
Abstract
Background
Central pontine myelinolysis (CPM) is a non-inflammatory demyelinating lesion of the pons. CPM and extrapontine demyelination (EPM) are together termed osmotic demyelination syndrome (ODS), a known and serious complication of acute correction of hyponatremia. Conversely, hyperglycemic hyperosmolarity syndrome (HHS) develops in patients with type 2 diabetes who still have some insulin secretory ability due to infection, non-compliance with treatment, drugs, and coexisting diseases, and is often accompanied by ketosis. HHS represents a life-threatening endocrine emergency (mortality rate, 10–50%) associated with marked hyperglycemia and severe dehydration. HHS may develop ODS, and some cases have been associated with hypernatremia.
Case presentation
The patient was an 87-year-old woman with hyperglycemia, dehydration, malnutrition, and potential thrombus formation during long-term bed rest. HHS was suspected to have developed due to progression of hyperglycemia and dehydration caused by pneumonia. Furthermore, ketoacidosis developed from ketosis and prerenal renal failure associated with circulating hypovolemia shock, which was also associated with disseminated intravascular coagulation. Treatment was started with continuous intravenous injection of fast-acting insulin and low-sodium replacement fluid. In addition, ceftriaxone sodium hydrate, heparin sodium, thrombomodulin α, human serum albumin, and dopamine hydrochloride were administered. Blood glucose, serum sodium, serum osmolality, and general condition (including vital, infection/inflammatory findings, and disseminated intravascular coagulation) improved promptly, but improvements in disturbance of consciousness were poor. Diffusion-weighted imaging of the brain 72 h after starting treatment showed no obvious abnormalities, but high-intensity signals in the midline of the pons became apparent 30 days later, leading to definitive diagnosis of CPM.
Conclusions
Fluctuation of osmotic pressure by treatment from hyperosmolarity due to hyperglycemia and hypernatremia in the presence of risk factors such as malnutrition, severe illness, and metabolic disorders may be a cause of CPM onset. When treating HHS with risk factors, the possibility of progression to ODS needs to be kept in mind.
Similar content being viewed by others


Background
Central pontine myelinolysis (CPM) with extrapontine demyelination (EPM) is called osmotic demyelination syndrome (ODS), and is now recognized as a serious complication following acute correction of hyponatremia [1]. It is believed that When osmotic pressure is rapidly increased by correction of low sodium, the blood–brain barrier (BBB) is thought to be destroyed and cytotoxic factors in the blood cause demyelination and subsequent ODS [2, 3]. However, alongside hyponatremia, many other factors are considered to be involved in the onset of ODS [4,5,6,7]. CPM is a non-inflammatory demyelinating lesion of the pons, as first reported by Adams et al. in 3 cases of chronic alcoholism and 1 case of malnutrition [4], and EPM was reported later [8].
On the other hand, hyperglycemic hyperosmolarity syndrome (HHS) develops in type 2 diabetic patients who still have some degree of insulin secretory ability due to infections, non-compliance with treatment, drugs, or coexisting diseases (endocrine diseases, cancer, etc.), and is often accompanied by ketosis. In addition, HHS is a life-threatening endocrine emergency (mortality rate, 10–50%) associated with marked hyperglycemia and severe dehydration [9]. HHS may develop to ODS [10,11,12,13,14,15,16,17,18,19,20,21,22], and some cases have been reported in association with hypernatremia [10,11,12, 15, 16].
Here, we report a rare case of ODS develo** during treatment of HHS with marked hypernatremia.
Case presentation
The patient was an 87-year-old woman with a history of venous stasis dermatitis in both lower legs. She had no history of either diagnosis of or treatment for diabetes, but hemoglobin (Hb)A1c had been recorded as 6.8% about 1 year before this presentation. She had been admitted to a psychiatric hospital for about 1 year, due to exacerbations of both depression and Alzheimer-type dementia that had developed 10 years earlier and 12 years earlier, respectively. About 2 months before presentation, her dietary intake decreased and infusion of glucose, electrolytes and water was started. She had been in a bedridden state with no speech and almost no appetite from about 1 month before presentation. At that point, hyperglycemia and hypernatremia were inferred to have already been present for a long time. Two days before presentation, sudden high fever (38 °C) and involuntary movements of the trunk and upper limbs appeared. One day later, she entered a coma. A blood glucose level (BG) of 1000 mg/dL and a serum sodium (Na) level of 179 mmol/L (glucose-corrected Na level: 194 mmol/L) were confirmed, and the patient was referred to our department for emergency hospitalization.
Glasgow coma scale score was 3 (eye opening, 1; best verbal response, 1; best motor response, 1), the pupils were 3 mm on both sides, and light reflex was rather dull, accompanied by involuntary movements of the whole body. Body temperature was 37.6 °C, blood pressure was 57/40 mmHg, heart rate was 114 beats/min, and peripheral oxygen saturation was maintained at 95% under mask administration of oxygen at 10 L/min. The tongue was very dry, and turgor of the skin was low. No abnormalities were observed in other physical findings except for the presence of moist rales at the end of inspiration in bilateral lower lung fields. Drugs being administered were limaprost alfadex at 5 mg/day, furosemide at 10 mg/day, and paroxetine at 5 mg/day.
Results of blood and biochemical examinations and blood gas analysis are shown in Table 1. Negative results were obtained for anti-glutamic acid decarboxylase antibodies (< 5.0 U/mL) (Table 1). Computed tomography of the chest showed infiltrative shadows in both lower lung fields (image not shown). This patient with hyperglycemia, dehydration, malnutrition, and potential thrombus formation during long-term bed rest was suspected to have developed into HHS and ketosis due to progression of hyperglycemia and dehydration caused by pneumonia. Furthermore, ketoacidosis had developed from ketosis and prerenal renal failure associated with circulating hypovolemia shock, which was also associated with disseminated intravascular coagulation (DIC).
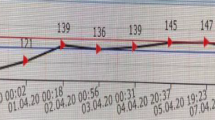
Treatment was started with intravenous infusion of fast-acting insulin (Humalin R; Eli Lilly, Kobe, Japan) (starting at 4 units/h and gradually decreasing) and low-sodium replacement fluid [23]. In the first 24 h, 6000 mL of replacement fluid (95.8 g of glucose, 0.3% Na) was added, and 2000 mL of replacement fluid (20.8 g of glucose, 0.2% Na) was administered within the period of 24–48 h. At 48–72 h, 1000 mL of replacement solution (75 g of glucose, 0.1% Na) was administered, and combined use of tube feeding was started (Fig. 1). Correction of K was performed appropriately. Although BG was ≥ 1000 mg/dL at 8 h after starting treatment, Na improved to 149.5 mmol/L (glucose-corrected Na level: 164.4 mmol/L). After 24 h, although BG, Na and sOsm had decreased to 716 mg/dL, 154.0 mmol/L and 402.3 mOsm/kg H2O, respectively, glucose-corrected Na level remained almost unchanged (164.2 mmol/L). At 48 h later, BG had improved to 110 mg/dL, Na to 154 mmol/L, and sOsm to 370.0 mOsm/kg H2O. However, glucose-corrected Na level (166.0 mmol/L) was not showing improvement. At 72 h later, BG had improved to 283 mg/dL, Na to 150 mmol/L (glucose-corrected Na level: 152.5 mmol/L), and sOsm to 345.5 mOsm/kg H2O (Table 2). In addition, ceftriaxone sodium hydrate at 1 g/day, heparin sodium at 8000 units/day, thrombomodulin α at 6400 units/day, total human serum albumin at 62.5 g, and dopamine hydrochloride at 3 μg/kg were administered. General condition, including vital signs, infection/inflammatory findings, and DIC improved promptly (Fig. 1). After 72 h, the patient opened her eyes. However, because the state of no response to the stimulus and involuntary movements continued for 7 days, ODS was suspected and magnetic resonance imaging (MRI) of the brain was performed. No clear abnormalities were evident on diffusion-weighted imaging (DWI) (Fig. 2a), and no definitive diagnosis was reached. The patient was subsequently able to move in response to instructions, but we could not exclude the possibility of pseudobulbar paralysis associated with ODS, as she could barely speak and showed no improvement of dysphagia. A high-intensity signal in the pons was identified on DWI of the brain at 30 days after starting treatment (Fig. 2b), leading to definitive diagnosis of CPM. After the life-threatening state was averted and general condition improved, she was transferred to a long-term care facility. As of about 1 year after onset, we obtained information that the patient had recovered to the point that she could speak spontaneously and responded to conversation in a manner reflective of a good condition.

Clinical course. The clinical course up to 60 h after admission. Closed circles and solid lines show changes in blood glucose levels, and closed triangles and dotted lines show changes in serum sodium. Down arrow indicates infusion of 6400 units of thrombomodulin α. Closed and open arrowheads indicate infusions of 1 g of ceftriaxone sodium hydrate and 12.5 g of human serum albumin, respectively. Closed bar and numbers in brackets indicate serum osmolality. The line graph at the bottom shows insulin usage. Numbers in parentheses indicate, from left to right, the number of insulin units used just before breakfast, just before lunch, and just before dinner. G, glucose content; Na, sodium concentration; W, amount of water; Lis, insulin lispro; Gla, biosimilar insulin glargine

Diffusion-weighted imaging of the brain. a Image 72 h after admission. b Image 30 days after admission. Arrowheads indicate high-intensity regions in the pons
Discussion and Conclusions
Although ODS has been reported to rarely develop in HHS [10,11,12,13,14,15,16,17,18,19,20,21,22], fewer reports have described development of ODS in HHS with hypernatremia [10,11,12, 15, 16], and causes of ODS development have yet to be clarified in terms of marked hyperosmolarity or changes in osmotic pressure associated with treatment. Within 7 days of onset, findings of ODS are not detectable on MRI [24, 25]. On the other hand, Ruzek et al. reported DWI as extremely useful for early diagnosis of CPM, based on the possibility of diagnosing CPM by MRI 24 h after onset [26]. MRI findings, including DWI, are also known to be observed 24 h after onset [25, 27]. In this case, no clear abnormality was observed on DWI at 72 h after symptom onset. Relatively rapid improvement (fluctuation) of osmotic pressure by treatment for hyperosmolarity due to hyperglycemia and hypernatremia was thought to be the cause of CPM onset in this case. A similar case in which CPM and EPM developed due to rapid improvement of hypernatremia was reported by Go et al. [28].
The issue that we would have changed in our treatment of this case was the performance of dehydration correction using a hypotonic solution. We speculated that rapid changes in osmotic pressure might have been avoidable using physiological saline or at least half-saline. On the other hand, slowly reducing blood glucose may be a meaningful strategy from the perspective of preventing the onset of ODS. Regarding the pathogenesis of ODS, rapid changes in osmotic pressure presumably induce apoptosis of astrocytes [2] and disrupt the blood–brain barrier. As a result, cytotoxic factors in blood become able to enter the brain, injuring oligodendrocytes and leading to demyelination [3]. Furthermore, microglia reportedly activate early in the onset of ODS and accumulate in the demyelinating region, and may express inflammatory cytokines and participate in the progression of demyelination, leading to “myelin melting” [29]. With these mechanisms, dexamethasone reportedly acts to prevent breakdown of the blood–brain barrier, while minocycline may prevent the onset and development of ODS by suppressing the expression of inflammatory cytokines from microglia and migration and accumulation of microglia to demyelinated parts; that is, by suppressing microglial activation [30]. Dexamethasone and minocycline may thus have potential as clinical therapeutic agents for ODS in the future. However, the use of dexamethasone in patients with severe hyperglycemia requires careful consideration and may not represent a suitable first-line option for the treatment of DOC. Further, alcohol poisoning, liver diseases including liver transplantation, malnutrition, malignant tumors, severe diseases or sepsis during pregnancy or postpartum, adrenal insufficiency, and metabolic disorders have been mentioned as risk factors for the development of ODS [7]. Our patient also showed malnutrition, severe illness, and metabolic disorders as risk factors.
A key limitation of this report was that the condition of the patient before admission to our hospital could not be accurately gauged due to a lack of data from the referring facility. In addition, the interval to follow-up MRI was about 1 month, providing a weak basis for estimating the time of ODS onset.
In conclusion, we have reported a rare case involving an 87-year-old woman with CPM during treatment of HHS with marked hypernatremia. Fluctuations in osmotic pressure with treatment for hyperosmolarity due to hyperglycemia and hypernatremia in the presence of risk factors such as malnutrition, severe illness, and metabolic disorders were considered as the causes of CPM onset. When treating HHS with risk factors, the possibility of progression to ODS should always be kept in mind.
Availability of data and materials
Not applicable.
Abbreviations
- CPM:
-
Central pontine myelinolysis
- EPM:
-
Extrapontine demyelination
- ODS:
-
Osmotic demyelination syndrome
- HHS:
-
Hyperglycemic hyperosmolarity syndrome
- BG:
-
Blood glucose level
- Na:
-
Sodium
- K:
-
Potassium
- sOsm:
-
Serum osmolality
- DIC:
-
Disseminated intravascular coagulation
- MRI:
-
Magnetic resonance imaging
- DWI:
-
Diffusion-weighted imaging
References
Sterns RH, Riggs JE, Schochet SS Jr. Osmotic demyelination syndrome following correction of hyponatremia. N Engl J Med. 1986;314:1535–42.
Kengne FG, Nicaise C, Soupart A, Boom A, Schiettecatte J, Pochet R, Brion JP, Decaux G. Astrocytes are an early target in osmotic demyelination syndrome. J Am Soc Nephrol. 2011;22:1834–45.
Murase T, Sugimura Y, Takefuji S, Oiso Y, Murata Y. Mechanisms and therapy of osmotic demyelination. Am J Med. 2006;119:S69–73.
Adams RD, Victor M, Mancall EL. Central pontine myelinolysis: a hitherto undescribed disease occurring in alcoholic and malnourished patients. AMA Arch Neurol Psychiatry. 1959;81:154–72.
Ayus JC, Krothapalli RK, Arieff AI. Treatment of symptomatic hyponatremia and its relation to brain damage. A prospective study. N Engl J Med. 1987;317:1190–5.
McKee AC, Winkelman MD, Banker BQ. Central pontine myelinolysis in severely burned patients: relationship to serum hyperosmolality. Neurology. 1988;38:1211–7.
Shah MK, Mandayam S, Adrogué HJ. Osmotic demyelination unrelated to hyponatremia. Am J Kidney Dis. 2018;71:436–40.
Wright DG, Laureno R, Victor M. Pontine and extrapontine myelinolysis. Brain. 1979;102:361–85.
Stoner GD. Hyperosmolar hyperglycemic state. Am Fam Phys. 2017;96:729–36.
Landers JW, Chason JL, Samuel VN. Central pontine myelinolysis: a pathogenic hypothesis. Neurology. 1965;15:968–71.
Kusuyama Y, Tanaka S, Sakatsuji K, Nishihara T, Saito K, Ikeda K, Inui J, Iwahashi Y. Central pontine myelinolysis: an immunofluorescent study. Acta Pathol Jpn. 1982;32:725–32.
McComb RD, Pfeiffer RF, Casey JH, Wolcott G, Till DJ. Lateral pontine and extrapontine myelinolysis associated with hypernatremia and hyperglycemia. Clin Neuropathol. 1989;8:284–8.
O’Malley G, Moran C, Draman MS, King T, Smith T, Thompson CJ, Agha A. Central pontine myelinolysis complicating treatment of the hyperglycaemic hyperosmolar state. Ann Clin Biochem. 2008;45:440–3.
Burns JD, Kosa SC, Wijdicks EF. Central pontine myelinolysis in a patient with hyperosmolar hyperglycemia and consistently normal serum sodium. Neurocrit Car. 2009;11:251–4.
Mao S, Liu Z, Ding M. Central pontine myelinolysis in a patient with epilepsia partialis continua and hyperglycaemic hyperosmolar state. Ann Clin Biochem. 2011;48:79–82.
Hegazi MO, Mashankar A. Central pontine myelinolysis in the hyperosmolar hyperglycaemic state. Med Princ Pract. 2013;22:96–9.
Guerrero WR, Dababneh H, Nadeau SE. Hemiparesis, encephalopathy, and extrapontine osmotic myelinolysis in the setting of hyperosmolar hyperglycemia. J Clin Neurosci. 2013;20:894–6.
Rodríguez-Velver KV, Soto-Garcia AJ, Zapata-Rivera MA, Montes-Villarreal J, Villarreal-Pérez JZ, Rodríguez-Gutiérrez R. Osmotic demyelination syndrome as the initial manifestation of a hyperosmolar hyperglycemic state. Case Rep Neurol Med. 2014;2014:652523. https://doi.org/10.1155/2014/652523.
Saini M, Mamauag MJ, Singh R. Central pontine myelinolysis: a rare presentation secondary to hyperglycaemia. Singapore Med J. 2015;56:e71–3.
Donnelly H, Connor S, Quirk J. Central pontine myelinolysis secondary to hyperglycaemia. Pract Neurol. 2016;16:493–5.
Talluri S, Charumathi R, Khan M, Kissell K. Atypical presentation of central pontine myelinolysis in hyperglycemia. Endocrinol Diabetes Metab Case Rep. 2017;17:0064. https://doi.org/10.1530/EDM-17-0064.
Hirosawa T, Shimizu T. Osmotic demyelination syndrome due to hyperosmolar hyperglycemia. Cleve Clin J Med. 2018;85:511–3.
Blas-Macedo J, Blas-Soto V. Hypernatremia in hyperosmolar hyperglycemic syndrome. Rev Med Inst Mex Seguro Soc. 2011;49:335–7.
Graff-Radford J, Fugate JE, Kaufmann TJ, Mandrekar JN, Rabinstein AA. Clinical and radiologic correlations of central pontine myelinolysis syndrome. Mayo Clin Proc. 2011;86:1063–7.
Förster A, Nölte I, Wenz H, Al-Zghloul M, Kerl HU, Brockmann MA, Brockmann C, Groden C. Value of diffusion-weighted imaging in central pontine and extrapontine myelinolysis. Neuroradiology. 2013;55:49–56.
Ruzek KA, Campeau NG, Miller GM. Early diagnosis of central pontine myelinolysis with diffusion-weighted imaging. Am J Neuroradiol. 2004;25:210–3.
Chua GC, Sitoh YY, Lim CC, Chua HC, Ng PY. MRI findings in osmotic myelinolysis. Clin Radiol. 2002;57:800–6.
Go M, Amino A, Shindo K, Tsunoda S, Shiozawa Z. A case of central pontine myelinolysis and extrapontine myelinolysis during rapid correction of hypernatremia. Rinsho Shinkeigaku. 1994;34:1130–5.
Takefuji S, Murase T, Sugimura Y, Takagishi Y, Hayasaka S, Oiso Y, Murata Y. Role of microglia in the pathogenesis of osmotic-induced demyelination. Exp Neurol. 2007;204:88–94.
Suzuki H, Sugimura Y, Iwama S, Suzuki H, Suzuki H, Ozaki N, Nagasaki H, Arima H, Sawada M, Oiso Y. Minocycline prevents osmotic demyelination syndrome by inhibiting the activation of microglia. J Am Soc Nephrol. 2010;21:2090–8.
Acknowledgements
We wish to thank the patient’s family for their permission to publish this manuscript. Furthermore, we would like to thank Forte Science Communications (Tokyo, Japan) for providing medical editorial services.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
K.K., N.Koji., and M.I. attended the patient; K.K. and N.Kori wrote the manuscript; N.Koji., N.Kori., M.I., and Y.N. provided conceptual advice. N.Kori supervised management of the case and contributed to writing and editing the manuscript. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Written, informed consent was obtained from the patient’s family for publication of this case report and all accompanying images.
Competing interests
Yoshihiko Nishio has received honoraria for scientific lectures from Eli Lilly, Novo Nordisk Pharma, and Sanofi, and a scholarship donation from Novo Nordisk Pharma. Koshi Kusumoto, Nobuyuki Koriyama, Nami Kojima and Maki Ikeda have nothing to disclose.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Kusumoto, K., Koriyama, N., Kojima, N. et al. Central pontine myelinolysis during treatment of hyperglycemic hyperosmolar syndrome: a case report. Clin Diabetes Endocrinol 6, 23 (2020). https://doi.org/10.1186/s40842-020-00111-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40842-020-00111-6