazawae, V. parvula, V. ratti, V. rodentium, V. rogosae, V. seminalis, V. tobetsuensis) have been reported, and detailed information on their isolation and pathogenicity is listed in Table 1. The majority of these species have been detected in humans, with only three species, namely V. caviae, V. criceti, and V. magna, being reported only in animals to date (Fu et al. 2022; Jiang et al. 2016; Kraatz and Taras 2008). However, it is worth noting that V. ratti was also previously thought to be present only in animals, but was later detected in the human intestine and reproductive tract (Marchandin et al. 2005; Pham et al. 2017).
Veillonella spp. share similar properties, including utilizing lactic acid, in preference to carbohydrates, as an energy source in the host organism. On the one hand, the lactic acid produces short-chain fatty acids, such as acetic acid and propionic acid, to maintain the health of the organism (Mashima et al. 2021), and on the other hand, it produces toxic LPS and H2S, which pose a risk of disease in immunocompromised individuals (Zhan et al. 2022). The specific metabolites of Veillonella have led to controversy among researchers regarding the outcome of their elevated abundance. Some studies have suggested that the elevated abundance of lactic acid-using Veillonella may decrease lactate accumulation during exercise and thus elevate the body’s endurance (Scheiman et al. 2019). However, many studies have suggested that elevated abundance of Veillonella can cause a range of diseases. For example, V. parvula has been detected in blood, bone, the oral cavity, urinary tract, heart valves, cerebrospinal fluid, respiratory tract, and gastrointestinal tract, causing multiple diseases including bacteremia, intervertebral discitis, dental caries, urinary tract infection, endocarditis, meningitis, and inflammatory bowel disease (IBD) (Chang et al. 2023; Rojas-Tapias et al. 2022; Li et al. 2017; Bhatti and Frank 2000; Ke et al. 2022; Berenger et al. 2015; Chen et al. 2021; Washio et al. 2014). Under normal conditions, Veillonella is not present in the blood, bones, heart valves, or cerebrospinal fluid, and if detected at these sites, it is considered a potential pathogen. However, in the oral cavity, respiratory tract, and gastrointestinal tract, where Veillonella is already present under natural conditions, its level of abundance is critical to the disease state. The quantification of Veillonella abundance in patient samples by molecular techniques is therefore of great significance for disease diagnosis.
Previous studies involving the molecular detection of Veillonella were mainly focused at the species level. For example, Rôças and Siqueira Jr (2006) reported the detection of V. parvula by nested PCR, and Mashima and Nakazawa reported the detection of V. tobetsuensis and V. infantium (Mashima and Nakazawa 2013; Mashima et al. 2018a) by amplification of the DnaK gene. Although the specificity of species-level detection is high, the detection spectrum is narrow in clinical practice and difficult to apply to the detection of samples containing unknown pathogens. Rinttilä et al. proposed a qPCR assay for the detection of Veillonella at the genus level, but the specificity was poor, partly because the assay system incorporated an insufficient number of strains (Rinttilä et al. 2004). A new detection method for Veillonella is therefore required. In this study, we designed a pair of primers and a probe that were highly conserved within, and highly specific to, genera to establish a rapid and objective quantitative assay involving multiplexed sequence alignment. We then analyzed the applicability of this assay using clinical samples.
Materials and methods
Ethical approval
This study was approved by the medical ethics committee of the capital institute of pediatrics, Bei**g, China.
Bacterial strains
Four species of Veillonella and seven common clinical pathogens were used to evaluate the test system, including V. dispar, V. nakazawae, V. parvula, V. rodentium, Enterococcus faecalis, Klebsiella pneumoniae, Morganella morganii, Escherichia coli, Proteus mirabilis, Salmonella enteritidis, and Shigella dysenteriae. V. parvula (ATCC 10790), V. Nakazawa (CCUG 74597T), V. dispar (ATCC 17745), and V. rodentium (ATCC 17743) are the standard strains, and the rest of the strains have been identified and stored in the Bacteriology Research Laboratory of the Capital Institute of Pediatrics (Feng et al. 2023).
Strain culture and DNA extraction
V. dispar, V. nakazawae, V. parvula, and V. rodentium were incubated for 48 h under anaerobic conditions at 37 °C in brain heart infusion (BHI) supplemented with 5% defibrinated sheep blood and 2% sodium lactate. E. faecalis, K. pneumoniae, M. morganii, E. coli, P. mirabilis, S. enteritidis, and S. dysenteriae were incubated for 24 h under aerobic conditions at 37 °C in BHI.
The bacterial solution was collected from the cultured bacteria, and the optical density of it measured at a wavelength of 600 nm (OD600) was monitored by spectrophotometer, and the OD600 was adjusted to 1.0 with PBS buffer. The total bacterial DNA was extracted from the collected bacterial solution and stored at −80 °C according to the instruction of TIANamp Bacteria DNA Kit (TIANGEN, China).
Multiple sequence matching
The primer and probe were designed at the genus level according to the 16S rRNA gene sequences. The 16S rRNA gene data of 15 species of Veillonella were downloaded from EZbiocloud database, and the gene sequences of 6 other genera of Veillonellaceae and 9 common genera of the intestine were also searched, and the corresponding sequences of the model strains of each genus were selected for download (Yoon et al. 2017). Multiple sequence alignment was performed using MEGA and Genedoc to identify conserved regions within the genus Veillonella, and then, these conserved regions were compared with the gene sequences of other genera to obtain intergenera-specific loci and regions, which were recorded as SNP loci and high specificity regions.
Primer and probe design
The 16S rRNA gene sequences were used for the primer and probe design. According to the principles of primer and probe design, a pair of primers was designed in the highly conserved region, and the specificity of primer and probe were analyzed using NCBI Primer-BLAST, and Primer Primier 5 was used to predict the formation of primer dimer and secondary structure. The primer and probe were synthesized and purified using high-performance liquid chromatography (Sangon Biotech, Shanghai, China).
Construction of recombinant plasmids
The conserved sequence of V. parvula 16S rRNA gene (431–567) was amplified using the designed primers according to the manufacturer’s instructions, and the PCR amplification product was cloned into vector pGM-T (Tiangen Biotech, Bei**g, China). The plasmids were detected using a NanoDrop spectrophotometer. Then, standard recombinant plasmids were prepared at 10-fold dilutions ranging from 108 to 100 copies/μL for sensitivity analysis. Plasmid concentration and copy number were determined using the formula: DNA copy number (copies/μL) = [6.02 × 1023 × plasmid concentration (ng/μL) × 10−9]/[DNA length (nucleotides) × 660]. The length of DNA (3152 bp) was the sum of plasmid fragment length (137 bp) and vector length (3015 bp).
Real-time quantitative PCR assay
The qPCR assay was performed in a reaction volume of 20 μL using the premixed reaction solution TIANGEN SuperReal PreMix (TIANGEN, China). PCR conditions were as follows: 10 μL Supermix for probes, 0.6 μL upstream primer, 0.6 μL downstream primer, 0.4 μL probe, 6.4 μL ddH2O, 2 μL DNA; temperature cycling: predenaturing at 95 °C for 15 min, 40 cycles of PCR at 95 °C for 3 s and at 60 °C for 30 s.
Droplet digital PCR assay
The ddPCR assay was performed in a reaction volume of 20 μL using a commercial ddPCR kit (**nyi, Bei**g, China). PCR mixture containing 15 μL Supermix for probes, 2.4 μL upstream primer, 2.4 μL downstream primer, 0.75 μL probe, 8.45 μL ddH2O, and 1 μL DNA. Twenty microliters of PCR mixture and 180 μL Droplet generation oil were mixed, and droplet generation was performed using the test instrument Drop Maker M1. The droplet emulsion was thermally cycled in the following conditions: predenaturing at 95 °C for 10 min, 40 cycles of PCR at 94 °C for 30 s, and at 60 °C for 1 min, and the test instrument was cooled at 12 °C for 5 min, and then, the reaction was finished. The completed reaction was removed from the small centrifuge tube, put into the microdrop detection chip. Of droplet detection oil, 430 μL and 500 μL were added into the oil wells and put into the Chip Reader Biochip Analyzer for analysis. ChipReaderR1 software was used to determine the nucleic acid copy number.
Simulation of sample preparation
After V. parvula were incubated for 48 h under anaerobic conditions at 37 °C in BHI, the bacterial solution was 10-fold diluted with BHI and spread it on Columbia agar containing 2% sodium lactate. After overnight anaerobic culture, observe the growth of V. parvula in Columbia agar, count the colony formation units, and dilute the bacterial solution according to the results. One milliliter of the V. parvula solution was diluted to the appropriate concentration, and genomic DNA was extracted according to the guidelines of TIANamp Bacteria DNA Kit (TIANGEN, China), and qPCR and ddPCR were performed after gradient dilution. Another 1 mL of V. parvula solution was diluted to the appropriate concentration and added to 200 mg of inactivated stool. Inactivated feces were sterilized using an autoclave at 121 °C for 30 min. Fecal genomic DNA was extracted with reference to the TIANamp stool DNA kit (TIANGEN, China), and gradient dilution was used for qPCR and ddPCR assay.
Assessing clinical testing capabilities of assay systems using isolation methods
A stool sample from a patient with IBD was used for the detection of Veillonella. Of the sample, 200 mg was accurately weighed for DNA extraction, and the abundance of Veillonella in the sample was calculated by qPCR. A gradient dilution of 1 g of stool sample was applied to OVSA medium. For each liter of OVSA medium containing 40 g TSA, 21 mL sodium lactate, 50 mL sheep blood, 16 mg vancomycin, and 1 mg oxacillin sodium, sheep blood and antibiotics were added after the basal medium was sterilized and cooled to 50 °C. The sample was incubated anaerobically for 48 h. A monoclonal colony was selected for thermal lysis and the lysed DNA was used for PCR amplification. The amplified samples were sent to Biotech Bioengineering (Shanghai, China) for sequencing. The ability of the assay system to detect clinical samples was evaluated by isolation and enumeration of Veillonella.
Results
Multiple sequence alignment and primer design
The full-length 16S rRNA gene sequences of 30 genera were aligned and five single nucleotide polymorphisms (SNPs) (at positions 452, 1026, 1037, 1142, and 1172) were found to be conserved among 15 species of Veillonella but specific to other genera of the Veillonellaceae and other common intestinal genera. Furthermore, three highly specific regions were found (at positions 171–173, 503–505, and 1017–1019), which could be used as alternative sites for primer and probe design (Fig. S1; Table 2).
After evaluation, it was found that SNP sites 452 and 1172, and high-specificity regions 503–505 and 1017–1019, were of sufficient length to constitute a conserved Veillonella sequence. According to the guidelines for primer and probe design (provide by TargetingOne Corporation, Bei**g, China), an amplified fragment should be less than 150 bp. Site 1172 was located too far from the high-specificity region 1017–1019 to satisfy this condition. Instead, SNP 452 was selected as the 3' end of the upstream primer, high-specificity region 503–505 was selected as the central region of the probe, and the conserved sequence 549–567 was selected as the downstream primer. We thereby obtained a pair of primers and a probe with both intra-genus conservation and inter-genus specificity for Veillonella (Fig. 1). The Primer-BLAST tool (https://www.ncbi.nlm.nih.gov/tools/primer-blast/) predicted no cross-reactivity with other species and high specificity. A pair of primers and a probe were used for subsequent analysis (Table 3).

The primer and probe sequences of Veillonella. Asterisk denotes the site that determines the multiple subtypes of Veillonella species (i.e., A for V. montpellierensis, G for the other 14 species)
Evaluation of the specificity and sensitivity of the qPCR assay
Four species of Veillonella and seven clinically common intestinal pathogens, E. faecalis, K. pneumoniae, M. morganii, E. coli, P. mirabilis, S. enteritidis, and S. dysenteriae, were detected to verify the specificity of the qPCR test. The results showed that the amplification curves of Veillonella and other strains could be clearly distinguished. The Ct value of H2O was 36.2 when a fluorescence value of 1000 was used as the baseline, which indicated that some environmental factors interfered with the identification of Veillonella (e.g., aerosols containing Veillonella DNA). To fully exclude environmental factors from interfering with the identification of Veillonella, Ct = 35 was chosen to distinguish between positive and negative fluorescence signals. Using this threshold, only V. dispar, V. nakazawae, V. parvula, and V. rodentium produced positive fluorescent signals, while the other species tested produced negative fluorescent signals (Fig. 2a).

The sensitivity and specificity of the qPCR assay. a The specificity of qPCR. b The sensitivity of qPCR. c Standard curve for qPCR sensitivity detection. The results of qPCR are shown by an amplification curve, and the legend is arranged according to the Ct values from smallest to largest. The dotted line is the fluorescence threshold
The qPCR sensitivity was tested using standard plasmids. The concentration of plasmid template determined by the Nanodrop spectrophotometer was 11.0 ng/μL, the conversion copy number was 3.234 × 109 copies/μL, and the concentration of recombinant plasmid at a 10-fold serial dilution was 108–100 copies/μL. The results of sensitivity analysis are shown in Fig. 2b. The Ct value was 11.99 when the plasmid concentration was 108 copies/μL and 33.98 when the plasmid concentration was 102 copies/μL. A concentration range from 108 to 102 copies/μL conformed to linearity with an R2 value of 0.9994 (Fig. 2c). Furthermore, the Ct value was 35.56 when the plasmid concentration was 101 copies/μL, which exceeded the specificity test determined threshold and was not represented on the standard curve; therefore, the lower limit of qPCR detection was determined to be 100 copies/μL.
Evaluation of the specificity and sensitivity of the ddPCR assay
To verify the specificity of the ddPCR assay, the assay was conducted to detect V. parvula, V. nakazawae, and the common clinical pathogens, K. pneumoniae, S. enteritidis, P. mirabilis, E. faecalis, and E. coli. Among them, V. parvula and V. nakazawae produced high intensity positive fluorescent signals with copy numbers of 153,614.4 copies/μL and 170,915.1 copies/μL, respectively, while all other strains tested produced lower intensity fluorescent signals of 1.3 copies/μL for K. pneumoniae, 2.4 copies/μL for S. enteritidis, 0 copies/μL for P. mirabilis, and 3.5 copies/μL for E. faecalis, and the copy numbers of all other test organisms were fewer than 5 copies/μL (Fig. 3a). The number of droplets generated for each sample exceeded 30,000 microdroplets, and the threshold amplification number of greater than 3000 basically excluded the interference of other genera and environmental factors on the identification of Veillonella.

The sensitivity and specificity of the ddPCR assay. a The specificity of ddPCR. b The sensitivity of ddPCR. The results of ddPCR are shown by an FAM scatter graph. c Standard curve for ddPCR sensitivity detection
The same plasmid template as that used for qPCR was then applied to test the sensitivity of ddPCR. The plasmid copy number was 3.234 × 109 copies/μL, and the concentration of recombinant plasmid at 10-fold serial dilutions was 106–100 copies/μL. The ddPCR method was used to detect the Veillonella 16S rRNA gene, and the results of sensitivity analysis are shown in Fig. 3b. When the plasmid concentration was greater than 104 copies/μL, which was beyond the detection range of ddPCR, the copy number data were not quantitatively significant. The copy number was 11,055 copies/μL at a plasmid concentration of 104 copies/μL and 11.3 copies/μL at a plasmid concentration of 101 copies/μL. The log-linear R2 of the copy number results was 0.9986 at plasmid concentrations of 104–101 copies/μL, and the copy number was detected when the plasmid concentration was 101 copies/μL (Fig. 3c). When the plasmid concentration was 100 copy/μL, the detected copy number was 0 copy/μL, which was lower than the negative control and was not on the standard curve, so the lower limit of ddPCR detection was determined to be 11.3 copies/μL.
Comparison of the detection capabilities of qPCR and ddPCR
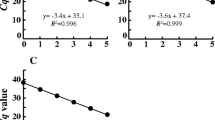
The number of bacterial colonies of 100μL V. parvula grown by overnight anaerobic culture on Columbia agar plates was 107 CFU, corresponding to a bacterial concentration in solution of 108 CFU/mL. Based on these results, the extracted genomic DNA of V. parvula was diluted across a gradient and assayed using qPCR and ddPCR to compare the ability of the two methods to detect V. parvula in the bacterial solution. The results of qPCR are shown in Fig. 4a. The Ct values were linear with an R2 value of 0.9960 at concentrations ranging from 108 to 102 CFU/mL. It was therefore concluded that qPCR was more accurate for the detection of 102–108 CFU/mL of Veillonella in a bacterial solution (Fig. 4b). The results of ddPCR are shown in Fig. 4c. The logarithm of the corresponding copy number results was linear with an R2 value of 0.9912 when the concentration ranged from 104 to 101 CFU/mL. It was therefore concluded that ddPCR was more accurate for the detection of 101–104 CFU/mL of Veillonella in a bacterial solution (Fig. 4d).

Comparison of the detection capacities of qPCR and ddPCR for Veillonella. a The sensitivity of qPCR. b Standard curve for qPCR sensitivity detection. c The sensitivity of ddPCR. d Standard curve for ddPCR sensitivity detection
To further investigate the ability of qPCR and ddPCR to detect Veillonella in samples, we simulated clinical samples by adding bacterial suspension to inactivated stool samples. The extracted fecal genomic DNA corresponded to a concentration of 108 CFU/mL of V. parvula. qPCR and ddPCR were performed after gradient dilution of fecal genomic DNA to compare the detection abilities of the two methods using the simulated samples. The results of the qPCR assay are shown in Fig. 5a. The corresponding Ct values were linear with an R2 value of 0.9972 at concentrations ranging from 108 to 103 CFU/mL. It was therefore concluded that qPCR was more accurate for the detection of 103–108 CFU/mL of Veillonella in the samples (Fig. 5b). The results of ddPCR are shown in Fig. 5c. The logarithm of the corresponding copy number results was linear with an R2 value of 0.9944 at concentrations ranging from 104 to 101 CFU/mL. It was therefore concluded that ddPCR was more accurate for the detection of 101–104 CFU/mL of Veillonella in fecal samples (Fig. 5d).

Comparison of the detection capacities of qPCR and ddPCR for Veillonella in simulated samples. a The sensitivity of qPCR. b Standard curve for qPCR sensitivity detection. c The sensitivity of ddPCR. d Standard curve for ddPCR sensitivity detection
Evaluation of the clinical testing capability of the assay system by isolation and culture
The DNA concentration of a 200 mg clinical sample, as determined by the Nanodrop assay, was 98.3 ng/μL, and the Ct value was 17.86 (Fig. 6a) using qPCR to detect Veillonella, and the abundance was calculated to be 8.04 × 106 CFU/mL (Fig. 6b) according to the standard curve. Then, the content of Veillonella in 1 g of stool sample was found to be 4.02 × 107 CFU/mL, and the sample was diluted across a gradient according to qPCR and ddPCR. Then, 100 μL of each dilution (103–106) was speaded on Veillonella selection medium(OVSA), and a bacterial count of 3.9 × 107 CFU/mL was obtained after anaerobic incubation for 48 h, which was similar to the molecular detection result (Fig. 6c). Single clone was selected for sequencing, and sequencing data showed that V. dispar, V. nakazawae, and V. rodentium were identified.

Abundance testing of Veillonella spp. in clinical samples. a qPCR results, asterisk indicates samples. b The abundance of Veillonella was calculated from the qPCR results using the standard curve. c Spread plate to detect the abundance of Veillonella in the sample. Veillonella colonies are white and opaque and are boxed in yellow in the figure
Discussion
An abnormal abundance of bacteria is closely associated with human disease. Advances in sequencing technology have revealed perturbations in the populations of an increasing number of anaerobic bacteria, and the potential link with disease has attracted the attention of researchers. As a class of opportunistic pathogen, Veillonella has been reported to be potentially associated with numerous diseases. Because of the stringent culture conditions and long incubation time required for Veillonella growth, the detection of Veillonella in patient samples by molecular methods would significantly reduce the diagnosis time. Previous studies have mainly focused on detection of Veillonella at the species level. For example, Rôças et al. reported the accurate detection of V. parvula by PCR (Rôças and Siqueira Jr 2006), Izumi et al. reported the accurate detection of V. tobetsuensis (Mashima and Nakazawa 2013) and V. infantium (Mashima et al. 2018a) by DnaK gene detection, and Igarashi et al. (Igarashi et al. 2009) and Mashima et al. (Mashima et al. 2016) accurately detected a variety of oral Veillonella species based on the rpoB gene. Detection of Veillonella at the species level is essential because of the distinct roles played by different Veillonella in disease. However, in the testing of clinical samples with no clear source of infection, species-level testing has limitations in terms of its narrow detection spectrum and high workload. In addition, only 15 species of Veillonella have been reported to date, but unknown species of Veillonella are still to be explored by amplicon sequencing, which could be overlooked when using species-level methods. The establishment of a quantitative method for the detection of Veillonella at the genus level not only allows for efficient and rapid determination of the extent of Veillonella infection in patients and therefore enables effective and timely treatment, but also provides the potential for detecting more undefined Veillonella species. In previous studies, researchers have generally neglected the quantification and detection of Veillonella at the genus level. Therefore, new detection methods for Veillonella at the genus level were needed.
In this study, we developed a method to quantify the 16S rRNA genes of 15 species of Veillonella using qPCR and ddPCR, which offered advantages in terms of detection range and accuracy compared with previous reports. The results of sample testing using the two assays showed that qPCR was more accurate for the detection of 103–108 CFU/mL of Veillonella in samples, and ddPCR was more accurate for the detection of 101–104 CFU/mL of Veillonella in samples. This indicated that ddPCR was more suitable for the detection of Veillonella in low-abundance samples with higher detection sensitivity, and high-concentration samples could be further diluted until the detection range is achieved. By contrast, because Veillonella commonly colonizes the human oral cavity, qPCR for Veillonella is more suitable for clinical applications and can be used to supplement the quantification of Veillonella with ddPCR when a sample has a low Veillonella content that cannot be quantified by qPCR. The combination of these two methods provides accurate quantification of Veillonella in samples at concentrations ranging from 101 to 108 CFU/mL.
IBD is a group of nonspecific chronic inflammatory disease of the gastrointestinal tract in which the microbiota of the gut of affected patients differs significantly from that of healthy individuals and is accompanied by elevated nitrate levels and a decreased concentration of short-chain fatty acids (Schirmer et al. 2019). As a class of anaerobic bacteria that use nitrate as a redox receptor, elevated nitrate levels promote the increased abundance of Veillonella (Rojas-Tapias et al. 2022). During the course of our study, a stool specimen from a patient with IBD was collected and subjected to conventional smear and molecular assays. The results showed that the detected abundance of Veillonella was essentially the same between the two methods at 3.9 × 107 CFU/mL and 4.02 × 107 CFU/mL, respectively. The accuracy of the assay system was verified by the gold standard method of spread plate.
In this study, we isolated V. dispar, V. nakazawae, V. rodentium, and other species of Veillonella from clinical samples of patients with IBD using our newly developed assay system. The qPCR assay can rapidly detect both DNA and single colonies directly following thermal lysis, with accurate results and without the need for electrophoresis. Currently, this detection system can accurately detect Veillonella at the genus level but cannot further identify the corresponding species. In subsequent studies, we will combine it with molecular detection at the species level to obtain more detailed results. During laboratory testing, environmental factors, such as aerosols, can interfere with the experimental results, and negative pair setting is necessary to determine the threshold for a positive result to ensure accuracy.
In summary, the present study was conducted to establish a quantitative detection system for Veillonella spp. in simulated samples. This developed assay may be applied clinically to detect changes in Veillonella spp. in patients in a timely manner, as well as for research purposes to potentially aid the discovery of new species of Veillonella.
Data availability
All data generated or analyzed during this study are included in this published article and its supplementary information files.
References
Arif N, Do T, Byun R, Sheehy E, Clark D, Gilbert SC, Beighton D (2008) Veillonella rogosae sp. nov., an anaerobic, Gram-negative coccus isolated from dental plaque. Int J Syst Evol Microbiol 58(Pt 3):581. https://doi.org/10.1099/ijs.0.65093-0
Aujoulat F, Bouvet P, Jumas-Bilak E, Jean-Pierre H, Marchandin H (2014) Veillonella seminalis sp. nov., a novel anaerobic Gram-stain-negative coccus from human clinical samples, and emended description of the genus Veillonella. Int J Syst Evol Microbiol 64(Pt_10):3526–3531. https://doi.org/10.1099/ijs.0.064451-0
Berenger BM, Chui L, Borkent A, Lee M-C (2015) Anaerobic urinary tract infection caused by Veillonella parvula identified using cystine-lactose-electrolyte deficient media and matrix-assisted laser desorption ionization-time of flight mass spectrometry. IDCases 2(2):44–46. https://doi.org/10.1016/j.idcr.2015.02.002
Bhatti MA, Frank MO (2000) Veillonella parvula meningitis: case report and review of Veillonella infections. Clin Infect Dis 31(3):839–840. https://doi.org/10.1086/314046
Byun R, Carlier J-P, Jacques NA, Marchandin H, Hunter N (2007) Veillonella denticariosi sp. nov., isolated from human carious dentine. Int J Syst Evol Microbiol 57(12):2844–2848. https://doi.org/10.1099/ijs.0.65096-0
Chang X, Chen Y, Cui D, Wang Y, Zhou Y, Zhang X, Tang G (2023) Propionate-producing Veillonella parvula regulates the malignant properties of tumor cells of OSCC. Med Oncol 40(3):98. https://doi.org/10.1007/s12032-023-01962-6
Chen J, Kong L, Peng X, Chen Y, Ren B, Li M, Li J, Zhou X, Cheng L (2021) Core microbiota promotes the development of dental caries. Appl Sci 11(8):3638. https://doi.org/10.3390/app11083638
Cobo F, Pérez-Carrasco V, García-Salcedo JA, Navarro-Marí JM (2020) Bacteremia caused by Veillonella dispar in an oncological patient. Anaerobe 66:102285. https://doi.org/10.1016/j.anaerobe.2020.102285
Enaud R, Hooks KB, Barre A, Barnetche T, Hubert C, Massot M, Bazin T, Clouzeau H, Bui S, Fayon M (2019) Intestinal inflammation in children with cystic fibrosis is associated with Crohn’s-like microbiota disturbances. J Clin Med 8(5):645. https://doi.org/10.3390/jcm8050645
Feng J, Cui X, Du B, Zhao H, Feng Y, Cui J, Yan C, Gan L, Fan Z, Fu T, Xu Z, Yu Z, Zhang R, Du S, Tian Z, Zhang Q, Xue G, Yuan J (2023) Detection and quantification of Klebsiella pneumoniae in fecal samples using digital droplet PCR in comparison with real-time PCR. Microbiol Spectr. https://doi.org/10.1128/spectrum.04249-22
Fu S, Yuan Y, Tian X, Zhou L, Guo L, Zhang D, He J, Peng C, Qiu Y, Ye C (2022) Detection of colistin sulfate on piglet gastrointestinal tract microbiome alterations. Vet Sci 9(12):666. https://doi.org/10.3390/vetsci9120666
Gouze H, Noussair L, Padovano I, Salomon E, de Laroche M, Duran C, Felter A, Carlier R, Breban M, Dinh A (2019) Veillonella parvula spondylodiscitis. Med Mal Infect 49(1):54–58. https://doi.org/10.1016/j.medmal.2018.10.001
Igarashi E, Kamaguchi A, Fujita M, Miyakawa H, Nakazawa F (2009) Identification of oral species of the genus Veillonella by polymerase chain reaction. Oral Microbiol Immunol 24(4):310–313. https://doi.org/10.1111/j.1399-302X.2009.00513.x
Ito Y, Nakayama H, Niitsu Y, Kaneko N, Otsuka M, Sawada Y, Takeuchi Y, Sekido N (2022) The first case of Veillonella atypica bacteremia in a patient with renal pelvic tumor. Anaerobe 73:102491
Jang HJ, Choi JY, Kim K, Yong SH, Kim YW, Kim SY, Kim EY, Jung JY, Kang Y, Park MS (2021) Relationship of the lung microbiome with PD-L1 expression and immunotherapy response in lung cancer. Respir Res 22(1):1–10. https://doi.org/10.1016/j.anaerobe.2021.102491
Jiang XL, Yong S, W-y ZHU (2016) Fermentation characteristics of Megasphaera elsdenii J6 derived from pig feces on different lactate isomers. J Integr Agric 15(7):1575–1583. https://doi.org/10.1016/S2095-3119(15)61236-9
Jumas-Bilak E, Carlier J-P, Jean-Pierre H, Teyssier C, Gay B, Campos J, Marchandin H (2004) Veillonella montpellierensis sp. nov., a novel, anaerobic, Gram-negative coccus isolated from human clinical samples. Int J Syst Evol Microbiol 54(4):1311–1316. https://doi.org/10.1099/ijs.0.02952-0
Ke L, Chen L, Yaling Y, Can G, Jun L, Chuan Z (2022) Investigation on the pathological mechanism of frequent exacerbators with chronic obstructive pulmonary disease based on the characteristics of respiratory flora. Front Med 8:2895. https://doi.org/10.3389/fmed.2021.816802
Kraatz M, Taras D (2008) Veillonella magna sp. nov., isolated from the jejunal mucosa of a healthy pig, and emended description of Veillonella ratti. Int J Syst Evol Microbiol 58(12):2755–2761. https://doi.org/10.1099/ijs.0.2008/001032-0
Kumar P, Kumaresan M, Biswas R, Saxena SK (2023) Veillonella atypica causing retropharyngeal abscess: a rare case presentation. Anaerobe 81:102712. https://doi.org/10.1016/j.anaerobe.2023.102712
Li J, Chen P, Li J, Gao X, Chen X, Chen J (2017) A new treatment of sepsis caused by Veillonella parvula: a case report and literature review. J Clin Pharm Ther 42(5):649–652. https://doi.org/10.1111/jcpt.12559
Marchandin H, Teyssier C, Jumas-Bilak E, Robert M, Artigues A-C, Jean-Pierre H (2005) Molecular identification of the first human isolate belonging to the Veillonella ratti–Veillonella criceti group based on 16S rDNA and dnaK gene sequencing. Res Microbiol 156(4):603–607. https://doi.org/10.1016/j.resmic.2005.01.013
Mashima I, Djais AA, Haase EM, Scannapieco FA, Otomo M, Saitoh M, Nakazawa F (2018a) Establishment of a species-specific primer pair for detecting Veillonella infantium based on the 70 kDa heat shock protein gene dnaK. Anaerobe 52:79–82. https://doi.org/10.1016/j.anaerobe.2018.06.004
Mashima I, Liao Y-C, Miyakawa H, Theodorea CF, Thawboon B, Thaweboon S, Scannapieco FA, Nakazawa F (2018b) Veillonella infantium sp. nov., an anaerobic, Gram-stain-negative coccus isolated from tongue biofilm of a Thai child. Int J Syst Evol Microbiol 68(4):1101–1106. https://doi.org/10.1099/ijsem.0.002632
Mashima I, Nakazawa F (2013) Identification of Veillonella tobetsuensis in tongue biofilm by using a species-specific primer pair. Anaerobe 22:77–81. https://doi.org/10.1016/j.anaerobe.2013.04.015
Mashima I, Nakazawa F (2017) Role of an autoinducer-2-like molecule from Veillonella tobetsuensis in Streptococcus gordonii biofilm formation. J Oral Biosci 59(3):152–156. https://doi.org/10.1016/j.job.2017.06.002
Mashima I, Theodorea CF, Thaweboon B, Thaweboon S, Nakazawa F (2016) Identification of Veillonella species in the tongue biofilm by using a novel one-step polymerase chain reaction method. PLoS One 11(6):e0157516. https://doi.org/10.1371/journal.pone.0157516
Mashima I, Miyoshi-Akiyama T, Tomida J, Kutsuna R, Washio J, Takahashi N, Nakazawa F, Sato T, Kawamura Y (2019) Draft genome sequences of two Veillonella tobetsuensis clinical isolates from intraoperative bronchial fluids of elderly patients with pulmonary carcinoma. Microbiol Resour Announc 8(38):e00397–19. https://doi.org/10.1128/mra.00397-19
Mashima I, Theodorea CF, Djais AA, Kunihiro T, Kawamura Y, Otomo M, Saitoh M, Tamai R, Kiyoura Y (2021) Veillonella nakazawae sp. nov., an anaerobic Gram-negative coccus isolated from the oral cavity of Japanese children. Int J Syst Evol Microbiol 71(1):004583. https://doi.org/10.1099/ijsem.0.004583
Meng H, Wang S, Tang X, Guo J, Xu X, Wang D, ** F, Zheng M, Yin S, He C (2022) Respiratory immune status and microbiome in recovered COVID-19 patients revealed by metatranscriptomic analyses. Front Cell Infect Microbiol 12:1561. https://doi.org/10.3389/fcimb.2022.1011672
Pham VT, Lacroix C, Braegger CP, Chassard C (2017) Lactate-utilizing community is associated with gut microbiota dysbiosis in colicky infants. Sci Rep 7(1):11176. https://doi.org/10.1038/s41598-017-11509-1
Qudeimat MA, Alyahya A, Karched M, Behbehani J, Salako NO (2021) Dental plaque microbiota profiles of children with caries-free and caries-active dentition. J Dent 104:103539. https://doi.org/10.1016/j.jdent.2020.103539
Rinttilä T, Kassinen A, Malinen E, Krogius L, Palva A (2004) Development of an extensive set of 16S rDNA-targeted primers for quantification of pathogenic and indigenous bacteria in faecal samples by real-time PCR. J Appl Microbiol 97(6):1166–1177. https://www.x-mol.com/paperRedirect/3249290. Accessed 10 May 2023
Rôças I, Siqueira J Jr (2006) Culture-independent detection of Eikenella corrodens and Veillonella parvula in primary endodontic infections. J Endod 32(6):509–512. https://doi.org/10.1016/j.joen.2005.07.004
Rojas-Tapias DF, Brown EM, Temple ER, Onyekaba MA, Mohamed AM, Duncan K, Schirmer M, Walker RL, Mayassi T, Pierce KA (2022) Inflammation-associated nitrate facilitates ectopic colonization of oral bacterium Veillonella parvula in the intestine. Nat Microbiol 7(10):1673–1685. https://doi.org/10.1038/s41564-022-01224-7
Rotjanapan P, Jaroensukrungruang A, Pisitkun P, Ngamjanyaporn P, Manonai J, Sawaswong V, Chanchaem P, Payungporn S (2021) Vaginal microbiota affects urinary tract infection risk in women with systemic lupus erythematosus: a pilot cross-sectional study from Thailand. Lupus Sci Med 8(1):e000551. https://doi.org/10.1136/lupus-2021-000551
Rovery C, Etienne A, Foucault C, Berger P, Brouqui P (2005) Veillonella montpellierensis endocarditis. Emerg Infect Dis 11(7):1112. https://doi.org/10.3201/eid1107.041361
Scheiman J, Luber JM, Chavkin TA, MacDonald T, Tung A, Pham L-D, Wibowo MC, Wurth RC, Punthambaker S, Tierney BT (2019) Meta-omics analysis of elite athletes identifies a performance-enhancing microbe that functions via lactate metabolism. Nat Med 25(7):1104–1109. https://doi.org/10.1038/s41591-019-0485-4
Schirmer M, Garner A, Vlamakis H, Xavier RJ (2019) Microbial genes and pathways in inflammatory bowel disease. Nat Rev Microbiol 17(8):497–511. https://doi.org/10.1038/s41579-019-0213-6
Shih C-T, Yeh Y-T, Lin C-C, Yang L-Y, Chiang C-P (2020) Akkermansia muciniphila is negatively correlated with hemoglobin A1c in refractory diabetes. Microorganisms 8(9):1360. https://doi.org/10.3390/microorganisms8091360
Son J-H, Kim JH, Chang HS, Park J-S, Park C-S (2020) Relationship of microbial profile with airway immune response in eosinophilic or neutrophilic inflammation of asthmatics. Allergy Asthma Immunol Res 12(3):412. https://doi.org/10.1016/j.jaci.2019.12.876
Toprak NU, Bozan T, Yılmaz S, Buyukbayrak EE, Tigen ET (2022) Polymicrobial bacteremia due to Lactobacillus jensenii and Veillonella montpellierensis in a pregnant patient; case report and review of literature. Anaerobe 75:102576. https://doi.org/10.1016/j.anaerobe.2022.102576
Washio J, Shimada Y, Yamada M, Sakamaki R, Takahashi N (2014) Effects of pH and lactate on hydrogen sulfide production by oral Veillonella spp. Appl Environ Microbiol 80(14):4184–4188. https://doi.org/10.1128/AEM.00606-14
Yang K, Wang Y, Zhang S, Zhang D, Hu L, Zhao T, Zheng H (2021) Oral microbiota analysis of tissue pairs and saliva samples from patients with oral squamous cell carcinoma–a pilot study. Front Microbiol 12:719601. https://doi.org/10.3389/fmicb.2021.719601
Yoon S-H, Ha S-M, Kwon S, Lim J, Kim Y, Seo H, Chun J (2017) Introducing EzBioCloud: a taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int J Syst Evol Microbiol 67(5):1613. https://doi.org/10.1099/ijsem.0.001755
Yuan H, Qiu J, Zhang T, Wu X, Zhou J, Park S (2021) Quantitative changes of Veillonella, Streptococcus, and Neisseria in the oral cavity of patients with recurrent aphthous stomatitis: a systematic review and meta-analysis. Arch Oral Biol 129:105198. https://doi.org/10.1016/j.archoralbio.2021.105198
Zhan Z, Liu W, Pan L, Bao Y, Yan Z, Hong L (2022) Overabundance of Veillonella parvula promotes intestinal inflammation by activating macrophages via LPS-TLR4 pathway. Cell Death Discov 8(1):251. https://doi.org/10.1038/s41420-022-01015-3
Funding
This work was supported by National Natural Science Foundation of China (82272352), Bei**g Natural Science Foundation (7222014), and Research Foundation of Capital Institute of Pediatrics (JHYJ-2023-02).
Author information
Authors and Affiliations
Contributions
JY, YL, and XZ designed the study and revised the manuscript. ZD, JC, GX, CY, LG, ZF, YF, HZ, ZX, ZY, TF, RZ, BD, XC, ZT, JC, YC, and ZL performed the experiments. QZ and JF collected the clinical samples. ZD and JC analyzed the results. ZD and JC wrote the manuscript. All authors read and approved the manuscript.
Corresponding authors
Ethics declarations
Ethical statement
This study was approved by the medical ethics committee of the capital institute of pediatrics, Bei**g, China.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
ESM 1
(PDF 981 kb)
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Ding, Z., Cui, J., Zhang, Q. et al. Detecting and quantifying Veillonella by real-time quantitative PCR and droplet digital PCR. Appl Microbiol Biotechnol 108, 45 (2024). https://doi.org/10.1007/s00253-023-12861-1
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00253-023-12861-1