
Abstract
Background
Avian influenza viruses (AIVs) have been identified from more than 100 different species of wild birds around the globe. Wild migratory birds can act as potential spreaders for AIVs to domestic birds between different countries. Egypt is situated on important migratory flyways for wild birds between different continents. While much is known about circulation of zoonotic potential H5N1 and H9N2 AIVs in domestic poultry in Egypt, little is known about the pivotal role of migratory birds in the maintenance and transmission of the viruses in Egypt.
Methods
Targeted AIV surveillance has been conducted in 2017 in different wetlands areas in Northern and Eastern Egypt.
Results
AIV of subtype H5 was detected in two bird species. In addition, a novel reassortant strain of the H6N2 subtype was identified which reveals the continuous risk of new influenza virus(es) introduction into Egypt. This novel virus possesses a reassortant pattern originating from different AIV gene pools.
Conclusions
Intervention control strategies should be performed to minimize the possible contact of domestic birds with wild birds to lower the risk of virus transmission at this interface. In addition, constant monitoring of AIVs in migratory birds is essential in the early detection of influenza virus introduction into Egypt.
Similar content being viewed by others

Background
Avian influenza virus (AIV), based on the antigenic properties of the hemagglutinin (HA) and neuraminidase (NA) proteins, has 16 HA subtypes and 9 NA subtypes (Krammer et al. 2018). Furthermore, AIVs are classified, according to their virulence in chickens, into low pathogenic avian influenza viruses (LPAIVs) and highly pathogenic avian influenza viruses (HPAIVs) (Chatziprodromidou et al. 2018). The classification into LPAIV vs. HPAIV is based on the amino acid sequence at the cleavage site of the HA0 precursor protein (multiple basic amino acid cleavage site for HPAIVs), and/or the lethality of the virus when injected into specific pathogen-free (SPF) chickens (HPAIV has an intravenous pathogenicity index > 1.2) (OIE 2018). Moreover, different AIV subtypes—including H5N1, H5N6, H6N1, H7N2, H7N3, H7N4, H7N7, H7N9, H9N2, H10N7, and H10N8 have crossed species barriers and caused human infections (To et al. 2012; Wei et al. 2013; Chen et al. 2014; Freidl et al. 2014).
Wild birds are natural reservoirs of AIVs, and most HA and NA subtypes have been detected in wild birds, notably waterfowl (i.e. ducks, geese and swans) (Olsen et al. 2006). LPAIV subtypes H3, H4, H5, H6 and H11 are the most frequently reported from apparently healthy migrating birds (Munster et al. 2007; Kuiken 2013; Latorre-Margalef et al. 2014). LPAIVs of the H7 and H9 subtypes are the most frequently detected subtypes in domestic birds, causing considerable economic losses in poultry production (Fusaro et al. 2011; Sun and Liu 2015; Thuy et al. 2016). The HPAIV infection of domestic birds, in particular of the order Galliformes, is associated with a high mortality rate reaching up to 100%, while HPAIV infections can be asymptomatic in some wild bird species (Pantin-Jackwood and Swayne 2009).
Egypt is identified as one of the countries endemically infected with HPAIV H5N1 since 2006 which repeatedly spilled over to humans since its first report (Abdelwhab et al. 2016). Wild birds were implicated as the main source of the introduction of HPAIV in Egypt in 2005 (Abdelwhab et al. 2016; Naguib et al. 2019), and this theory was supported by the massive spread of the disease throughout Egypt within few weeks, and similar observation was made with HPAIV H5N8 in late 2016 (Kandeil et al. 2017; Selim et al. 2017; Yehia et al. 2018). In parallel, LPAIV of H9N2 is endemic in domestic birds in Egypt since its first recorded incidence in 2010 (Monne et al. 2013; Naguib et al. 2017). The simultaneous existence of the three AIV subtypes among different poultry populations in Egypt rates this country as a hotspot for the generation of new subtypes and genotypes of AIVs by reassortment (Naguib and Harder 2018), where a reassortant HPAIV H5N2 was recently reported in Egypt as a result of natural reassortment between the Egyptian LPAIV H9N2 and HPAIV H5N8 viruses (Hagag et al. 2019). The aim of this study was to define AIVs prevalence in wild birds in Egypt in 2017 by collecting samples from the main avian stopover and wintering places.
Methods
Samples collection
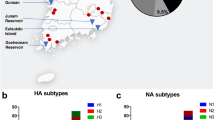
In November and December 2017, 168 swab samples (pooled oropharyngeal and cloacal swab) were collected from 84 migrating birds comprising 6 different bird species namely Eurasian Coot (Fulica atra), Common Quail (Coturnix coturnix), Eurasian Teal (Anas crecca), Turtle Dove (Streptopelia turtur), Northern Shoveler (Spatula clypeata), and Great Cormorant (Phalacrocorax carbo). Samples were collected from live bird markets (LBMs) near wetlands and lakes in Sharkia, Behira, and Ismailia Governorates in Egypt during a targeted surveillance of wild birds conducted by General Organisation for Veterinary services (GOVs), and more details are presented in Table 1 and Fig. 1. Such migratory birds were captured earlier by hunters near different wetlands and lakes prior to be sold to consumers in live bird markets for human consumption. Samples were shipped to the National Laboratory for veterinary Quality control on Poultry production (NLQP), Animal Health Research Institute (AHRI), Giza, Egypt for virus screening and identification.

Map of Egypt showing different geographic locations and bird species involved in this study
Virus screening and isolation
Viral RNA was extracted from pooled tracheal and cloacal swabs of individual birds following QIAamp viral RNA mini kit (Qiagen Gmbh, Hilden, Germany) instructions. RNAs were screened for the Matrix (M) gene of influenza A viruses using AgPath-ID one step Reverse transcription polymerase chain reaction (RT-PCR) kit (Applied Biosystems, Foster City, CA, USA) in 25 µL volume/reaction containing specific primers and probe as described previously (Yehia et al. 2018) using the real-time PCR Mx3005P QPCR System (Agilent, Santa Clara, CA, USA). Samples with a quantitation cycle (cq) value ≤ 38 were considered positive. Positive AIV RNAs were hemagglutinin (HA)-subtyped for H5, H7, and H9 subtypes and neuraminidase (NA) subtyped using specific subty** RT-qPCR (Slomka et al. 2007, 2009; Ben Shabat et al. 2010). All other RNA positive samples that were found to be Matrix positive but not H5, H7 or H9 positive, were tested against a panel including all H1–15 (Lee et al. 2001; Lednicky and Loeb 2013) and N1–N9 subty** (Fereidouni et al. 2009). Virus isolation was attempted on all samples with a Ct ≤ 35 through allantoic sac inoculation of 10-day-old specific-pathogen-free (SPF) embryonated chicken eggs according to the OIE diagnostic manual standard protocols (OIE 2018).
Genetic and phylogenetic analyses
Amplification attempts for all influenza M-positive RNA samples were performed using SuperScript III One-Step RT-PCR System (Invitrogen, Carlsbad, CA, USA) and primers described previously (Hoffmann et al. 2001) for HA and NA genes and as by (Naguib et al. 2015) for the remaining gene segments. The gene-specific RT-PCR amplicons were size-separated by agarose gel electrophoresis, excised and purified from gels using the QIAquick Gel Extraction Kit (Qiagen, Hilden, Germany). Further, purified PCR products were used directly for cycle sequencing reactions using BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA). Reaction products were purified using Centrisep spin column (Thermo Fisher Scientific, Waltham, MA, USA) and sequenced on an ABI PRISM® 3100 Genetic Analyzer (Life Technologies, Carlsbad, CA, USA).
Sequences were visualized and assembled using the Geneious software, version 11.0.5 (Kearse et al. 2012). A Basic Local Alignment Search Tool (BLAST) search was performed by using Global Initiative on Sharing All Influenza Data (GISAID) platform, and sequences established in this study have been submitted to the GISAID database (accession numbers: EPI1370641-8). In addition, genetic sequences of representative strains were retrieved from the GISAID platform and the influenza research database (IRD). Alignment and identity matrix analyses were conducted by using Multiple Alignment using Fast Fourier Transform (MAFFT) (Katoh and Standley 2013) and Geneious software, version 11.0.5 (Kearse et al. 2012). Phylogenetic analyses were based on maximum likelihood methodology based on Akaike criterion after selection of the best-fit models by using IQ-TREE software version 1.1.3 (Nguyen et al. 2015). Trees were viewed and edited with FigTree v1.4.2 software (http://tree.bio.ed.ac.uk/software/figtree/).
To estimate the source and time of emergence of the Egyptian LPAIV H6N2, a total of 68 representative sequence data of the HA gene segment were obtained from the GISAID and IRD databases. The Markov chain Monte Carlo (MCMC) method implemented in the BEAST (Bayesian Evolutionary Analysis Sampling Trees) package (version 1.8.0) (Drummond et al. Identity analysis To trace the origin of the Egyptian LPAIV EG/P2-29, a comparative identity analysis of all gene segments was conducted using the BLAST tool in the GISAID platform against all published sequences. The HA and M gene segments of the EG/P2-29 virus shared sequence identities at the nucleotide level of 97% and 99% with the LPAIV H6N1 (A/mallard duck/Georgia/7/2015 (H6N1)). The NA gene segment revealed highest similarity of 98% with the NA gene of the A/mallard/Markeev/16-3-11/2016 (H5N2) strain. The polymerase basic 2 (PB2) and polymerase basic 1 (PB1) genes segments shared a homology of 98% with HPAIV H5N5 A/wigeon/Italy/16VIR9616-3/2016. The polymerase acidic (PA) gene segment of the EG/P2-29 was very closely related to H7N9 virus isolated from the Netherlands (A/chicken/Netherlands/16007311-037041/2016) with 99% identity. Further, the nucleoprotein (NP) gene displayed an identity of 97% with recent H5N8 Korean viruses (Additional file 1: Table S1). The nucleocapsid (NS) gene segment possessed a high homology 98% with A/tufted duck/Georgia/1/2012 (H2N3) virus and similar identity was noted with A/domestic duck/Georgia/12/2016 (H4N6) virus. Different gene segments and their highest-identity viruses are listed in more detail in Additional file 1: Table S1. Taken together, these results indicate that the EG/P2-29 has been reassorted from different AIV subtypes. The HA gene of the EG/P2-29 virus possessed only one basic amino acid, arginine in the cleavage site of HA1 and HA2 “PQIETRGLF” indicating low pathogenicity of this virus. The HA carries P186, E190, Q226, and G228 amino acid residues at the receptor binding sites suggesting a higher affinity for avian receptor (α2‐3‐SA). However, the EG/P2-29 virus exhibits T160A amino acid mutation in the HA protein (Additional file 1: Table S2). Additionally, for the potential N‐linked glycosylation sites, eight sites were presented in the HA coding protein (positions 26, 27, 39, 182, 306, 311, 498 and 557). The NA fragment possessed a complete NA stalk (no 9aa deletion). No mutations associated with resistance to NA inhibitors were observed in the NA protein (Samson et al. 2013). Moreover, the Egyptian EG/P2-29 possessed no substitutional mutations at known molecular features associated with virulence, like E627 and D701 in PB2; I368V and L598P in PB1; V100A and in PA (Additional file 1: Table S2) (Stubbs and Te Velthuis 2014). Nevertheless, V291I substitutional mutation was observed in the PB2 encoded protein. The PB1‐F2 protein was encoded by 90aa; where an N66S substitution was found, and this substitution is associated with the increased virulence (Conenello et al. 2007). The N30D and T215A changes in the M1 protein (Fan et al. 2009) and the P42S and N205S changes in the NS1 protein suggested that the viruses could exhibit increased virulence in mammals (Kamal et al. 2014). Furthermore, S31 was found in M2 proteins, indicating no resistance to amantadine (Gleed et al. 2015). The maximum clade credibility (MCC) phylogenetic tree of the HA gene segment revealed that the Egyptian EG/P2-29 virus isolated in this study clustered closely together with the H6N1 virus previously detected in Georgia 2015 (Fig. 2). The NA gene segment possessed a close phylogenetic relationship with AIV H5N2 viruses that were previously isolated in Ukraine (A/mallard/Markeev/16-3-11/2016) in 2016 (Fig. 3), where they shared a common ancestor with A/mallard duck/Netherlands/18/2012 (H4N2). Maximum clade credibility (MCC) phylogenetic tree of the HA gene by BEAST analysis. The tree was scaled to time using the collection dates (year) of all samples. The scale bar at the bottom indicates the most recent sampling time. Taxa are coloured according to virus subtype. Egyptian H6N2 virus is indicated with a black dot Maximum clade credibility (MCC) phylogenetic tree of the NA gene by BEAST analysis. Taxa are coloured according to virus subtype. Egyptian H6N2 virus is indicated with a black dot Based on the phylogenetic trees, the PB2 and PB1 segments of the Egyptian EG/P2-29 virus were closely related to H5N5 and H5N8 viruses reported in Europe 2016 and Saudi Arabia 2017, respectively (Fig. 4a, b) which located within clade 2.3.4.4. The PA gene of EG/P2-29 showed a close relationship with H7N9 and H3N2 viruses detected in the Netherlands (Fig. 4c). The NP gene was grouped with the Korean H5N8 viruses (Fig. 4d). The M segment of the Egyptian virus was found phylogenetically related to the H10N7 virus from China and viruses from Bangladesh and the same Georgian virus A/mallard duck/Georgia/7/2015; while the NS showed close phylogenetic relatedness to H6N1 and H7N1 from Bangladesh (Fig. 4e, f). Phylogenetic trees of the nucleotide sequences of the PB2 (a), PB1 (b), PA (c), NP (d), M (e), and NS (f) gene segments. Maximum likelihood calculations were done with the IQTree software (Nguyen et al. 2015) under the best fit model according to the Bayesian criterion. Egyptian H6N2 virus is coloured red and indicated with a black dotGenetic characterization of the reassortant H6N2
Phylogenetic analysis of reassortant H6N2



Discussion
Wild migratory birds are the natural reservoirs of avian influenza viruses, in which they are generally found as low-pathogenic subtypes (Olsen et al. 2006). Egypt is a particularly important country for wild birds. This is due to the very strategic location of the country at the narrow boarder between the African and Asian continents (BirdLife 2018). Egypt is situated on the path of the Mediterranean-Black Sea and East-Africa West-Asia flyways of wild migratory birds, where wide variety of bird species are using these flyways each year particularly of the order Anseriformes and Charadriiformes (Ibrahim 2011; BirdLife 2018; Naguib et al. 2019). The surveillance of AIVs in domestic poultry in Egypt has been maintained for more than a decade and has undergone remarkable progress, but studies on wild birds are limited.
Reassortment is considered the main mechanism which allows new influenza virus to evolve through different gene segment(s) exchanges (Steel and Lowen 2014). The ability of migratory waterfowl to move over large geographic distances, combined with the substantial AIV prevalence and the diversity, can offer the opportunity for novel reassortant viruses to emerge through co-infection events (Lu et al. 2014). Avian influenza virus of H6 subtype has been found in wild birds with different NA combinations (NA1-9) (Peng et al. 2014; Gerloff et al. 2016; Verhagen et al. 2017; Li et al. In summary, the genetic and phylogenetic analyses of the Egyptian EG/P2-29 virus indicated that a novel virus has emerged through a reassortment event that has been occurred between H6 subtype and non‐H6 subtypes in the AIV gene pool. The human adaptation mutations observed among the HA, as well as the novel genetic reassortment pattern, might have a higher affinity toward human‐type receptor. Hence, further studies into the pathogenicity and transmission of this virus in mammalian animal model are required. Continual surveillance of AIVs in migratory birds in Egypt is particularly important to better understand the role of wild birds in the transmission of AIVs to domestic poultry population; in addition to the early detection of reassortant viruses that might likely harbour novel characteristics with significant threats for human health.Conclusions
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Abdelwhab EM, Hassan MK, Abdel-Moneim AS, Naguib MM, Mostafa A, Hussein ITM, et al. Introduction and enzootic of A/H5N1 in Egypt: virus evolution, pathogenicity and vaccine efficacy ten years on. Infect Genet Evol. 2016;40:80–90.
Al-Ghadeer H, Chu DKW, Rihan EMA, Abd-Allah EA, Gu H, Chin AWH, et al. Circulation of influenza A(H5N8) virus, Saudi Arabia. Emerg Infect Dis. 2018;24:1961–4.
Ben Shabat M, Meir R, Haddas R, Lapin E, Shkoda I, Raibstein I, et al. Development of a real-time TaqMan RT-PCR assay for the detection of H9N2 avian influenza viruses. J Virol Methods. 2010;168:72–7.
BirdLife International. Migratory soaring birds project. 2018. https://www.birdlife.org/worldwide/programmes/migratory-birds. Accessed 15 May 2019.
Chatziprodromidou IP, Arvanitidou M, Guitian J, Apostolou T, Vantarakis G, Vantarakis A. Global avian influenza outbreaks 2010–2016: a systematic review of their distribution, avian species and virus subtype. Syst Rev. 2018;7:17.
Chen H, Yuan H, Gao R, Zhang J, Wang D, **ong Y, et al. Clinical and epidemiological characteristics of a fatal case of avian influenza A H10N8 virus infection: a descriptive study. Lancet. 2014;383:714–21.
Conenello GM, Zamarin D, Perrone LA, Tumpey T, Palese P. A single mutation in the PB1-F2 of H5N1 (HK/97) and 1918 influenza A viruses contributes to increased virulence. PLoS Pathog. 2007;3:1414–21.
Darriba D, Taboada GL, Doallo R, Posada D. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods. 2012;9:772.
Drummond AJ, Suchard MA, **e D, Rambaut A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 2012;29:1969–73.
Fan S, Deng G, Song J, Tian G, Suo Y, Jiang Y, et al. Two amino acid residues in the matrix protein M1 contribute to the virulence difference of H5N1 avian influenza viruses in mice. Virology. 2009;384:28–32.
Fereidouni SR, Starick E, Grund C, Globig A, Mettenleiter TC, Beer M, et al. Rapid molecular subty** by reverse transcription polymerase chain reaction of the neuraminidase gene of avian influenza A viruses. Vet Microbiol. 2009;135:253–60.
Freidl GS, Meijer A, de Bruin E, de Nardi M, Munoz O, Capua I, et al. Influenza at the animal–human interface: a review of the literature for virological evidence of human infection with swine or avian influenza viruses other than A (H5N1). Eurosurveillance. 2014;19:8–26.
Fusaro A, Monne I, Salviato A, Valastro V, Schivo A, Amarin NM, et al. Phylogeography and evolutionary history of reassortant H9N2 viruses with potential human health implications. J Virol. 2011;85:8413–21.
Gerloff NA, Khan SU, Zanders N, Balish A, Haider N, Islam A, et al. Genetically diverse low pathogenicity avian influenza A virus subtypes co-circulate among poultry in Bangladesh. PLoS ONE. 2016;11:e0152131.
Gleed ML, Ioannidis H, Kolocouris A, Busath DD. Resistance-mutation (N31) effects on drug orientation and channel hydration in amantadine-bound influenza A M2. J Phys Chem B. 2015;119:11548–59.
Hagag NM, Erfan AM, El-Husseiny M, Shalaby AG, Saif MA, Tawakol MM, et al. Isolation of a novel reassortant highly pathogenic avian influenza (H5N2) virus in Egypt. Viruses. 2019;11:565.
Hoffmann E, Stech J, Guan Y, Webster RG, Perez DR. Universal primer set for the full-length amplification of all influenza A viruses. Arch Virol. 2001;146:2275–89.
Ibrahim WAL. An overview of bird migration studies in Egypt. Ring. 2011;33:55–75.
Kamal RP, Katz JM, York IA. Molecular determinants of influenza virus pathogenesis in mice. Curr Top Microbiol. 2014;385:243–74.
Kandeil A, Kayed A, Moatasim Y, Webby RJ, McKenzie PP, Kayali G, et al. Genetic characterization of highly pathogenic avian influenza A H5N8 viruses isolated from wild birds in Egypt. J Gen Virol. 2017;98:1573–86.
Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–80.
Kayed AS, Kandeil A, Gomaa MR, El-Shesheny R, Mahmoud S, Hegazi N, et al. Surveillance for avian influenza viruses in wild birds at live bird markets, Egypt, 2014–2016. Influenza Other Resp. 2019;13:407–14.
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, et al. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28:1647–9.
Krammer F, Smith GJD, Fouchier RAM, Peiris M, Kedzierska K, Doherty PC, et al. Influenza. Nat Rev Dis Primers. 2018;4:3.
Kuiken T. Is low pathogenic avian influenza virus virulent for wild waterbirds? Proc Biol Sci. 2013;280:20130990.
Latorre-Margalef N, Tolf C, Grosbois V, Avril A, Bengtsson D, Wille M, et al. Long-term variation in influenza A virus prevalence and subtype diversity in migratory mallards in northern Europe. Proc Biol Sci. 2014;281:20140098.
Lednicky JA, Loeb JC. Detection and isolation of airborne influenza A H3N2 virus using a sioutas personal cascade impactor sampler. Influenza Res Treat. 2013;2013:656825.
Lee MS, Chang PC, Shien JH, Cheng MC, Shieh HK. Identification and subty** of avian influenza viruses by reverse transcription-PCR. J Virol Methods. 2001;97:13–22.
Li J, Quan C, **e Y, Ke C, Nie Y, Chen Q, et al. Continued reassortment of avian H6 influenza viruses from Southern China, 2014–2016. Transbound Emerg Dis. 2019;66:592–8.
Lu L, Lycett SJ, Leigh Brown AJ. Reassortment patterns of avian influenza virus internal segments among different subtypes. BMC Evol Biol. 2014;14:16.
Monne I, Hussein HA, Fusaro A, Valastro V, Hamoud MM, Khalefa RA, et al. H9N2 influenza A virus circulates in H5N1 endemically infected poultry population in Egypt. Influenza Other Resp. 2013;7:240–3.
Munster VJ, Baas C, Lexmond P, Waldenstrom J, Wallensten A, Fransson T, et al. Spatial, temporal, and species variation in prevalence of influenza A viruses in wild migratory birds. PLoS Pathog. 2007;3:e61.
Naguib MM, Arafa AS, El-Kady MF, Selim AA, Gunalan V, Maurer-Stroh S, et al. Evolutionary trajectories and diagnostic challenges of potentially zoonotic avian influenza viruses H5N1 and H9N2 co-circulating in Egypt. Infect Genet Evol. 2015;34:278–91.
Naguib MM, Arafa AS, Parvin R, Beer M, Vahlenkamp T, Harder TC. Insights into genetic diversity and biological propensities of potentially zoonotic avian influenza H9N2 viruses circulating in Egypt. Virology. 2017;511:165–74.
Naguib MM, Harder T. Endemic situation of multiple avian influenza strains in poultry in Egypt:a continuing nightmare. Zoonoses Public Hlth. 2018;65:908–10.
Naguib MM, Verhagen JH, Samy A, Eriksson P, Fife M, Lundkvist Å, et al. Avian influenza viruses at the wild–domestic bird interface in Egypt. Infect Ecol Epidemiol. 2019;9:1575687.
Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015;32:268–74.
OIE. Avian influenza (Infection with avian influenza viruses). OIE (World Organisation for Animal Health) terrestrial manual 2018. Chapter 3.4.4. 2018. https://www.oie.int/fileadmin/Home/eng/Health_standards/tahm/3.03.04_AI.pdf. Accessed 15 May 2019.
Olsen B, Munster VJ, Wallensten A, Waldenstrom J, Osterhaus AD, Fouchier RA. Global patterns of influenza a virus in wild birds. Science. 2006;312:384–8.
Pantin-Jackwood MJ, Swayne DE. Pathogenesis and pathobiology of avian influenza virus infection in birds. Rev Sci Tech. 2009;28:113–36.
Peng J, Yang H, Jiang H, Lin YX, Lu CD, Xu YW, et al. The origin of novel avian influenza A (H7N9) and mutation dynamics for its human-to-human transmissible capacity. PLoS ONE. 2014;9:e93094.
Samson M, Pizzorno A, Abed Y, Boivin G. Influenza virus resistance to neuraminidase inhibitors. Antivir Res. 2013;98:174–85.
Selim AA, Erfan AM, Hagag N, Zanaty A, Samir AH, Samy M, et al. Highly pathogenic avian influenza virus (H5N8) clade 2.3.4.4 infection in migratory birds. Egypt. Emerg Infect Dis. 2017;23:1048–51.
Slomka MJ, Pavlidis T, Banks J, Shell W, McNally A, Essen S, et al. Validated H5 Eurasian real-time reverse transcriptase-polymerase chain reaction and its application in H5N1 outbreaks in 2005-2006. Avian Dis. 2007;51:373–7.
Slomka MJ, Pavlidis T, Coward VJ, Voermans J, Koch G, Hanna A, et al. Validated RealTime reverse transcriptase PCR methods for the diagnosis and pathoty** of Eurasian H7 avian influenza viruses. Influenza Other Resp. 2009;3:151–64.
Steel J, Lowen AC. Influenza A virus reassortment. Curr Top Microbiol. 2014;385:377–401.
Stubbs TM, Te Velthuis AJ. The RNA-dependent RNA polymerase of the influenza A virus. Future Virol. 2014;9:863–76.
Sun Y, Liu J. H9N2 influenza virus in China: a cause of concern. Protein Cell. 2015;6:18–25.
Thuy DM, Peacock TP, Bich VTN, Fabrizio T, Hoang DN, Tho ND, et al. Prevalence and diversity of H9N2 avian influenza in chickens of Northern Vietnam, 2014. Infect Genet Evol. 2016;44:530–40.
To KK, Ng KH, Que TL, Chan JM, Tsang KY, Tsang AK, et al. Avian influenza A H5N1 virus: a continuous threat to humans. Emerg Microbes Infect. 2012;1:e25.
Verhagen JH, Lexmond P, Vuong O, Schutten M, Guldemeester J, Osterhaus AD, et al. Discordant detection of avian influenza virus subtypes in time and space between poultry and wild birds; towards improvement of surveillance programs. PLoS ONE. 2017;12:e0173470.
Wei SH, Yang JR, Wu HS, Chang MC, Lin JS, Lin CY, et al. Human infection with avian influenza A H6N1 virus: an epidemiological analysis. Lancet Respir Med. 2013;1:771–8.
Yehia N, Naguib MM, Li R, Hagag N, El-Husseiny M, Mosaad Z, et al. Multiple introductions of reassorted highly pathogenic avian influenza viruses (H5N8) clade 2.3.4.4b causing outbreaks in wild birds and poultry in Egypt. Infect Genet Evol. 2018;58:56–65.
Acknowledgements
The authors acknowledge the colleagues in GOVs for their valuable support during sample collections. We are grateful to submitters of sequences in the GISAID database.
Funding
This work was funded by an internal project of the Reference Laboratory for Veterinary Quality Control on Poultry Production, Animal Health Research Institute. This work was supported in part by the Swedish Research Council VR (Grant Numbers 2016-02596 and 2018-02569) to MMN.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Supplementary information
Additional file 1: Table S1.
Nucleotide sequence identities between the EG/P2-29 (H6N2) virus and nearest homologues in the GenBank and GISAID database. Table S2. Molecular determinants within the Egyptian (EG/P2-29) H6N2 virus (H3 numbering).
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zanaty, A.M., Erfan, A.M., Mady, W.H. et al. Avian influenza virus surveillance in migratory birds in Egypt revealed a novel reassortant H6N2 subtype. Avian Res 10, 41 (2019). https://doi.org/10.1186/s40657-019-0180-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40657-019-0180-7