
Abstract
The cellular and molecular mechanisms that drive neurodegeneration remain poorly defined. Recent clinical trial failures, difficult diagnosis, uncertain etiology, and lack of curative therapies prompted us to re-examine other hypotheses of neurodegenerative pathogenesis. Recent reports establish that mitochondrial and calcium dysregulation occur early in many neurodegenerative diseases (NDDs), including Alzheimer's disease, Parkinson’s disease, Huntington's disease, and others. However, causal molecular evidence of mitochondrial and metabolic contributions to pathogenesis remains insufficient. Here we summarize the data supporting the hypothesis that mitochondrial and metabolic dysfunction result from diverse etiologies of neuropathology. We provide a current and comprehensive review of the literature and interpret that defective mitochondrial metabolism is upstream and primary to protein aggregation and other dogmatic hypotheses of NDDs. Finally, we identify gaps in knowledge and propose therapeutic modulation of mCa2+ exchange and mitochondrial function to alleviate metabolic impairments and treat NDDs.
Similar content being viewed by others

Introduction
The brain consumes 20% of the body’s ATP at rest, although it accounts for only 2% of body mass [1]. The high-energy requirements of the brain support neurotransmission, action potential firing, synapse development, maintenance of brain cells, neuronal plasticity, and cellular activities required for learning and memory [2, 3]. In neurons, most of the energy is consumed for synaptic transmission. Action potential signaling represents the second-largest metabolic need, and it is estimated that ~ 400–800 million ATP molecules are used to reestablish the electrochemical gradient (Na+ out, K+ in, at the plasma membrane) after production of the single action potential [4]. The energetic demand of neurons results in a substantial dependence on mitochondria for ATP production through oxidative phosphorylation (OxPhos) [4]. Any dysfunction in mitochondria can lessen the energetic capacity of OxPhos and may elicit a metabolic switch from OxPhos to glycolysis (Warburg-like effect) as a compensatory attempt to maintain cellular ATP in the context of neurodegenerative stress [5, 6]. However, a long-term OxPhos-to-glycolysis shift can result in a bioenergetic crisis and make neurons more vulnerable to oxidative stress and neuronal cell death [7, 8].
Neurodegenerative diseases (NDDs) are characterized by numerous cellular features, including the loss of neurons, neuronal dysfunction in specific brain regions, aggregation of distinct protein(s), impaired protein clearance, mitochondrial dysfunction, oxidative stress, neuroinflammation, axonal transport defects and cell death. The myriad of cellular pathologies suggest that there are common/central molecular mechanisms driving NDDs [9, 10]. In addition to ATP production, the mitochondrion is an epicenter of many metabolic pathways and important cellular functions, including the fine-tuning of intracellular calcium (iCa2+) signaling, regulation of cell death, lipid synthesis, ROS signaling, and cellular quality control [11]. Disruption in mitochondrial function and metabolism appears to underlie several NDDs such as Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), and others [12, 13]. At present, most therapies for NDDs provide only symptomatic relief, and there remain no drugs to inhibit neurodegeneration [14,15,16]. Mitochondrial alterations/impaired brain energetics are thought to present in the asymptomatic stage of disease prior to the onset of clinical symptoms [14, 17, 18]. This supports the notion that mitochondrial metabolic defects may be drivers or even initiators of the neurodegenerative process. In addition, several therapeutics that improve mitochondrial function have been reported to be efficacious in NDD models [19,20,21].
Mitochondrial calcium (mCa2+) is a critical regulator of mitochondrial function. In the matrix, mCa2+ tightly regulates TCA cycle activity and augments metabolic output. However, an excess of mCa2+ can impair mitochondrial respiration, enhance reactive oxygen species (ROS) production and activate cell death [22]. Here, we hypothesize that dysfunction in mCa2+ is an early common cellular event that impairs mitochondrial metabolism and drives and exacerbates neuropathology. Defining the molecular basis of mitochondrial function and metabolism in NDDs will help define novel cellular events and pathways and their temporal occurrence in NDD progression to identify new therapeutic targets for various neurological conditions. Here, we review recent advancements in our understanding of the essential role of mitochondrial metabolism and discuss how impaired mCa2+ signaling may be causal and central in neurodegeneration.
Evidence for impaired mitochondrial metabolism in NDDs
Strategies to combat NDDs have generally been unsuccessful and are focused on reducing symptoms and disease modification. Both clinical and experimental studies suggest that impaired energy metabolism correlates with various neurological deficits, highlighting new therapeutic opportunities [14]. Here we outline various mitochondrial metabolic defects that are strongly linked to the progression of neurodegeneration.
Alzheimer’s disease (AD)
AD is the most common form of dementia and is characterized by irreversible memory loss due to neuronal dysfunction, dysconnectivity, and cell death. Familial AD (FAD) is caused by pathogenic mutations in amyloid precursor protein (APP) or presenilin (PS1 and PS2) that lead to overproduction, improper cleavage, and the accumulation of amyloid-beta (Aβ). Prognostic disease phenotypes are associated with the formation of Aβ plaques, neurofibrillary tangles (NFTs, consisting of the microtubule protein tau), synaptic failure, reduced synthesis of the neurotransmitter acetylcholine, and chronic inflammation [9]. Most therapeutic strategies have been focused on Aβ metabolism and clearance due to extensive preclinical and clinical data in support of a causal role in AD progression [23, 24]. According to the “amyloid cascade hypothesis,” Aβ aggregation can initiate a series of events, including tau pathology, oxidative stress, inflammation, neuronal calcium (Ca2+) dysregulation, and metabolic alterations, which culminate in neuronal cell loss and AD pathogenesis [25]. However, this hypothesis does not fully explain the etiology of sporadic forms of AD (SAD) that account for 90–95% of AD-associated dementia.
An alternative hypothesis is that the microtubule-associated protein tau becomes hyperphosphorylated, resulting in axonal transport defects of organelles (including mitochondria), synaptic dysfunction, and cell death [26]. In cortical brain tissue from AD patients and mouse models, tau is reported to interact with mitochondrial transporters and complexes, resulting in mitochondrial dysfunction and AD pathology [27, 28]. However, there appears to be a limited correlation between the severity of cognitive decline and amyloid or tau plaque formation [29, 30], suggesting Aβ/tau metabolism and processing may not be the cause, or at least the singular cause, of disease. Consistent with previous studies, RNA-sequencing data from AD patients also suggest that Aβ and tau accumulation may not be mediators of the disease [31, 32]. Also, clinical trials of therapies targeting Aβ/tau production, metabolism, and clearance have universally shown little efficacy making it likely that other proximal mechanisms of AD pathogenesis exist [33, 34].
Mitochondrial dysfunction appears to be a primary occurrence in AD that precedes Aβ deposition, synaptic degeneration, and NFTs formation. In support of this concept, cytoplasmic hybrid cells (cybrids) generated from platelet mitochondria of SAD patients were reported to have a deficiency in complex I and complex IV of the electron transport chain (ETC), reduced mitochondrial membrane potential (Δψm), altered mitochondrial morphology, increased Aβ generation and tau oligomerization (reviewed in [35]). Transmission electron microscopy (TEM) showed smaller mitochondria with altered cristae structure and a decrease in mitochondrial content both in AD mice and patients [36,37,38]. Furthermore, fibroblasts derived from SAD patients also showed impaired mitochondrial dynamics, bioenergetics, and Ca2+ dysregulation [17, 39]. This change in mitochondria morphology in AD may be due to a shift in the mitochondrial fission/fusion balance and a decrease in biogenesis [40].
Importantly, experimental evidence suggests that bioenergetic alterations in AD precede the formation of Aβ plaques [41]. Data supporting metabolic deficits in AD were first published in the early 1980s from 2-[18F] fluoro-2-deoxy-D-glucose (FDG) positron emission tomography (PET) studies, which showed reduced glucose metabolism in the parietal, temporal and frontal cortex of AD patients [42,43,44]. Postmortem brain tissue isolated from AD patients displays reduced mitochondrial metabolic enzyme activity for pyruvate dehydrogenase (PDH) [45, 46], alpha-ketoglutarate dehydrogenase (α-KGDH) [46], isocitrate dehydrogenase (ICDH) [47], and complex IV or cytochrome-c-oxidase (COX) [48,49,50]. In addition, succinate dehydrogenase (SDH) and malate dehydrogenase (MDH) activity are increased in AD patient's brains [51]. Microarray data [52] and bioinformatics analysis of four transcriptome datasets [53] suggests a significant downregulation in nuclear-encoded OxPhos genes in the hippocampus of AD patients. More recent data confirm impaired ATP synthase activity due to loss of the oligomycin sensitive conferring protein subunit in the brain of FAD and SAD patients [54].
Diminished PDH function, as noted in AD, limits the shuttling of pyruvate into the TCA cycle, causing pyruvate accumulation and favoring anaerobic metabolism. Anaerobic metabolism leads to the production of lactic acid and further reduces acetyl-CoA availability, which subsequently decreases OxPhos. These observations suggest a metabolic shift from OxPhos to glycolysis may occur with AD progression. This shift is perhaps a compensatory response to enhance energy production through glycolysis, which is noteworthy in the context of mitochondrial dysfunction [5, 6]. Interestingly, PDH, α-KGDH, and ICDH activity are all reported to be calcium-controlled, suggesting a clear link between mCa2+ levels and AD pathogenesis, which will be discussed in the upcoming section. A recent study also indicates that reduced mitochondrial pyruvate uptake in FAD-PS2-expressing cells may elicit impairments in bioenergetics and mitochondrial ATP synthesis [13]. The mechanism for defective mitochondrial pyruvate flux is associated with the hyper-activation of glycogen-synthase-kinase-3β (GSK3β), which decreases hexokinase 1 association with mitochondria and destabilizes the mitochondrial pyruvate carrier complexes [13]. Similarly, α-KGDH is sensitive to oxidative stress, and its reduced activity in PS1 mutant (M146L) fibroblasts suggests a possible mechanism for ROS-dependent metabolic deficiencies [18, 55]. Oxidative stress, as seen in AD brains [56], is reported to increase the expression of SDHA (one of the four nuclear-encoded subunits of complex II, SDH) [57, 58], and the activity of MDH [59]. In summation, alterations in key metabolic enzymes may compromise the neurons’ ability to generate ATP via OxPhos and be an early driver of cellular stress in AD.
Beyond energetic compromise, diminished acetyl-CoA supply caused either by a reduction in glucose metabolism or by reduced PDH activity impairs the synthesis of the neurotransmitter acetylcholine (ACh). ACh is generated from choline and acetyl-CoA by choline acetyltransferase. After synthesis, ACh is transported via an ATP-consuming process and stored in synaptic vesicles [60]. The loss of ACh synthesis in AD results in defective cholinergic neurotransmission [61, 62]. This provides another tangible link between energetic compromise and neuronal dysfunction in AD.
Several of the mitochondrial dehydrogenases mentioned above (PDH, α-KGDH, and ICDH) are known to be regulated by the Ca2+ concentration within the mitochondrial matrix [63,64,65]. The reactions catalyzed by the Ca2+-regulated mitochondrial dehydrogenases are rate-limiting steps in the TCA cycle, and therefore free-Ca2+ content in the mitochondrial matrix is a major regulator of metabolic output. PDH activity increases upon dephosphorylation of its E1α subunit, which is mediated by the Ca2+-sensitive phosphatase (PDP1) [64]. In neurons, Ca2+ influx through voltage-dependent Ca2+ channels is required for the fusion of synaptic vesicles with the plasma membrane and release of neurotransmitters at the synaptic cleft [66, 67]. Neuronal communication through synaptic transmission is an energy-demanding process, and mitochondria have a critical role in this process by providing ATP (via OxPhos) and by buffering synaptic Ca2+/iCa2+ to modulate neurotransmitter release [68]. The efficient regulation and buffering of iCa2+ is critical to prevent neuronal excitotoxicity. Mitochondria and the endoplasmic reticulum (ER) both are significant modulators of iCa2+ signaling and the role of ER in neuronal iCa2+ buffering is well known [69, 70]. However, our understanding of mCa2+ buffering in neurons is limited and evolving. Ca2+ enters the mitochondrial matrix through the mitochondrial calcium uniporter channel (mtCU) [71, 72] and is extruded via the mitochondrial Na+/Ca2+ exchanger (NCLX) [73, 74]. Any dysfunction in mCa2+ exchange or matrix buffering capacity can lead to impairments in mitochondrial Ca2+ homeostasis resulting in mCa2+ overload, oxidative stress, metabolic dysfunction, and cell death that can cause or precede AD-pathology [75,76,77,78]. We and others have reported that mitochondrial and metabolic dysfunction is a primary contributor to AD pathogenesis, with dysfunction observable before the appearance of Aβ aggregates and NFTs [18, 77, 79, 80]. We found alterations in the expression of mCa2+ handling genes in samples isolated from the brains of SAD patients post-mortem and in the triple transgenic mouse model of AD (3xTg-AD) prior to observable AD pathology [77]. Our observations suggest mCa2+ overload caused by an age-dependent remodeling of mCa2+ exchange machinery contributes to the progression of AD by promoting metabolic and mitochondrial dysfunction. We also found a decrease in OxPhos capacity in APPswe cell lines (K670N, M671L Swedish mutation), providing further evidence of impaired mitochondrial metabolism in AD [77]. Importantly, the genetic rescue of neuronal mCa2+ efflux capacity by expression of NCLX in 3xTg-AD mice was sufficient to block age-dependent AD-like pathology [77]. Employing quantitative comparative proteomics strategies in AD mice, other groups have reported significant alterations in the mitochondrial proteome, including the citric acid cycle, OxPhos, pyruvate metabolism, glycolysis, oxidative stress, ion transport, apoptosis, and mitochondrial protein synthesis well before the onset of the AD phenotype [79,80,81]. Further evidence of mCa2+ dysregulation is from metabolomics in an Aβ-transgenic C. elegans model (GRU102), wherein the authors showed a reduction in TCA cycle flux before the appearance of significant Aβ deposition, with the greatest reduction observed in α-KGDH activity. Knockdown of α-KGDH in control worms elicited reductions in both basal and maximal respiration like that observed in the AD worm model [18]. These observations suggest that reduced α-KGDH activity alone is sufficient to recapitulate the metabolic deficits observed in AD and is in line with a study by Yao et al. [46] wherein 3-month old 3xTg-AD mice were found to have reduced mitochondrial respiration and PDH activity, coupled with increased ROS generation [46]. Altogether, these data indicate that mCa2+ dysregulation is likely an early event in AD.
Mitochondria are highly dynamic, and exhibit cell type-specific metabolism in the brain [37, 82]. Axonal mitochondria appear small and sparse whereas dendritic mitochondria are elongated and more densely packed [82]. To ensure appropriate energy supply, especially in distal regions of the axons, mitochondria must be properly positioned. Indeed, mitochondria undergo bi-directional axonal transport including anterograde transport (from cell body to axon) and retrograde transport (from axon to cell body) [83, 84]. Axonal transport is mediated by ATP‐hydrolyzing motor proteins (kinesin‐I for anterograde and dynein for retrograde) to move cargo along microtubule tracks [85] and defects in transport seem to present before evident AD hallmarks [86, 87]. Defects in anterograde transport result in an insufficient supply of ATP at the synapse, resulting in synaptic starvation and dysfunction, an early pathological feature of AD [36]. Similarly, defective retrograde transport can lead to the accumulation of damaged mitochondria, which can compromise mitochondrial quality control mechanisms, which is also noted to occur in AD [88]. Recently, data from the APP-PS1 mouse model showed a reduction in neuronal mitochondria density around amyloid plaques, suggesting impaired mitochondrial transport and/or quality control in AD [37]. Further, several studies indicate that axonal transport of AD-associated proteins becomes defective early in disease progression, resulting in the accumulation of toxic cargo which can elicit protein aggregation, axonal swellings, and neuronal dysfunction [36, 87]. The mechanisms regulating axonal transport are not completely understood but some studies suggest that it is mediated by the interaction of kinesin motor protein with the mitochondrial adaptor proteins, Miro and Milton (known as trafficking kinesin protein (TRAK) family) [89]. Miro is a GTPase with two Ca2+ binding EF-hand domains and is localized to the outer mitochondrial membrane (OMM) and has an essential role in Ca2+-dependent regulation of mitochondrial transport. Intriguingly, Miro1 may also serve as a cytoplasmic Ca2+ sensor and may increase mCa2+ uptake via interaction with MCU’s N-terminal domain [90, 91]. An increase in mCa2+ has been shown to inhibit mitochondrial axonal transport and blocking mCa2+ influx into mitochondria by direct MCU inhibition enhances mitochondrial trafficking in axons [90].
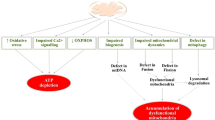
While multiple molecular mechanisms likely contribute to AD pathogenesis, the data suggest that neuronal mCa2+ overload is a primary mediator of AD progression, causing impaired mitochondrial metabolism and ATP production, mitochondrial transport, and increased mitochondrial permeability transition pore (mPTP) opening (Fig. 1). This in turn results in loss of synaptic function, amyloid deposition, tau pathology, and cell death.

Hypothetical mechanisms of mCa2+ overload-induced cellular dysfunction in AD progression. Loss of NCLX and remodeling of the mtCU causes mCa2+ overload that leads to mPTP opening, loss of ATP, and interrupted axonal transport, resulting in AD progression
Parkinson’s disease (PD)
PD is the second most common NDD afflicing ~ 1% of the population above 60 years of age [92]. It is clinically characterized by both motor dysfunction such as tremor (involuntary shaking), bradykinesia (slowness of movements), rigidity (resistance to movement), and akinesia, as well as non-motor disturbances such as depression, anxiety, fatigue, and dementia. These symptoms are caused by a diminishment of the neurotransmitter dopamine due to degeneration of dopaminergic neurons in the pars compacta of the substantia nigra in the midbrain and the deposition of intraneuronal proteinaceous inclusions known as Lewy bodies that are mainly composed of α-synuclein [93]. Most PD cases are sporadic with no known singular cause. Familial PD is associated with mutations in many genes including: SNCA (α-synuclein) [94], PRKN (parkin) [95], PARK7 (DJ-1) [96], LRRK2 (leucine-rich repeat kinase 2) [97], and PINK1 (phosphatase and tensin homologue (PTEN)-induced kinase 1) [98]. Studies suggest that homozygous mutations in Parkin are the most common cause of juvenile PD, but their role in idiopathic PD is unclear. Mutations in Parkin are not reliably associated with Lewy body pathology. Post-mortem examination of patients with Parkin mutations shows a clinical phenotype of dopaminergic neuronal loss and gliosis but lacking Lewy body pathology. However, this remains controversial as a few case reports demonstrate the presence of Lewy pathology in patients with Parkin mutations. Further studies are needed to define if parkin and Lewy body pathology are in linear pathways (reviewed in [99]).
Drug therapy for PD is limited and is primarily focused on enhancing dopamine levels via administration of l-3,4-dihydroxyphenylalanine (L-DOPA or Levodopa), which is metabolized to dopamine after crossing the blood–brain barrier [100, 101]. However, this therapy is only effective in the early stages of disease, and provides symptomatic relief with many adverse side effects, and is insufficient to block the progression of PD [15, 102], suggest a crucial need for new, effective therapies [103, 104]. Although the exact mechanisms of PD pathogenesis are not clear, many possible molecular events have been proposed to contribute to this process.
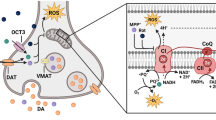
Mitochondrial dysfunction and impaired cellular bioenergetics have emerged as likely mechanisms driving PD pathogenesis in several studies [105, 106]. Dopaminergic neurons consume ~ 20-times more energy as compared to other neurons because of their anatomical structure (extensive long and branched axons), greater number of transmitter release sites, and their pacemaking activity [107]. The high-energetic demand of dopaminergic neurons makes them more susceptible to mitochondrial dysfunction and eventually to cell death in comparison to other neuronal cells [108, 109]. Defects in mitochondrial respiration are supported by findings of reduced glucose utilization in PD patients [110], as well as reduced pyruvate oxidation in fibroblasts derived from PD patients [111], which suggest reduced acetyl-CoA entry into the TCA cycle. The first study showing that defects in mitochondrial respiration may be causal in PD came in the early 1980s. In this study, experimental inhibition of complex I (NADH-ubiquinone reductase) of the ETC was sufficient to cause parkinsonism [112, 113]. This is consistently supported by observations of a profound reduction in ETC activity, mostly complex I, in the substantia nigra, platelets, and skeletal muscle of PD patients [114]. Furthermore, inhibitors of complex I, such as MPP+ (1-methyl-4-phenylpyridinium), 6-hydroxydopamine, rotenone and annonacin all elicit PD-like phenotypes, suggesting that mitochondrial dysfunction is sufficient to promote neuronal dysfunction in PD [115,116,117]. Complex I is a key entry point for electrons into the respiratory chain and is responsible for ~ 40% of mitochondrial ATP production [118, 119]. In addition to complex I, a reduction in complex II and III activity and the mitochondrial DNA (mtDNA) transcription factor, TFAM, has also been reported in PD patients [120,121,122]. Reduced ETC capacity in PD may cause a significant reduction in ATP [123] resulting in a cellular energy crisis that can impact various processes including: (1) ATP-dependent proton pumps that drive vesicular accumulation of dopamine [124, 125]; (2) axonal transport of cargo [126]; (3) mitochondrial dynamics (fusion, fission, turnover, biogenesis and transport) [127, 128]; and (4) ATP-dependent protein degradation systems (e.g. ubiquitin–proteasome and autophagy) [129, 130]. In addition, complex I and III deficiency in PD is linked with increased production of free radicals that further impair mitochondria function, drive protein aggregation and culminate in cell death [131,132,133]. Dopamine is very unstable and sequestered inside synaptic vesicles via the ATP-dependent vesicular monoamine transporter. If not sequestered, it is metabolized by monoamine oxidase to the toxic dopamine metabolite 3,4 dihydroxyphenylacetaldehyde, which contributes to oxidative stress, mPTP opening, and dopaminergic neuronal cell death [134]. Over the past decades, many PD-associated genetic mutations have been found to elicit changes in mitochondrial function and metabolism, supporting the notion that mitochondrial dysfunction is implicated in neuronal cell loss associated with familial PD and vice versa [98]. Mutant α-synuclein localizes to the inner mitochondrial membrane [135] and inhibits complex I activity, and promotes oxidative stress [136]. The interaction of α-synuclein with mitochondria can result in cytochrome c release, increased mCa2+ levels, changes in mitochondrial morphology, and a decline in mitochondrial respiration. α-synuclein-mitochondrial interplay may also inhibit autophagic clearance and increase its aggregation propensity (reviewed in [137]).
A recent study suggested that mitochondrial impairments occur with Lewy body formation [138]. Furthermore, loss of function mutations in DJ-1 caused impairments in OxPhos, and complex I assembly resulting in decreased ATP production, oxidative stress, and increased glycolysis [139, 140]. These findings raise the possibility that mitochondrial dysfunction is causal in maladaptive protein aggregation. Furthermore, Parkin, as an E3 ubiquitin ligase, is directly involved in the proteasomal degradation of protein aggregates. It localizes to mitochondria and prevents cytochrome c release, mitochondrial swelling, and the accumulation of α-synuclein, which may protect dopaminergic neurons from mitochondrial and neuronal dysfunction [141,142,143].
Parkin and PINK1 are required for mitochondrial quality control [144, 145]; thus, loss of Parkin/PINK1 function is hypothesized to cause the accumulation of dysfunctional mitochondria that impair neuronal function. Previous work revealed that PINK1 deficient neurons display reduced NCLX-dependent mCa2+ efflux resulting in matrix Ca2+ overload and subsequent mPTP opening, mitochondrial oxidative stress, lower Δψm, and diminished OxPhos [146]. Furthermore, fibroblasts derived from patients with PINK1 mutations also exhibited impaired mitochondrial metabolism, low Δψm, and low respiration, which was linked to reduced substrate availability [147]. In addition, the activation of NCLX via protein kinase A (PKA)-dependent phosphorylation of serine 258, a putative NCLX regulatory site, increases mCa2+ efflux and protects PINK-1 deficient neurons from mitochondrial dysfunction and cell death [148]. This paradigm fits with previous reports where mCa2+ overload caused by increased mCa2+ uptake (via ERK1/2-dependent upregulation of MCU) caused dendritic degeneration in a late-onset familial PD model (mutation in Leucine-Rich Repeat Kinase 2) [149], and a report of MCU overexpression eliciting excitotoxic cell death [78]. Along the same line, inhibition of MCU is protective in zebrafish models of PD [150, 151]. These findings suggest mCa2+ overload is a contributor to PD progression.
In summary, increasing evidence supports the centrality of impaired mitochondrial function and metabolism in both sporadic and familial PD, resulting in oxidative stress, ETC dysfunction, defective mitochondrial quality control, protein aggregation, progressive cellular dysfunction, and neurodegeneration.
Huntington's disease (HD)
HD is an autosomal-dominant neurodegenerative disease resulting from an expansion of cytosine–adenine–guanine (CAG) repeats (> 35 bp) within the coding sequence of the huntingtin gene (HTT). Mutant huntingtin protein (mHtt) is prone to proteolytic cleavage, misfolding, and aggregation. Clinically, HD is characterized by progressive motor, cognitive, and behavioral dysfunction largely due to the loss of γ-aminobutyric acid (GABAergic) medium spiny neurons in the striatum [152]. The energy impairment hypothesis of HD was first proposed in the early 1980s from clinical observations, which revealed deficits in brain glucose utilization and weight loss in HD patients [153, 154]. Consistently, compelling evidence from PET studies suggests decreased glucose utilization in HD brains [155, 156], suggesting a defect in metabolism. In addition, compared to a control population, pre-symptomatic HD children, with no manifest symptoms, revealed a lower body mass index suggesting energy dysregulation and impairments in anabolic growth [157].
In HD patients, many key enzymes of the TCA cycle and ETC display reduced expression, including PDH, SDH, complex II, III, and IV [158]. In addition, HD patients increase lactate production in the pre-symptomatic phase of HD, indicating a possible reduction in oxidative mitochondrial metabolism and metabolic shift from OxPhos to glycolysis [159,160,161,162]. Irreversible inhibition of SDH by chronic administration of 3-nitropropionic acid in both rodents and non-human primates elicited regional lesions in the striatum accompanied by HD-like pathology [163,164,165]. These results suggest that defects in key TCA cycle enzymes are sufficient to drive HD-pathology. Furthermore, treatment of an HD mouse model with coenzyme Q and creatine for energy supplementation resulted in increased longevity and improved motor function [166, 167], suggesting that improving mitochondrial function and cellular bioenergetics is a viable therapeutic approach to treat HD.
Various other changes in mitochondrial function have been reported in HD. Recently, an examination of HD patient-derived induced pluripotent stem cells (iPSCs) and differentiated neural stem cells revealed altered mitochondria morphology (round and fragmented structure), lower mitochondrial respiration, decreased ATP levels and complex III activity, activation of apoptosis, and increased glycolysis [168]. Proteomic analysis in undifferentiated human HD embryonic stem cells found a decrease in key proteins involved in the ETC before observable differences in huntingtin protein [169]. These studies suggest that mitochondrial function is impaired early in HD pathogenesis. Also, mitochondrial dysfunction is linked with glutamate-mediated excitotoxicity in HD, and this is linked to defects in mCa2+ homeostasis. Studies indicate early abnormalities in mCa2+ that contribute to HD pathology [170]. For example, mitochondria from HD patients have an increased probability of mPTP opening, mitochondrial swelling, oxidative stress, and mCa2+ overload [170, 171]. As in other NDDs, impaired axonal transport is also reported in HD [172] and may be caused by mitochondrial dysfunction and impaired ATP production. Overall, these findings support a prominent role for mitochondrial and metabolic defects in HD pathogenesis.
Altogether, numerous studies support that mitochondrial dysfunction and energy impairments occur before overt pathological symptoms and appear to be central in driving the progression of various NDDs. We hypothesize that metabolic and mitochondrial dysfunction is a result of iCa2+ dysfunction and remodeling of the mCa2+ exchange machinery, which, although initially meant to be compensatory, causes a series of events that culminate in neurodegeneration (Fig. 2).

Mitochondrial and metabolic dysfunction in neurodegeneration. Mitochondrial dysfunction and energy impairments are central events in neurodegeneration
Molecular mechanisms of altered metabolism in NDDs
Above we outlined experimental evidence linking impaired energy metabolism to the initiation or progression of NDDs. This has led to the hypothesis that defects in mitochondrial energy production initiate a cascade of events that causes the neuronal cell death observed in NDDs [173]. However, additional work has raised the possibility that primary defects in other cellular processes may secondarily impair mitochondrial bioenergetics and contribute to NDD pathogenesis [10, 174]. Potential mechanisms that may alter metabolism in NDDs (Fig. 3), and the significance of such altered metabolism for NDDs etiology, are discussed below.

Calcium-centric view of impaired mitochondrial metabolism in NDDs. (1–2) An increase in intracellular calcium by different Ca2+ transport systems in the plasma membrane and the endoplasmic reticulum promotes its entry into the mitochondrial matrix via the mtCU. (3) mCa2+ enhances the activity of key TCA enzymes, leading to elevated OxPhos and ATP generation. On the other side, insufficient or excessive mCa2+ content can impair mitochondrial metabolism in NDDs. The ER plays a crucial role in regulating cellular energetics via the regulated release of Ca2+ near sites of ER-mitochondrial contact to support ATP production. (4) The changes in mitochondrial dynamics alter respiratory complex assembly and affect the coupling between respiration and ATP synthesis. (5–8) The production of ROS and activation of AMPK signaling by Ca2+ and insulin signaling also constitute the diverse array of signaling pathways that elicit transcription regulation of energy metabolism genes
Calcium signaling
Calcium signaling is required for neuronal function and regulates a range of processes, including neuronal excitability, neurotransmitter release, mitochondrial metabolism, and cell death. Tight control over iCa2+ flux is therefore essential for coordinated activity and neuronal homeostasis. As discussed above, altered Ca2+ homeostasis has been reported in NDDs and may contribute to neuronal dysfunction and death (reviewed in [175]). This section discusses the impact of altered Ca2+ handling in various subcellular compartments and its impact on metabolism.
Intracellular calcium
Perturbation of global iCa2+ homeostasis alters Ca2+ content in compartments, including the ER and mitochondria. Both organelles are implicated in the pathophysiology of NDDs, thus altered iCa2+ levels may contribute to NDD progression. Indeed, iCa2+ overload is a widely accepted feature of NDDs and is a likely cause of dysfunction and death of the neuronal populations affected by these diseases [176].
Elevated iCa2+ content is a common feature of AD and is especially pronounced in neurons containing NFTs [177]. Elevated iCa2+ in AD can exert detrimental effects by altering Ca2+-dependent signaling. Two examples of Ca2+-dependent proteins in neurons are the phosphatase calcineurin and Ca2+/calmodulin-dependent protein kinase II (CaMKII). Altered calcineurin and CaMKII signaling have been linked to memory impairment, synaptic loss, and neurodegeneration, all features of AD progression [178]. Such findings have inspired the “Ca2+ hypothesis of AD,” which proposes that cellular Ca2+ dysregulation is a central driver of disease progression [179, 180].
While Ca2+ dysregulation likely precedes neurodegeneration, several reports describe mechanisms by which Aβ directly elevates iCa2+ content, suggesting a vicious positive feedback loop that reinforces Ca2+ overload. First, Aβ can promote ROS production and subsequent oxidation of membrane lipids that can disrupt cellular ion transport [181]. Second, Aβ peptides may form Ca2+-permeable pores in the plasma membrane, allowing for direct influx of Ca2+ into the neuron [182]. This idea is supported by the observation that neurites with more Aβ have greater levels of iCa2+ [183]. Aβ is also proposed to stimulate Ca2+ uptake through L-type voltage-gated Ca2+ channels [184], but this notion is still debated [185]. Finally, Aβ may hyperactivate the NMDA receptor, leading to cellular Ca2+ overload [186]. Dysregulated Ca2+ handling is also implicated in the pathophysiology of PD [187]. Neurons with α-synuclein mutations have increased plasma membrane ion permeability, possibly due to the formation of pores by mutant α-synuclein [188]. Pharmacologic inhibition of Cav1.3 L-type Ca2+ channels is protective in animal models of PD [189], suggesting that increased ion channel activity contributes to excess iCa2+ entry. Store-operated calcium entry is also impaired in PD and leads to the depletion of ER Ca2+ content [175]. Likewise, neuronal Ca2+ dysregulation is a common feature of HD [190]. mHtt can stimulate NMDA receptors in medium spiny striatal neurons, potentially leading to excess iCa2+ [176]. Also, mHtt binds to and potentiates IP3 receptor signaling, enhancing Ca2+ release from the ER [191]. These combined effects all tend to deplete the ER of Ca2+ and can ultimately enhance store-operated Ca2+ entry [192], setting up a continuous cycle that promotes increased iCa2+ load.
ER calcium
Alterations in iCa2 handling in NDDs can cause secondary changes in ER Ca2+ load. The ER plays a critical role in regulating cellular energetics via the regulated release of Ca2+ near sites of ER-mitochondrial contact. In brief, these discrete sites of ER-mitochondrial apposition (examined further below under “MAMs” or mitochondrial associated membranes) create a microdomain where Ca2+ concentration can rise to levels as much as 20 × greater than in the bulk cytosol [193, 194]. This localized, high Ca2+ concentration is required for the activation of the mCa2+ uptake machinery (gating of the mtCU) and efficient ER-to-mitochondria Ca2+ transfer [195]. Inter-organelle Ca2+ transport is especially important in regulating iCa2+ homeostasis in neurons and is implicated not only in energetic homeostasis but also vesicle trafficking and neurotransmitter release [196, 197]. Thus, any structural disruption in ER-mitochondrial contact sites in NDDs and subsequent perturbation in ER-mitochondrial Ca2+ transfer has the potential to exacerbate iCa2+ stress and accelerate disease progression. Moreover, altered ER Ca2+ content and ER Ca2+ release will affect mCa2+ content. As discussed in the next section, either insufficient or excessive mCa2+ content can impair mitochondrial metabolism and signaling, thus underlying the significance of altered ER Ca2+ handling for cellular bioenergetics in NDDs.
Mitochondrial calcium
Given the central role of mCa2+ in regulating cellular metabolism and survival, it is not surprising that altered mCa2+ handling is reported in cellular NDD models. NDDs are universally associated with mCa2+ overload, which can impair cellular metabolism by inducing oxidative stress, which itself can impair OxPhos; and by inducing mPTP, which compromises ATP production by collapsing Δψm [198, 199].
In AD, mCa2+ overload can result from excessive ER-to-mitochondrial Ca2+ transfer induced by Aβ oligomers [200]. There are also reports that Aβ accumulates in mitochondria and interacts with the matrix mPTP regulator cyclophilin D, thus increasing permeability transition [201, 202] and impairing mitochondrial energetics in a Ca2+-independent manner. More recent results from our laboratory indicate that mCa2+ efflux is compromised in AD, due to downregulation of NCLX, which further promotes mCa2+ overload [77].
Signs of mCa2+ overload are observed in cellular models of PD induced by expression of mutant α-synuclein. These include loss of Δψm, cristae structure, and ATP content, features that are exacerbated by simultaneous expression of mutant PINK1 and rescued by pharmacologic blockade of mCa2+ uptake [203]. α-synuclein can accumulate within mitochondria and increases mCa2+ content, leading to increased ROS production [204]. However, conflicting reports [205] suggest that the effects of α-synuclein on mCa2+ homeostasis may be more nuanced. In some cases, α-synuclein may be beneficial by promoting ER-mitochondrial contacts to enhance ER-to-mitochondrial Ca2+ transfer and support mitochondrial bioenergetics [206]. Altered mCa2+ handling has been suggested in HD, but existing reports have yielded disparate conclusions on this point. The reader is referred to a recent review by Cali et al. [198] for a more detailed discussion.
Mitochondrial-associated membranes
Mitochondrial-associated membranes (MAMs) are regions where the ER is in close proximation with the outer mitochondrial membrane to allow crosstalk between these organelles. MAMs are particularly important for the exchange of Ca2+ and phospholipids, both of which impact ER/mitochondrial function and thus have profound effects on cellular metabolism and overall homeostasis (reviewed in [207]). MAMs are required for the synthesis of lipids such as phosphatidylcholine [208], with the mitochondrion serving as the site of phosphatidylethanolamine (PE) generation, an intermediate in phosphatidylcholine production. In turn, PE is crucial for overall mitochondrial morphology and function [209]. MAMs are also enriched for proteins involved in mitochondrial fission and fusion [210, 211], and so can influence mitochondrial dynamics, morphology, and biogenesis. Likewise, MAMs are important sites for the regulation of mitophagy and the clearance of defective mitochondria [212].
MAMs are often found at synapses, where they may modulate synaptic activity [213]. Efficient ER-to-mitochondria Ca2+ transfer is necessary for ATP production and may be especially important for meeting the high energetic demands of synaptic transmission [214] and/or serve as an important mechanism to buffer synaptic Ca2+. The ER and mitochondrial membranes are held in apposition at MAMs via a network of tether proteins [215, 216], some of which have been implicated in NDDs.
ER-mitochondrial tethers include the ER-mitochondria encounter structure (ERMES), which was identified in yeast [217]. Mammalian counterparts to the ERMES complex are still being validated, but may include the IP3 receptor, phosphofurin acidic cluster sorting protein-2 (PACS-2), B-cell receptor associated protein 31 (Bap31), PDZD8 in the ER, the mitochondrial fission protein Fis1, and the outer mitochondrial membrane protein VDAC [218]. PDZD8 is required for ER-mitochondria tethering, and loss of PDZD8 is sufficient to impact ER-mitochondrial Ca2+ dynamics in mammalian neurons [219]. Mitofusin 2 has also been proposed as a MAM tether [220], but this idea remains controversial [221, 222]. Additional proposed tethers include the oxysterol binding-related proteins ORP5 and ORP8, which can interact with mitochondrial protein tyrosine phosphatase interacting protein 51 (PTPIP51) [223]. The OMM protein synaptojanin 2 binding protein (SYNJ2BP) and the ER protein ribosome-binding protein 1 (RRBP1) are proposed to mediate specific interactions between the rough ER and mitochondria [224]. Finally, a tethering complex that may have particular importance in NDDs is comprised of the ER vesicle-associated membrane proteins-associated protein B (VAPB) and mitochondrial PTPIP51 [225, 226].
Altered ER-mitochondrial contacts in NDDs may contribute to disease pathology [227, 228]. Loss of MAM tethers can disrupt ER-mitochondrial Ca2+ transfer and so impair mitochondrial metabolism, leading to cellular energy depletion and the activation of autophagy [229, 230]. MAM disruption in NDDs could also lead to energetic compromise by impairing the synthesis of phospholipids important for mitochondrial membranes, such as cardiolipin [208, 231, 232]. This species is enriched in the mitochondrial inner membrane and is critical for proper ETC and ATP synthase function [233,234,235,236]. Finally, ER-mitochondrial associations regulate a number of processes that are commonly disrupted in NDDs such as Ca2+ handling, inflammation, axonal transport, and mitochondrial function [237]. These observations support the hypothesis that altered ER-mitochondrial communication is a common mechanism underlying NDDs.
The AD-related proteins APP and γ-secretase are all enriched at MAMs [238]. Observations of altered lipid metabolism and Ca2+ handling in both FAD and SAD suggest that these proteins may be associated with MAM dysfunction [228, 239]. Altered iCa2+ handling in AD could result from enhanced ER-mitochondrial Ca2+ transfer. The finding that ER Ca2+ concentration is increased in AD supports this view. Finally, altered lipid homeostasis resulting from dysfunctional ER/mitochondrial tethering may also impair mitochondrial energetics in AD. The MAMs of AD brain tissue and cells exhibit increased sphingomyelin hydrolysis by sphingomyelinase, which leads to increased ceramide content [240]. Increased ceramide content in AD appears sufficient to impair mitochondrial respiration [241, 242], as pharmacologic reduction of ceramide levels in AD models can rescue mitochondrial respiration [240]. Specific mechanisms by which elevated ceramide content in mitochondrial membranes may impair respiratory function and cellular bioenergetics have been detailed elsewhere [173].
Furthermore, altered ER-mitochondrial contacts and signaling are reported in PD, leading some to propose that disrupted MAMs are a significant contributor to PD pathogenesis [228, 237]. Proteins that are implicated in familial PD such as α-synuclein, PINK1, and Parkin all alter ER-mitochondrial signaling [243,244,245]. However, the specific consequences of these alterations on PD pathology are still the subject of active investigation [207].
The protein α-synuclein localizes to MAMs [245] and is thought to influence Ca2+ signaling [205, 206] and lipid metabolism [246], ultimately leading to defective ER and mitochondrial function [206]. Whereas wild-type α-synuclein promotes ER-mitochondrial contacts [206], the association of familial PD mutant α-synuclein with MAMs is disrupted. This change may represent one mechanism for compromised MAM structure and function in PD [246]. However, conflicting data suggest that overexpression of either wild-type or mutant α-synuclein can disrupt ER-mitochondrial contacts by binding to VAPB on the ER membrane and interfering with VAPB-PTPIP51 interactions [245]. Disruption of this tether complex can impair mitochondrial energetics because it compromises Ca2+ exchange between the two organelles [245]. Similar mechanisms may explain how DJ-1 mutations contribute to early-onset PD [247]. DJ-1 is normally localized to MAMs where it promotes ER-mitochondrial association and facilitates mCa2+ uptake [248]. Mutant DJ-1, as seen in PD, may disrupt MAM structure, ER-mitochondrial contacts, mCa2+ uptake, and mitochondrial bioenergetics [249]. In addition, mutations in Parkin and PINK1 may initiate PD pathogenesis via effects at MAMs. PINK and Parkin are recruited to sites of contact between ER and defective mitochondria to coordinate their autophagic clearance [244, 250]. Thus, defective PINK or Parkin may disrupt mitochondrial quality control mechanisms that rely on MAM interactions. Over time, this could impair cellular metabolism and contribute to PD pathology due to the accumulation of dysfunctional mitochondria.
Mitochondrial structural defects
Mitochondrial structure is determined by a precise balance between mitochondrial fusion and fission and membrane dynamics that are mediated by several proteins including mitofusin 1 (MFN1), mitofusin 2 (MFN2), optic atrophy 1 (OPA1), dynamin-related protein 1 (DRP1), mitochondrial fission factor (MFF), and fission 1 protein [251]. During fasting or starvation mitochondria tend to fuse [252] due to inhibition of Drp1 by PKA and AMPK [253, 254]. These changes in mitochondrial structure alter respiratory complex assembly and affect the coupling between respiration and ATP synthesis [252], thereby increasing ATP production efficiency when fuel is scarce.
Defective mitochondrial fission and fusion have been implicated in NDDs [251], and abnormal mitochondrial structure and morphology are reported in AD, PD, and HD [319]. Variants in insulin signaling pathway genes, such as AKT [1), and determining the precise temporal order of pathological cellular events is of paramount importance to development of novel therapeutic targets to combat NDDs.
Abbreviations
- NDDs:
-
Neurodegenerative diseases
- AD:
-
Alzheimer’s disease
- PD:
-
Parkinson’s disease
- HD:
-
Huntington’s disease
- PET:
-
Positron emission tomography
- Aβ:
-
Amyloid-beta
- NFTs:
-
Neurofibrillary tangles
- OxPhos:
-
Oxidative phosphorylation
- ETC:
-
Electron transport chain
- PDH:
-
Pyruvate dehydrogenase
- α-KGDH:
-
Alpha-ketoglutarate dehydrogenase
- ICDH:
-
Isocitrate dehydrogenase
- SDH:
-
Succinate dehydrogenase
- MDH:
-
Malate dehydrogenase
- Δψm:
-
Mitochondrial membrane potential
- COX:
-
Cytochrome-c-oxidase
- iCa2 + :
-
Intracellular calcium
- mtCU:
-
Mitochondrial calcium uniporter channel
- mCa2 + :
-
Mitochondrial calcium
- NCLX:
-
Mitochondrial Na+/Ca2+ exchanger
- MCU:
-
Mitochondrial calcium uniporter
- mPTP:
-
Mitochondrial permeability transition pore
- PMCA:
-
Plasma membrane Ca2+ ATPase
- NCX:
-
Na+/Ca2+ exchanger
- TRP:
-
The transient receptor potential
- VDCC:
-
Voltage-gated calcium channels
- AMPAR:
-
α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor
- mGluR:
-
Metabotropic glutamate receptors
- NMDAR:
-
N-Methyl-D-aspartate receptor
- NCX:
-
Na+/Ca2+ exchanger
- SOCE:
-
Store-operated calcium entry
- IP3R:
-
Inositol 1,4,5-trisphosphate receptor
- RYR:
-
Ryanodine receptor
- SERCA:
-
Sarco/endoplasmic reticulum Ca2+-ATPase
- TCA:
-
Tricarboxylic acid cycle
- MAMs:
-
Mitochondrial-associated membranes
- ROS:
-
Reactive oxygen species
- RNS:
-
Reactive nitrogen species
- AMPK:
-
AMP-activated protein kinase
- PGC-1α:
-
Peroxisome proliferator-activated receptor (PPAR)-γ co-activator 1α
- mtDNA:
-
Mitochondrial DNA
References
Kety SS (1957) The general metabolism of the brain in vivo. Metabolism of the nervous system. Elsevier, pp 221–237
Oyarzabal A, Marin-Valencia I (2019) Synaptic energy metabolism and neuronal excitability, in sickness and health. J Inherit Metab Dis 42:220–236. https://doi.org/10.1002/jimd.12071
Vergara RC, Jaramillo-Riveri S, Luarte A, Moenne-Loccoz C, Fuentes R, Couve A, Maldonado PE (2019) The energy homeostasis principle: neuronal energy regulation drives local network dynamics generating behavior. Front Comput Neurosci 13:49. https://doi.org/10.3389/fncom.2019.00049
Bordone MP, Salman MM, Titus HE, Amini E, Andersen JV, Chakraborti B, Diuba AV, Dubouskaya TG, Ehrke E, Espindola de Freitas A et al (2019) The energetic brain—a review from students to students. J Neurochem 151:139–165. https://doi.org/10.1111/jnc.14829
Formentini L, Pereira MP, Sanchez-Cenizo L, Santacatterina F, Lucas JJ, Navarro C, Martinez-Serrano A, Cuezva JM (2014) In vivo inhibition of the mitochondrial H+-ATP synthase in neurons promotes metabolic preconditioning. EMBO J 33:762–778. https://doi.org/10.1002/embj.201386392
Motori E, Atanassov I, Kochan SMV, Folz-Donahue K, Sakthivelu V, Giavalisco P, Toni N, Puyal J, Larsson NG (2020) Neuronal metabolic rewiring promotes resilience to neurodegeneration caused by mitochondrial dysfunction. Sci Adv 6:eaba8271. https://doi.org/10.1126/sciadv.aba8271
Burmistrova O, Olias-Arjona A, Lapresa R, Jimenez-Blasco D, Eremeeva T, Shishov D, Romanov S, Zakurdaeva K, Almeida A, Fedichev PO et al (2019) Targeting PFKFB3 alleviates cerebral ischemia-reperfusion injury in mice. Sci Rep 9:11670. https://doi.org/10.1038/s41598-019-48196-z
Herrero-Mendez A, Almeida A, Fernandez E, Maestre C, Moncada S, Bolanos JP (2009) The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C-Cdh1. Nat Cell Biol 11:747–752. https://doi.org/10.1038/ncb1881
Jack CR Jr, Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, Holtzman DM, Jagust W, Jessen F, Karlawish J et al (2018) NIA-AA research framework: toward a biological definition of Alzheimer’s disease. Alzheimer’s Dementia 14:535–562. https://doi.org/10.1016/j.jalz.2018.02.018
Mattson MP, Pedersen WA, Duan W, Culmsee C, Camandola S (1999) Cellular and molecular mechanisms underlying perturbed energy metabolism and neuronal degeneration in Alzheimer’s and Parkinson’s diseases. Ann NY Acad Sci 893:154–175. https://doi.org/10.1111/j.1749-6632.1999.tb07824.x
Spinelli JB, Haigis MC (2018) The multifaceted contributions of mitochondria to cellular metabolism. Nat Cell Biol 20:745–754. https://doi.org/10.1038/s41556-018-0124-1
Golpich M, Amini E, Mohamed Z, Azman Ali R, Mohamed Ibrahim N, Ahmadiani A (2017) Mitochondrial dysfunction and biogenesis in neurodegenerative diseases: pathogenesis and treatment. CNS Neurosci Ther 23:5–22. https://doi.org/10.1111/cns.12655
Rossi A, Rigotto G, Valente G, Giorgio V, Basso E, Filadi R, Pizzo P (2020) Defective mitochondrial pyruvate flux affects cell bioenergetics in Alzheimer’s disease-related models. Cell Rep 30(2332–2348):e2310. https://doi.org/10.1016/j.celrep.2020.01.060
Cunnane SC, Trushina E, Morland C, Prigione A, Casadesus G, Andrews ZB, Beal MF, Bergersen LH, Brinton RD, de la Monte S et al (2020) Brain energy rescue: an emerging therapeutic concept for neurodegenerative disorders of ageing. Nat Rev Drug Discov 19:609–633. https://doi.org/10.1038/s41573-020-0072-x
McFarthing K, Buff S, Rafaloff G, Dominey T, Wyse RK, Stott SRW (2020) Parkinson’s disease drug therapies in the clinical trial pipeline: 2020. J Parkinsons Dis 10:757–774. https://doi.org/10.3233/JPD-202128
Travessa AM, Rodrigues FB, Mestre TA, Ferreira JJ (2017) Fifteen years of clinical trials in Huntington’s disease: a very low clinical drug development success rate. J Huntington’s Dis 6:157–163. https://doi.org/10.3233/JHD-170245
Perez MJ, Ponce DP, Aranguiz A, Behrens MI, Quintanilla RA (2018) Mitochondrial permeability transition pore contributes to mitochondrial dysfunction in fibroblasts of patients with sporadic Alzheimer’s disease. Redox Biol 19:290–300. https://doi.org/10.1016/j.redox.2018.09.001
Teo E, Ravi S, Barardo D, Kim HS, Fong S, Cazenave-Gassiot A, Tan TY, Ching J, Kovalik JP, Wenk MR et al (2019) Metabolic stress is a primary pathogenic event in transgenic Caenorhabditis elegans expressing pan-neuronal human amyloid beta. Elife. https://doi.org/10.7554/eLife.50069
Fao L, Rego AC (2020) Mitochondrial and redox-based therapeutic strategies in Huntington’s disease. Antioxid Redox Signal. https://doi.org/10.1089/ars.2019.8004
Mi Y, Qi G, Brinton RD, Yin F (2020) Mitochondria-targeted therapeutics for Alzheimer’s disease: the good, the bad, the potential. Antioxid Redox Signal. https://doi.org/10.1089/ars.2020.8070
Weissig V (2020) Drug development for the therapy of mitochondrial diseases. Trends Mol Med 26:40–57. https://doi.org/10.1016/j.molmed.2019.09.002
Britti E, Delaspre F, Tamarit J, Ros J (2018) Mitochondrial calcium signalling and neurodegenerative diseases. . , Neuronal Signal. https://doi.org/10.1042/NS20180061
Mehta D, Jackson R, Paul G, Shi J, Sabbagh M (2017) Why do trials for Alzheimer’s disease drugs keep failing? A discontinued drug perspective for 2010–2015. Expert Opin Investig Drugs 26:735–739. https://doi.org/10.1080/13543784.2017.1323868
Panza F, Lozupone M, Watling M, Imbimbo BP (2019) Do BACE inhibitor failures in Alzheimer patients challenge the amyloid hypothesis of the disease? Expert Rev Neurother 19:599–602. https://doi.org/10.1080/14737175.2019.1621751
Selkoe DJ, Hardy J (2016) The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med 8:595–608. https://doi.org/10.15252/emmm.201606210
Cai Q, Tammineni P (2017) Mitochondrial aspects of synaptic dysfunction in Alzheimer’s disease. J Alzheimers Dis 57:1087–1103. https://doi.org/10.3233/JAD-160726
Liu C, Song X, Nisbet R, Gotz J (2016) Co-immunoprecipitation with tau isoform-specific antibodies reveals distinct protein interactions and highlights a putative role for 2N tau in disease. J Biol Chem 291:8173–8188. https://doi.org/10.1074/jbc.M115.641902
Manczak M, Reddy PH (2012) Abnormal interaction of VDAC1 with amyloid beta and phosphorylated tau causes mitochondrial dysfunction in Alzheimer’s disease. Hum Mol Genet 21:5131–5146. https://doi.org/10.1093/hmg/dds360
Giannakopoulos P, Herrmann FR, Bussiere T, Bouras C, Kovari E, Perl DP, Morrison JH, Gold G, Hof PR (2003) Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer’s disease. Neurology 60:1495–1500. https://doi.org/10.1212/01.wnl.0000063311.58879.01
Guillozet AL, Weintraub S, Mash DC, Mesulam MM (2003) Neurofibrillary tangles, amyloid, and memory in aging and mild cognitive impairment. Arch Neurol 60:729–736. https://doi.org/10.1001/archneur.60.5.729
Mostafavi S, Gaiteri C, Sullivan SE, White CC, Tasaki S, Xu J, Taga M, Klein HU, Patrick E, Komashko V et al (2018) A molecular network of the aging human brain provides insights into the pathology and cognitive decline of Alzheimer’s disease. Nat Neurosci 21:811–819. https://doi.org/10.1038/s41593-018-0154-9
Neff RA, Wang M, Vatansever S, Guo L, Ming C, Wang Q, Wang E, Horgusluoglu-Moloch E, Song WM, Li A et al (2021) Molecular subty** of Alzheimer’s disease using RNA sequencing data reveals novel mechanisms and targets. Sci Adv. https://doi.org/10.1126/sciadv.abb5398
Cummings J, Lee G, Ritter A, Sabbagh M, Zhong K (2020) Alzheimer’s disease drug development pipeline: 2020. Alzheimers Dement (NY) 6:e12050. https://doi.org/10.1002/trc2.12050
Jansen IE, Savage JE, Watanabe K, Bryois J, Williams DM, Steinberg S, Sealock J, Karlsson IK, Hagg S, Athanasiu L et al (2020) Author Correction: Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat Genet 52:354. https://doi.org/10.1038/s41588-019-0573-x
Swerdlow RH (2020) The mitochondrial hypothesis: dysfunction, bioenergetic defects, and the metabolic link to Alzheimer’s disease. Int Rev Neurobiol 154:207–233. https://doi.org/10.1016/bs.irn.2020.01.008
Calkins MJ, Manczak M, Mao P, Shirendeb U, Reddy PH (2011) Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer’s disease. Hum Mol Genet 20:4515–4529. https://doi.org/10.1093/hmg/ddr381
Fecher C, Trovo L, Muller SA, Snaidero N, Wettmarshausen J, Heink S, Ortiz O, Wagner I, Kuhn R, Hartmann J et al (2019) Cell-type-specific profiling of brain mitochondria reveals functional and molecular diversity. Nat Neurosci 22:1731–1742. https://doi.org/10.1038/s41593-019-0479-z
Schmukler E, Solomon S, Simonovitch S, Goldshmit Y, Wolfson E, Michaelson DM, Pinkas-Kramarski R (2020) Altered mitochondrial dynamics and function in APOE4-expressing astrocytes. Cell Death Dis 11:578. https://doi.org/10.1038/s41419-020-02776-4
Perez MJ, Ponce DP, Osorio-Fuentealba C, Behrens MI, Quintanilla RA (2017) Mitochondrial bioenergetics is altered in fibroblasts from patients with sporadic Alzheimer’s disease. Front Neurosci 11:553. https://doi.org/10.3389/fnins.2017.00553
Manczak M, Kandimalla R, Yin X, Reddy PH (2018) Hippocampal mutant APP and amyloid beta-induced cognitive decline, dendritic spine loss, defective autophagy, mitophagy and mitochondrial abnormalities in a mouse model of Alzheimer’s disease. Hum Mol Genet 27:1332–1342. https://doi.org/10.1093/hmg/ddy042
Hardy JA, Higgins GA (1992) Alzheimer’s disease: the amyloid cascade hypothesis. Science 256:184–185. https://doi.org/10.1126/science.1566067
de Leon MJ, Ferris SH, George AE, Christman DR, Fowler JS, Gentes C, Reisberg B, Gee B, Emmerich M, Yonekura Y et al (1983) Positron emission tomographic studies of aging and Alzheimer disease. AJNR Am J Neuroradiol 4:568–571
Ferris SH, de Leon MJ, Wolf AP, Farkas T, Christman DR, Reisberg B, Fowler JS, Macgregor R, Goldman A, George AE et al (1980) Positron emission tomography in the study of aging and senile dementia. Neurobiol Aging 1:127–131. https://doi.org/10.1016/0197-4580(80)90005-6
Foster NL, Chase TN, Fedio P, Patronas NJ, Brooks RA, Di Chiro G (1983) Alzheimer’s disease: focal cortical changes shown by positron emission tomography. Neurology 33:961–965. https://doi.org/10.1212/wnl.33.8.961
Sorbi S, Bird ED, Blass JP (1983) Decreased pyruvate dehydrogenase complex activity in Huntington and Alzheimer brain. Ann Neurol 13:72–78. https://doi.org/10.1002/ana.410130116
Yao J, Irwin RW, Zhao L, Nilsen J, Hamilton RT, Brinton RD (2009) Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA 106:14670–14675. https://doi.org/10.1073/pnas.0903563106
Gibson GE, Starkov A, Blass JP, Ratan RR, Beal MF (2010) Cause and consequence: mitochondrial dysfunction initiates and propagates neuronal dysfunction, neuronal death and behavioral abnormalities in age-associated neurodegenerative diseases. Biochim Biophys Acta 1802:122–134. https://doi.org/10.1016/j.bbadis.2009.08.010
Fisar Z, Hansikova H, Krizova J, Jirak R, Kitzlerova E, Zverova M, Hroudova J, Wenchich L, Zeman J, Raboch J (2019) Activities of mitochondrial respiratory chain complexes in platelets of patients with Alzheimer’s disease and depressive disorder. Mitochondrion 48:67–77. https://doi.org/10.1016/j.mito.2019.07.013
Kish SJ, Bergeron C, Rajput A, Dozic S, Mastrogiacomo F, Chang LJ, Wilson JM, DiStefano LM, Nobrega JN (1992) Brain cytochrome oxidase in Alzheimer’s disease. J Neurochem 59:776–779. https://doi.org/10.1111/j.1471-4159.1992.tb09439.x
Parker WD Jr, Filley CM, Parks JK (1990) Cytochrome oxidase deficiency in Alzheimer’s disease. Neurology 40:1302–1303. https://doi.org/10.1212/wnl.40.8.1302
Bubber P, Haroutunian V, Fisch G, Blass JP, Gibson GE (2005) Mitochondrial abnormalities in Alzheimer brain: mechanistic implications. Ann Neurol 57:695–703. https://doi.org/10.1002/ana.20474
Mastroeni D, Khdour OM, Delvaux E, Nolz J, Olsen G, Berchtold N, Cotman C, Hecht SM, Coleman PD (2017) Nuclear but not mitochondrial-encoded oxidative phosphorylation genes are altered in aging, mild cognitive impairment, and Alzheimer’s disease. Alzheimer’s Dementia 13:510–519. https://doi.org/10.1016/j.jalz.2016.09.003
Zhang L, Guo XQ, Chu JF, Zhang X, Yan ZR, Li YZ (2015) Potential hippocampal genes and pathways involved in Alzheimer’s disease: a bioinformatic analysis. Genet Mol Res (GMR) 14:7218–7232. https://doi.org/10.4238/2015.June.29.15
Beck JS, Mufson EJ, Counts SE (2016) Evidence for mitochondrial UPR gene activation in familial and sporadic Alzheimer’s disease. Curr Alzheimer Res 13:610–614. https://doi.org/10.2174/1567205013666151221145445
Gibson GE, Park LC, Zhang H, Sorbi S, Calingasan NY (1999) Oxidative stress and a key metabolic enzyme in Alzheimer brains, cultured cells, and an animal model of chronic oxidative deficits. Ann NY Acad Sci 893:79–94. https://doi.org/10.1111/j.1749-6632.1999.tb07819.x
Arslan J, Jamshed H, Qureshi H (2020) Early detection and prevention of Alzheimer’s Disease: role of oxidative markers and natural antioxidants. Front Aging Neurosci 12:231. https://doi.org/10.3389/fnagi.2020.00231
Au HC, Scheffler IE (1998) Promoter analysis of the human succinate dehydrogenase iron-protein gene–both nuclear respiratory factors NRF-1 and NRF-2 are required. Eur J Biochem 251:164–174. https://doi.org/10.1046/j.1432-1327.1998.2510164.x
Miranda S, Foncea R, Guerrero J, Leighton F (1999) Oxidative stress and upregulation of mitochondrial biogenesis genes in mitochondrial DNA-depleted HeLa cells. Biochem Biophys Res Commun 258:44–49. https://doi.org/10.1006/bbrc.1999.0580
Shi Q, Gibson GE (2011) Up-regulation of the mitochondrial malate dehydrogenase by oxidative stress is mediated by miR-743a. J Neurochem 118:440–448. https://doi.org/10.1111/j.1471-4159.2011.07333.x
Varoqui H, Erickson JD (1996) Active transport of acetylcholine by the human vesicular acetylcholine transporter. J Biol Chem 271:27229–27232. https://doi.org/10.1074/jbc.271.44.27229
Bierer LM, Haroutunian V, Gabriel S, Knott PJ, Carlin LS, Purohit DP, Perl DP, Schmeidler J, Kanof P, Davis KL (1995) Neurochemical correlates of dementia severity in Alzheimer’s disease: relative importance of the cholinergic deficits. J Neurochem 64:749–760. https://doi.org/10.1046/j.1471-4159.1995.64020749.x
Stanciu GD, Luca A, Rusu RN, Bild V, Beschea Chiriac SI, Solcan C, Bild W, Ababei DC (2019) Alzheimer’s disease pharmacotherapy in relation to cholinergic system involvement. Biomolecules. https://doi.org/10.3390/biom10010040
Denton RM (2009) Regulation of mitochondrial dehydrogenases by calcium ions. Biochim Biophys Acta 1787:1309–1316. https://doi.org/10.1016/j.bbabio.2009.01.005
Denton RM, Randle PJ, Martin BR (1972) Stimulation by calcium ions of pyruvate dehydrogenase phosphate phosphatase. Biochem J 128:161–163. https://doi.org/10.1042/bj1280161
Denton RM, Richards DA, Chin JG (1978) Calcium ions and the regulation of NAD+-linked isocitrate dehydrogenase from the mitochondria of rat heart and other tissues. Biochem J 176:899–906. https://doi.org/10.1042/bj1760899
Sabatini BL, Regehr WG (1996) Timing of neurotransmission at fast synapses in the mammalian brain. Nature 384:170–172. https://doi.org/10.1038/384170a0
Sudhof TC (2012) Calcium control of neurotransmitter release. Cold Spring Harb Perspect Biol 4:a011353. https://doi.org/10.1101/cshperspect.a011353
Verstreken P, Ly CV, Venken KJ, Koh TW, Zhou Y, Bellen HJ (2005) Synaptic mitochondria are critical for mobilization of reserve pool vesicles at Drosophila neuromuscular junctions. Neuron 47:365–378. https://doi.org/10.1016/j.neuron.2005.06.018
Berridge MJ (1998) Neuronal calcium signaling. Neuron 21:13–26. https://doi.org/10.1016/s0896-6273(00)80510-3
Genovese I, Giamogante F, Barazzuol L, Battista T, Fiorillo A, Vicario M, D’Alessandro G, Cipriani R, Limatola C, Rossi D et al (2020) Sorcin is an early marker of neurodegeneration, Ca(2+) dysregulation and endoplasmic reticulum stress associated to neurodegenerative diseases. Cell Death Dis 11:861. https://doi.org/10.1038/s41419-020-03063-y
Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL et al (2011) Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476:341–345. https://doi.org/10.1038/nature10234
De Stefani D, Raffaello A, Teardo E, Szabo I, Rizzuto R (2011) A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476:336–340. https://doi.org/10.1038/nature10230
Luongo TS, Lambert JP, Gross P, Nwokedi M, Lombardi AA, Shanmughapriya S, Carpenter AC, Kolmetzky D, Gao E, van Berlo JH et al (2017) The mitochondrial Na(+)/Ca(2+) exchanger is essential for Ca(2+) homeostasis and viability. Nature 545:93–97. https://doi.org/10.1038/nature22082
Palty R, Silverman WF, Hershfinkel M, Caporale T, Sensi SL, Parnis J, Nolte C, Fishman D, Shoshan-Barmatz V, Herrmann S et al (2010) NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc Natl Acad Sci USA 107:436–441
Calvo-Rodriguez M, Hou SS, Snyder AC, Kharitonova EK, Russ AN, Das S, Fan Z, Muzikansky A, Garcia-Alloza M, Serrano-Pozo A et al (2020) Increased mitochondrial calcium levels associated with neuronal death in a mouse model of Alzheimer’s disease. Nat Commun 11:2146. https://doi.org/10.1038/s41467-020-16074-2
Granatiero V, Pacifici M, Raffaello A, De Stefani D, Rizzuto R (2019) Overexpression of mitochondrial calcium uniporter causes neuronal death. Oxid Med Cell Longev 2019:1681254. https://doi.org/10.1155/2019/1681254
Jadiya P, Kolmetzky DW, Tomar D, Di Meco A, Lombardi AA, Lambert JP, Luongo TS, Ludtmann MH, Pratico D, Elrod JW (2019) Impaired mitochondrial calcium efflux contributes to disease progression in models of Alzheimer’s disease. Nat Commun 10:3885. https://doi.org/10.1038/s41467-019-11813-6
Qiu J, Tan YW, Hagenston AM, Martel MA, Kneisel N, Skehel PA, Wyllie DJ, Bading H, Hardingham GE (2013) Mitochondrial calcium uniporter Mcu controls excitotoxicity and is transcriptionally repressed by neuroprotective nuclear calcium signals. Nat Commun 4:2034. https://doi.org/10.1038/ncomms3034
Chou JL, Shenoy DV, Thomas N, Choudhary PK, Laferla FM, Goodman SR, Breen GA (2011) Early dysregulation of the mitochondrial proteome in a mouse model of Alzheimer’s disease. J Proteomics 74:466–479. https://doi.org/10.1016/j.jprot.2010.12.012
Volgyi K, Badics K, Sialana FJ, Gulyassy P, Udvari EB, Kis V, Drahos L, Lubec G, Kekesi KA, Juhasz G (2018) Early presymptomatic changes in the proteome of mitochondria-associated membrane in the APP/PS1 mouse model of Alzheimer’s disease. Mol Neurobiol 55:7839–7857. https://doi.org/10.1007/s12035-018-0955-6
Evans AR, Gu L, Guerrero R Jr, Robinson RA (2015) Global cPILOT analysis of the APP/PS-1 mouse liver proteome. Proteomics Clin Appl 9:872–884. https://doi.org/10.1002/prca.201400149
Lewis TL Jr, Kwon SK, Lee A, Shaw R, Polleux F (2018) MFF-dependent mitochondrial fission regulates presynaptic release and axon branching by limiting axonal mitochondria size. Nat Commun 9:5008. https://doi.org/10.1038/s41467-018-07416-2
Mandal A, Drerup CM (2019) Axonal transport and mitochondrial function in neurons. Front Cell Neurosci 13:373. https://doi.org/10.3389/fncel.2019.00373
Saxton WM, Hollenbeck PJ (2012) The axonal transport of mitochondria. J Cell Sci 125:2095–2104. https://doi.org/10.1242/jcs.053850
Hirokawa N, Takemura R (2005) Molecular motors and mechanisms of directional transport in neurons. Nat Rev Neurosci 6:201–214. https://doi.org/10.1038/nrn1624
Sleigh JN, Rossor AM, Fellows AD, Tosolini AP, Schiavo G (2019) Axonal transport and neurological disease. Nat Rev Neurol 15:691–703. https://doi.org/10.1038/s41582-019-0257-2
Stokin GB, Lillo C, Falzone TL, Brusch RG, Rockenstein E, Mount SL, Raman R, Davies P, Masliah E, Williams DS et al (2005) Axonopathy and transport deficits early in the pathogenesis of Alzheimer’s disease. Science 307:1282–1288. https://doi.org/10.1126/science.1105681
Cai Q, Tammineni P (2016) Alterations in mitochondrial quality control in Alzheimer’s disease. Front Cell Neurosci 10:24. https://doi.org/10.3389/fncel.2016.00024
Wang X, Schwarz TL (2009) The mechanism of Ca2+ -dependent regulation of kinesin-mediated mitochondrial motility. Cell 136:163–174. https://doi.org/10.1016/j.cell.2008.11.046
Chang KT, Niescier RF, Min KT (2011) Mitochondrial matrix Ca2+ as an intrinsic signal regulating mitochondrial motility in axons. Proc Natl Acad Sci USA 108:15456–15461. https://doi.org/10.1073/pnas.1106862108
Niescier RF, Hong K, Park D, Min KT (2018) MCU interacts with Miro1 to modulate mitochondrial functions in neurons. J Neurosci Off J Soc Neurosci 38:4666–4677. https://doi.org/10.1523/JNEUROSCI.0504-18.2018
Dorsey ER, Bloem BR (2018) The Parkinson Pandemic-a call to action. JAMA Neurol 75:9–10. https://doi.org/10.1001/jamaneurol.2017.3299
Simon-Gozalbo A, Rodriguez-Blazquez C, Forjaz MJ, Martinez-Martin P (2020) Clinical characterization of Parkinson’s disease patients with cognitive impairment. Front Neurol 11:731. https://doi.org/10.3389/fneur.2020.00731
Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R et al (1997) Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276:2045–2047. https://doi.org/10.1126/science.276.5321.2045
Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N (1998) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392:605–608. https://doi.org/10.1038/33416
Bonifati V, Rizzu P, van Baren MJ, Schaap O, Breedveld GJ, Krieger E, Dekker MC, Squitieri F, Ibanez P, Joosse M et al (2003) Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 299:256–259. https://doi.org/10.1126/science.1077209
Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB et al (2004) Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 44:601–607. https://doi.org/10.1016/j.neuron.2004.11.005
Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG et al (2004) Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 304:1158–1160. https://doi.org/10.1126/science.1096284
Dawson TM, Dawson VL (2010) The role of parkin in familial and sporadic Parkinson’s disease. Mov Disord 25(Suppl 1):S32-39. https://doi.org/10.1002/mds.22798
LeWitt PA, Giladi N, Navon N (2019) Pharmacokinetics and efficacy of a novel formulation of carbidopa-levodopa (Accordion Pill((R))) in Parkinson’s disease. Parkinsonism Relat Disord 65:131–138. https://doi.org/10.1016/j.parkreldis.2019.05.032
LeWitt PA, Hauser RA, Pahwa R, Isaacson SH, Fernandez HH, Lew M, Saint-Hilaire M, Pourcher E, Lopez-Manzanares L, Waters C et al (2019) Safety and efficacy of CVT-301 (levodopa inhalation powder) on motor function during off periods in patients with Parkinson’s disease: a randomised, double-blind, placebo-controlled phase 3 trial. Lancet Neurol 18:145–154. https://doi.org/10.1016/S1474-4422(18)30405-8
Muller T, Mohr JD (2019) Recent clinical advances in pharmacotherapy for levodopa-induced dyskinesia. Drugs 79:1367–1374. https://doi.org/10.1007/s40265-019-01170-5
Espay AJ, Kalia LV, Gan-Or Z, Williams-Gray CH, Bedard PL, Rowe SM, Morgante F, Fasano A, Stecher B, Kauffman MA et al (2020) Disease modification and biomarker development in Parkinson disease: revision or reconstruction? Neurology 94:481–494. https://doi.org/10.1212/WNL.0000000000009107
Simuni T, Siderowf A, Lasch S, Coffey CS, Caspell-Garcia C, Jennings D, Tanner CM, Trojanowski JQ, Shaw LM, Seibyl J et al (2018) Longitudinal change of clinical and biological measures in early Parkinson’s disease: Parkinson’s progression markers initiative cohort. Mov Disord 33:771–782. https://doi.org/10.1002/mds.27361
Jansen IE, Ye H, Heetveld S, Lechler MC, Michels H, Seinstra RI, Lubbe SJ, Drouet V, Lesage S, Majounie E et al (2017) Discovery and functional prioritization of Parkinson’s disease candidate genes from large-scale whole exome sequencing. Genome Biol 18:22. https://doi.org/10.1186/s13059-017-1147-9
Milanese C, Payan-Gomez C, Galvani M, Molano Gonzalez N, Tresini M, Nait Abdellah S, van Roon-Mom WMC, Figini S, Marinus J, van Hilten JJ et al (2019) Peripheral mitochondrial function correlates with clinical severity in idiopathic Parkinson’s disease. Mov Disord 34:1192–1202. https://doi.org/10.1002/mds.27723
Mamelak M (2018) Parkinson’s disease, the dopaminergic neuron and gammahydroxybutyrate. Neurol Ther 7:5–11. https://doi.org/10.1007/s40120-018-0091-2
Juarez Olguin H, Calderon Guzman D, Hernandez Garcia E, Barragan Mejia G (2016) The role of dopamine and its dysfunction as a consequence of oxidative stress. Oxid Med Cell Longev 2016:9730467. https://doi.org/10.1155/2016/9730467
Kim TY, Leem E, Lee JM, Kim SR (2020) Control of reactive oxygen species for the prevention of Parkinson’s disease: the possible application of flavonoids. Antioxidants (Basel). https://doi.org/10.3390/antiox9070583
Martin WR, Hayden MR (1987) Cerebral glucose and dopa metabolism in movement disorders. Can J Neurol Sci 14:448–451. https://doi.org/10.1017/s0317167100037896
Mytilineou C, Werner P, Molinari S, Di Rocco A, Cohen G, Yahr MD (1994) Impaired oxidative decarboxylation of pyruvate in fibroblasts from patients with Parkinson’s disease. J Neural Transm Park Dis Dement Sect 8:223–228. https://doi.org/10.1007/bf02260943
Langston JW, Ballard P, Tetrud JW, Irwin I (1983) Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 219:979–980. https://doi.org/10.1126/science.6823561
Schapira AH, Cooper JM, Dexter D, Jenner P, Clark JB, Marsden CD (1989) Mitochondrial complex I deficiency in Parkinson’s disease. Lancet 1:1269. https://doi.org/10.1016/s0140-6736(89)92366-0
Perier C, Vila M (2012) Mitochondrial biology and Parkinson’s disease. Cold Spring Harb Perspect Med 2:a009332. https://doi.org/10.1101/cshperspect.a009332
Cannon JR, Greenamyre JT (2010) Neurotoxic in vivo models of Parkinson’s disease recent advances. Prog Brain Res 184:17–33. https://doi.org/10.1016/S0079-6123(10)84002-6
Jackson-Lewis V, Przedborski S (2007) Protocol for the MPTP mouse model of Parkinson’s disease. Nat Protoc 2:141–151. https://doi.org/10.1038/nprot.2006.342
Valdez LB, Zaobornyj T, Bandez MJ, Lopez-Cepero JM, Boveris A, Navarro A (2019) Complex I syndrome in striatum and frontal cortex in a rat model of Parkinson disease. Free Radic Biol Med 135:274–282. https://doi.org/10.1016/j.freeradbiomed.2019.03.001
Galkin A, Drose S, Brandt U (2006) The proton pum** stoichiometry of purified mitochondrial complex I reconstituted into proteoliposomes. Biochim Biophys Acta 1757:1575–1581. https://doi.org/10.1016/j.bbabio.2006.10.001
Wikstrom M (1984) Two protons are pumped from the mitochondrial matrix per electron transferred between NADH and ubiquinone. FEBS Lett 169:300–304. https://doi.org/10.1016/0014-5793(84)80338-5
Grunewald A, Rygiel KA, Hepplewhite PD, Morris CM, Picard M, Turnbull DM (2016) Mitochondrial DNA depletion in respiratory chain-deficient Parkinson disease neurons. Ann Neurol 79:366–378. https://doi.org/10.1002/ana.24571
Haas RH, Nasirian F, Nakano K, Ward D, Pay M, Hill R, Shults CW (1995) Low platelet mitochondrial complex I and complex II/III activity in early untreated Parkinson’s disease. Ann Neurol 37:714–722. https://doi.org/10.1002/ana.410370604
Shinde S, Pasupathy K (2006) Respiratory-chain enzyme activities in isolated mitochondria of lymphocytes from patients with Parkinson’s disease: preliminary study. Neurol India 54:390–393. https://doi.org/10.4103/0028-3886.28112
Chan P, DeLanney LE, Irwin I, Langston JW, Di Monte D (1991) Rapid ATP loss caused by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in mouse brain. J Neurochem 57:348–351. https://doi.org/10.1111/j.1471-4159.1991.tb02134.x
Anne C, Gasnier B (2014) Vesicular neurotransmitter transporters: mechanistic aspects. Curr Top Membr 73:149–174. https://doi.org/10.1016/B978-0-12-800223-0.00003-7
Lotharius J, Brundin P (2002) Impaired dopamine storage resulting from alpha-synuclein mutations may contribute to the pathogenesis of Parkinson’s disease. Hum Mol Genet 11:2395–2407. https://doi.org/10.1093/hmg/11.20.2395
Chu Y, Morfini GA, Langhamer LB, He Y, Brady ST, Kordower JH (2012) Alterations in axonal transport motor proteins in sporadic and experimental Parkinson’s disease. Brain 135:2058–2073. https://doi.org/10.1093/brain/aws133
Han H, Tan J, Wang R, Wan H, He Y, Yan X, Guo J, Gao Q, Li J, Shang S et al (2020) PINK1 phosphorylates Drp 1(S616) to regulate mitophagy-independent mitochondrial dynamics. EMBO Rep. https://doi.org/10.15252/embr.201948686
Van Laar VS, Berman SB (2013) The interplay of neuronal mitochondrial dynamics and bioenergetics: implications for Parkinson’s disease. Neurobiol Dis 51:43–55. https://doi.org/10.1016/j.nbd.2012.05.015
De Mattos EP, Wentink A, Nussbaum-Krammer C, Hansen C, Bergink S, Melki R, Kam**a HH (2020) Protein quality control pathways at the crossroad of synucleinopathies. J Parkinsons Dis 10:369–382. https://doi.org/10.3233/JPD-191790
Pantazopoulou M, Brembati V, Kanellidi A, Bousset L, Melki R, Stefanis L (2020) Distinct alpha-Synuclein species induced by seeding are selectively cleared by the Lysosome or the Proteasome in neuronally differentiated SH-SY5Y cells. J Neurochem. https://doi.org/10.1111/jnc.15174
Musgrove RE, Helwig M, Bae EJ, Aboutalebi H, Lee SJ, Ulusoy A, Di Monte DA (2019) Oxidative stress in vagal neurons promotes parkinsonian pathology and intercellular alpha-synuclein transfer. J Clin Investig 129:3738–3753. https://doi.org/10.1172/JCI127330
Perier C, Tieu K, Guegan C, Caspersen C, Jackson-Lewis V, Carelli V, Martinuzzi A, Hirano M, Przedborski S, Vila M (2005) Complex I deficiency primes Bax-dependent neuronal apoptosis through mitochondrial oxidative damage. Proc Natl Acad Sci USA 102:19126–19131. https://doi.org/10.1073/pnas.0508215102
Tapias V, McCoy JL, Greenamyre JT (2019) Phenothiazine normalizes the NADH/NAD(+) ratio, maintains mitochondrial integrity and protects the nigrostriatal dopamine system in a chronic rotenone model of Parkinson’s disease. Redox Biol 24:101164. https://doi.org/10.1016/j.redox.2019.101164
Kristal BS, Conway AD, Brown AM, Jain JC, Ulluci PA, Li SW, Burke WJ (2001) Selective dopaminergic vulnerability: 3,4-dihydroxyphenylacetaldehyde targets mitochondria. Free Radic Biol Med 30:924–931. https://doi.org/10.1016/s0891-5849(01)00484-1
Parihar MS, Parihar A, Fujita M, Hashimoto M, Ghafourifar P (2008) Mitochondrial association of alpha-synuclein causes oxidative stress. Cell Mol Life Sci (CMLS) 65:1272–1284. https://doi.org/10.1007/s00018-008-7589-1
Hu D, Sun X, Liao X, Zhang X, Zarabi S, Schimmer A, Hong Y, Ford C, Luo Y, Qi X (2019) Alpha-synuclein suppresses mitochondrial protease ClpP to trigger mitochondrial oxidative damage and neurotoxicity. Acta Neuropathol 137:939–960. https://doi.org/10.1007/s00401-019-01993-2
Faustini G, Bono F, Valerio A, Pizzi M, Spano P, Bellucci A (2017) Mitochondria and alpha-Synuclein: friends or foes in the pathogenesis of Parkinson’s disease? Genes. https://doi.org/10.3390/genes8120377
Mahul-Mellier AL, Burtscher J, Maharjan N, Weerens L, Croisier M, Kuttler F, Leleu M, Knott GW, Lashuel HA (2020) The process of Lewy body formation, rather than simply alpha-synuclein fibrillization, is one of the major drivers of neurodegeneration. Proc Natl Acad Sci USA 117:4971–4982. https://doi.org/10.1073/pnas.1913904117
Chen R, Park HA, Mnatsakanyan N, Niu Y, Licznerski P, Wu J, Miranda P, Graham M, Tang J, Boon AJW et al (2019) Parkinson’s disease protein DJ-1 regulates ATP synthase protein components to increase neuronal process outgrowth. Cell Death Dis 10:469. https://doi.org/10.1038/s41419-019-1679-x
Heo JY, Park JH, Kim SJ, Seo KS, Han JS, Lee SH, Kim JM, Park JI, Park SK, Lim K et al (2012) DJ-1 null dopaminergic neuronal cells exhibit defects in mitochondrial function and structure: involvement of mitochondrial complex I assembly. PLoS ONE 7:e32629. https://doi.org/10.1371/journal.pone.0032629
Bian M, Liu J, Hong X, Yu M, Huang Y, Sheng Z, Fei J, Huang F (2012) Overexpression of parkin ameliorates dopaminergic neurodegeneration induced by 1- methyl-4-phenyl-1,2,3,6-tetrahydropyridine in mice. PLoS ONE 7:e39953. https://doi.org/10.1371/journal.pone.0039953
Chung E, Choi Y, Park J, Nah W, Park J, Jung Y, Lee J, Lee H, Park S, Hwang S et al (2020) Intracellular delivery of Parkin rescues neurons from accumulation of damaged mitochondria and pathological alpha-synuclein. Sci Adv 6:eaba1193. https://doi.org/10.1126/sciadv.aba1193
Darios F, Corti O, Lucking CB, Hampe C, Muriel MP, Abbas N, Gu WJ, Hirsch EC, Rooney T, Ruberg M et al (2003) Parkin prevents mitochondrial swelling and cytochrome c release in mitochondria-dependent cell death. Hum Mol Genet 12:517–526. https://doi.org/10.1093/hmg/ddg044
Ge P, Dawson VL, Dawson TM (2020) PINK1 and Parkin mitochondrial quality control: a source of regional vulnerability in Parkinson’s disease. Mol Neurodegener 15:20. https://doi.org/10.1186/s13024-020-00367-7
Narendra D, Tanaka A, Suen DF, Youle RJ (2008) Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 183:795–803. https://doi.org/10.1083/jcb.200809125
Gandhi S, Wood-Kaczmar A, Yao Z, Plun-Favreau H, Deas E, Klupsch K, Downward J, Latchman DS, Tabrizi SJ, Wood NW et al (2009) PINK1-associated Parkinson’s disease is caused by neuronal vulnerability to calcium-induced cell death. Mol Cell 33:627–638. https://doi.org/10.1016/j.molcel.2009.02.013
Abramov AY, Gegg M, Grunewald A, Wood NW, Klein C, Schapira AH (2011) Bioenergetic consequences of PINK1 mutations in Parkinson disease. PLoS ONE 6:e25622. https://doi.org/10.1371/journal.pone.0025622
Kostic M, Ludtmann MH, Bading H, Hershfinkel M, Steer E, Chu CT, Abramov AY, Sekler I (2015) PKA phosphorylation of NCLX reverses mitochondrial calcium overload and depolarization, promoting survival of PINK1-deficient dopaminergic neurons. Cell Rep 13:376–386. https://doi.org/10.1016/j.celrep.2015.08.079
Verma M, Callio J, Otero PA, Sekler I, Wills ZP, Chu CT (2017) Mitochondrial calcium dysregulation contributes to dendrite degeneration mediated by PD/LBD-associated LRRK2 mutants. J Neurosci Off J Soc Neurosci 37:11151–11165. https://doi.org/10.1523/JNEUROSCI.3791-16.2017
Soman S, Keatinge M, Moein M, Da Costa M, Mortiboys H, Skupin A, Sugunan S, Bazala M, Kuznicki J, Bandmann O (2017) Inhibition of the mitochondrial calcium uniporter rescues dopaminergic neurons in pink1(-/-) zebrafish. Eur J Neurosci 45:528–535. https://doi.org/10.1111/ejn.13473
Soman SK, Bazala M, Keatinge M, Bandmann O, Kuznicki J (2019) Restriction of mitochondrial calcium overload by mcu inactivation renders a neuroprotective effect in zebrafish models of Parkinson’s disease. Biol Open. https://doi.org/10.1242/bio.044347
Anil M, Mason SL, Barker RA (2020) The clinical features and progression of late-onset versus younger-onset in an adult cohort of huntington’s disease patients. J Huntington’s Dis 9:275–282. https://doi.org/10.3233/JHD-200404
Kuhl DE, Phelps ME, Markham CH, Metter EJ, Riege WH, Winter J (1982) Cerebral metabolism and atrophy in Huntington’s disease determined by 18FDG and computed tomographic scan. Ann Neurol 12:425–434. https://doi.org/10.1002/ana.410120504
Sanberg PR, Fibiger HC, Mark RF (1981) Body weight and dietary factors in Huntington’s disease patients compared with matched controls. Med J Aust 1:407–409
Berent S, Giordani B, Lehtinen S, Markel D, Penney JB, Buchtel HA, Starosta-Rubinstein S, Hichwa R, Young AB (1988) Positron emission tomographic scan investigations of Huntington’s disease: cerebral metabolic correlates of cognitive function. Ann Neurol 23:541–546. https://doi.org/10.1002/ana.410230603
Morea V, Bidollari E, Colotti G, Fiorillo A, Rosati J, De Filippis L, Squitieri F, Ilari A (2017) Glucose transportation in the brain and its impairment in Huntington disease: one more shade of the energetic metabolism failure? Amino Acids 49:1147–1157. https://doi.org/10.1007/s00726-017-2417-2
Lee JK, Mathews K, Schlaggar B, Perlmutter J, Paulsen JS, Ep** E, Burmeister L, Nopoulos P (2012) Measures of growth in children at risk for Huntington disease. Neurology 79:668–674. https://doi.org/10.1212/WNL.0b013e3182648b65
Naseri NN, Bonica J, Xu H, Park LC, Arjomand J, Chen Z, Gibson GE (2016) Novel metabolic abnormalities in the tricarboxylic acid cycle in peripheral cells from Huntington’s disease patients. PLoS ONE 11:e0160384. https://doi.org/10.1371/journal.pone.0160384
Ciammola A, Sassone J, Sciacco M, Mencacci NE, Ripolone M, Bizzi C, Colciago C, Moggio M, Parati G, Silani V et al (2011) Low anaerobic threshold and increased skeletal muscle lactate production in subjects with Huntington’s disease. Mov Disord 26:130–137. https://doi.org/10.1002/mds.23258
Jenkins BG, Koroshetz WJ, Beal MF, Rosen BR (1993) Evidence for impairment of energy metabolism in vivo in Huntington’s disease using localized 1H NMR spectroscopy. Neurology 43:2689–2695. https://doi.org/10.1212/wnl.43.12.2689
Jenkins BG, Rosas HD, Chen YC, Makabe T, Myers R, MacDonald M, Rosen BR, Beal MF, Koroshetz WJ (1998) 1H NMR spectroscopy studies of Huntington’s disease: correlations with CAG repeat numbers. Neurology 50:1357–1365. https://doi.org/10.1212/wnl.50.5.1357
Martin WR, Wieler M, Hanstock CC (2007) Is brain lactate increased in Huntington’s disease? J Neurol Sci 263:70–74. https://doi.org/10.1016/j.jns.2007.05.035
Beal MF, Brouillet E, Jenkins BG, Ferrante RJ, Kowall NW, Miller JM, Storey E, Srivastava R, Rosen BR, Hyman BT (1993) Neurochemical and histologic characterization of striatal excitotoxic lesions produced by the mitochondrial toxin 3-nitropropionic acid. J Neurosci Off J Soc Neurosci 13:4181–4192
Brouillet E, Hantraye P, Ferrante RJ, Dolan R, Leroy-Willig A, Kowall NW, Beal MF (1995) Chronic mitochondrial energy impairment produces selective striatal degeneration and abnormal choreiform movements in primates. Proc Natl Acad Sci USA 92:7105–7109. https://doi.org/10.1073/pnas.92.15.7105
Wiprich MT, Zanandrea R, Altenhofen S, Bonan CD (2020) Influence of 3-nitropropionic acid on physiological and behavioral responses in zebrafish larvae and adults. Comp Biochem Physiol C Toxicol Pharmacol 234:108772. https://doi.org/10.1016/j.cbpc.2020.108772
Andreassen OA, Dedeoglu A, Ferrante RJ, Jenkins BG, Ferrante KL, Thomas M, Friedlich A, Browne SE, Schilling G, Borchelt DR et al (2001) Creatine increase survival and delays motor symptoms in a transgenic animal model of Huntington’s disease. Neurobiol Dis 8:479–491. https://doi.org/10.1006/nbdi.2001.0406
Yang L, Calingasan NY, Wille EJ, Cormier K, Smith K, Ferrante RJ, Beal MF (2009) Combination therapy with coenzyme Q10 and creatine produces additive neuroprotective effects in models of Parkinson’s and Huntington’s diseases. J Neurochem 109:1427–1439. https://doi.org/10.1111/j.1471-4159.2009.06074.x
Lopes C, Tang Y, Anjo SI, Manadas B, Onofre I, de Almeida LP, Daley GQ, Schlaeger TM, Rego ACC (2020) Mitochondrial and redox modifications in huntington disease induced pluripotent stem cells rescued by CRISPR/Cas9 CAGs targeting. Front Cell Dev Biol 8:576592. https://doi.org/10.3389/fcell.2020.576592
McQuade LR, Balachandran A, Scott HA, Khaira S, Baker MS, Schmidt U (2014) Proteomics of Huntington’s disease-affected human embryonic stem cells reveals an evolving pathology involving mitochondrial dysfunction and metabolic disturbances. J Proteome Res 13:5648–5659. https://doi.org/10.1021/pr500649m
Panov AV, Gutekunst CA, Leavitt BR, Hayden MR, Burke JR, Strittmatter WJ, Greenamyre JT (2002) Early mitochondrial calcium defects in Huntington’s disease are a direct effect of polyglutamines. Nat Neurosci 5:731–736. https://doi.org/10.1038/nn884
Cherubini M, Lopez-Molina L, Gines S (2020) Mitochondrial fission in Huntington’s disease mouse striatum disrupts ER-mitochondria contacts leading to disturbances in Ca(2+) efflux and Reactive Oxygen Species (ROS) homeostasis. Neurobiol Dis 136:104741. https://doi.org/10.1016/j.nbd.2020.104741
Gunawardena S, Her LS, Brusch RG, Laymon RA, Niesman IR, Gordesky-Gold B, Sintasath L, Bonini NM, Goldstein LS (2003) Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyQ proteins in Drosophila. Neuron 40:25–40. https://doi.org/10.1016/s0896-6273(03)00594-4
Area-Gomez E, Guardia-Laguarta C, Schon EA, Przedborski S (2019) Mitochondria, OxPhos, and neurodegeneration: cells are not just running out of gas. J Clin Investig 129:34–45. https://doi.org/10.1172/JCI120848
Camandola S, Mattson MP (2017) Brain metabolism in health, aging, and neurodegeneration. EMBO J 36:1474–1492. https://doi.org/10.15252/embj.201695810
Pchitskaya E, Popugaeva E, Bezprozvanny I (2018) Calcium signaling and molecular mechanisms underlying neurodegenerative diseases. Cell Calcium 70:87–94. https://doi.org/10.1016/j.ceca.2017.06.008
Mattson MP (2007) Calcium and neurodegeneration. Aging Cell 6:337–350. https://doi.org/10.1111/j.1474-9726.2007.00275.x
Popugaeva E, Pchitskaya E, Bezprozvanny I (2018) Dysregulation of intracellular calcium signaling in Alzheimer’s disease. Antioxid Redox Signal 29:1176–1188. https://doi.org/10.1089/ars.2018.7506
Popugaeva E, Pchitskaya E, Bezprozvanny I (2017) Dysregulation of neuronal calcium homeostasis in Alzheimer’s disease—a therapeutic opportunity? Biochem Biophys Res Commun 483:998–1004. https://doi.org/10.1016/j.bbrc.2016.09.053
Hypothesis AAC, W, (2017) Calcium Hypothesis of Alzheimer’s disease and brain aging: a framework for integrating new evidence into a comprehensive theory of pathogenesis. Alzheimer’s Dementia 13(178–182):e117. https://doi.org/10.1016/j.jalz.2016.12.006
Bezprozvanny I, Mattson MP (2008) Neuronal calcium mishandling and the pathogenesis of Alzheimer’s disease. Trends Neurosci 31:454–463. https://doi.org/10.1016/j.tins.2008.06.005
Mattson MP, Cheng B, Davis D, Bryant K, Lieberburg I, Rydel RE (1992) beta-Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J Neurosci Off J Soc Neurosci 12:376–389
Arispe N, Diaz JC, Simakova O (2007) Abeta ion channels. prospects for treating Alzheimer’s disease with Abeta channel blockers. Biochim Biophys Acta 1768:1952–1965. https://doi.org/10.1016/j.bbamem.2007.03.014
Kuchibhotla KV, Goldman ST, Lattarulo CR, Wu HY, Hyman BT, Bacskai BJ (2008) Abeta plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron 59:214–225. https://doi.org/10.1016/j.neuron.2008.06.008
Ueda K, Shinohara S, Yagami T, Asakura K, Kawasaki K (1997) Amyloid beta protein potentiates Ca2+ influx through L-type voltage-sensitive Ca2+ channels: a possible involvement of free radicals. J Neurochem 68:265–271. https://doi.org/10.1046/j.1471-4159.1997.68010265.x
Nimmrich V, Grimm C, Draguhn A, Barghorn S, Lehmann A, Schoemaker H, Hillen H, Gross G, Ebert U, Bruehl C (2008) Amyloid beta oligomers (A beta(1–42) globulomer) suppress spontaneous synaptic activity by inhibition of P/Q-type calcium currents. J Neurosci Off J Soc Neurosci 28:788–797. https://doi.org/10.1523/JNEUROSCI.4771-07.2008
Zhang Y, Li P, Feng J, Wu M (2016) Dysfunction of NMDA receptors in Alzheimer’s disease. Neurol Sci Off J Ital Neurol Soc Ital Soc Clin Neurophysiol 37:1039–1047. https://doi.org/10.1007/s10072-016-2546-5
Zaichick SV, McGrath KM, Caraveo G (2017) The role of Ca(2+) signaling in Parkinson’s disease. Dis Model Mech 10:519–535. https://doi.org/10.1242/dmm.028738
Furukawa K, Matsuzaki-Kobayashi M, Hasegawa T, Kikuchi A, Sugeno N, Itoyama Y, Wang Y, Yao PJ, Bushlin I, Takeda A (2006) Plasma membrane ion permeability induced by mutant alpha-synuclein contributes to the degeneration of neural cells. J Neurochem 97:1071–1077. https://doi.org/10.1111/j.1471-4159.2006.03803.x
Ilijic E, Guzman JN, Surmeier DJ (2011) The L-type channel antagonist isradipine is neuroprotective in a mouse model of Parkinson’s disease. Neurobiol Dis 43:364–371. https://doi.org/10.1016/j.nbd.2011.04.007
Raymond LA (2017) Striatal synaptic dysfunction and altered calcium regulation in Huntington disease. Biochem Biophys Res Commun 483:1051–1062. https://doi.org/10.1016/j.bbrc.2016.07.058
Tang TS, Tu H, Chan EY, Maximov A, Wang Z, Wellington CL, Hayden MR, Bezprozvanny I (2003) Huntingtin and huntingtin-associated protein 1 influence neuronal calcium signaling mediated by inositol-(1,4,5) triphosphate receptor type 1. Neuron 39:227–239. https://doi.org/10.1016/s0896-6273(03)00366-0
Wu J, Ryskamp DA, Liang X, Egorova P, Zakharova O, Hung G, Bezprozvanny I (2016) Enhanced store-operated calcium entry leads to striatal synaptic loss in a Huntington’s disease mouse model. J Neurosci Off J Soc Neurosci 36:125–141. https://doi.org/10.1523/JNEUROSCI.1038-15.2016
Csordas G, Thomas AP, Hajnoczky G (1999) Quasi-synaptic calcium signal transmission between endoplasmic reticulum and mitochondria. EMBO J 18:96–108. https://doi.org/10.1093/emboj/18.1.96
Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, Tuft RA, Pozzan T (1998) Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 280:1763–1766
Szabadkai G, Simoni AM, Rizzuto R (2003) Mitochondrial Ca2+ uptake requires sustained Ca2+ release from the endoplasmic reticulum. J Biol Chem 278:15153–15161. https://doi.org/10.1074/jbc.M300180200
Dolphin AC, Lee A (2020) Presynaptic calcium channels: specialized control of synaptic neurotransmitter release. Nat Rev Neurosci 21:213–229. https://doi.org/10.1038/s41583-020-0278-2
Pinton P, Giorgi C, Siviero R, Zecchini E, Rizzuto R (2008) Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene 27:6407–6418. https://doi.org/10.1038/onc.2008.308
Cali T, Ottolini D, Brini M (2012) Mitochondrial Ca(2+) and neurodegeneration. Cell Calcium 52:73–85. https://doi.org/10.1016/j.ceca.2012.04.015
Ryan KC, Ashkavand Z, Norman KR (2020) The role of mitochondrial calcium homeostasis in Alzheimer’s and related diseases. Int J Mol Sci. https://doi.org/10.3390/ijms21239153
Calvo-Rodriguez M, Hernando-Perez E, Nunez L, Villalobos C (2019) Amyloid beta oligomers increase ER-mitochondria Ca(2+) cross talk in young hippocampal neurons and exacerbate aging-induced intracellular Ca(2+) remodeling. Front Cell Neurosci 13:22. https://doi.org/10.3389/fncel.2019.00022
Caspersen C, Wang N, Yao J, Sosunov A, Chen X, Lustbader JW, Xu HW, Stern D, McKhann G, Yan SD (2005) Mitochondrial Abeta: a potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. FASEB J 19:2040–2041. https://doi.org/10.1096/fj.05-3735fje
Du H, Guo L, Fang F, Chen D, Sosunov AA, McKhann GM, Yan Y, Wang C, Zhang H, Molkentin JD et al (2008) Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat Med 14:1097–1105. https://doi.org/10.1038/nm.1868
Marongiu R, Spencer B, Crews L, Adame A, Patrick C, Trejo M, Dallapiccola B, Valente EM, Masliah E (2009) Mutant Pink1 induces mitochondrial dysfunction in a neuronal cell model of Parkinson’s disease by disturbing calcium flux. J Neurochem 108:1561–1574. https://doi.org/10.1111/j.1471-4159.2009.05932.x
Parihar MS, Parihar A, Fujita M, Hashimoto M, Ghafourifar P (2009) Alpha-synuclein overexpression and aggregation exacerbates impairment of mitochondrial functions by augmenting oxidative stress in human neuroblastoma cells. Int J Biochem Cell Biol 41:2015–2024. https://doi.org/10.1016/j.biocel.2009.05.008
Hettiarachchi NT, Parker A, Dallas ML, Pennington K, Hung CC, Pearson HA, Boyle JP, Robinson P, Peers C (2009) alpha-Synuclein modulation of Ca2+ signaling in human neuroblastoma (SH-SY5Y) cells. J Neurochem 111:1192–1201. https://doi.org/10.1111/j.1471-4159.2009.06411.x
Cali T, Ottolini D, Negro A, Brini M (2012) alpha-Synuclein controls mitochondrial calcium homeostasis by enhancing endoplasmic reticulum-mitochondria interactions. J Biol Chem 287:17914–17929. https://doi.org/10.1074/jbc.M111.302794
Gomez-Suaga P, Bravo-San Pedro JM, Gonzalez-Polo RA, Fuentes JM, Niso-Santano M (2018) ER-mitochondria signaling in Parkinson’s disease. Cell Death Dis 9:337. https://doi.org/10.1038/s41419-017-0079-3
Flis VV, Daum G (2013) Lipid transport between the endoplasmic reticulum and mitochondria. Cold Spring Harb Perspect Biol. https://doi.org/10.1101/cshperspect.a013235
Joshi AS, Thompson MN, Fei N, Huttemann M, Greenberg ML (2012) Cardiolipin and mitochondrial phosphatidylethanolamine have overlap** functions in mitochondrial fusion in Saccharomyces cerevisiae. J Biol Chem 287:17589–17597. https://doi.org/10.1074/jbc.M111.330167
Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK (2011) ER tubules mark sites of mitochondrial division. Science 334:358–362. https://doi.org/10.1126/science.1207385
Schon EA, Area-Gomez E (2013) Mitochondria-associated ER membranes in Alzheimer disease. Mol Cell Neurosci 55:26–36. https://doi.org/10.1016/j.mcn.2012.07.011
Bockler S, Westermann B (2014) ER-mitochondria contacts as sites of mitophagosome formation. Autophagy 10:1346–1347. https://doi.org/10.4161/auto.28981
Mironov SL, Symonchuk N (2006) ER vesicles and mitochondria move and communicate at synapses. J Cell Sci 119:4926–4934. https://doi.org/10.1242/jcs.03254
Rangaraju V, Calloway N, Ryan TA (2014) Activity-driven local ATP synthesis is required for synaptic function. Cell 156:825–835. https://doi.org/10.1016/j.cell.2013.12.042
Achleitner G, Gaigg B, Krasser A, Kainersdorfer E, Kohlwein SD, Perktold A, Zellnig G, Daum G (1999) Association between the endoplasmic reticulum and mitochondria of yeast facilitates interorganelle transport of phospholipids through membrane contact. Eur J Biochem 264:545–553. https://doi.org/10.1046/j.1432-1327.1999.00658.x
Csordas G, Renken C, Varnai P, Walter L, Weaver D, Buttle KF, Balla T, Mannella CA, Hajnoczky G (2006) Structural and functional features and significance of the physical linkage between ER and mitochondria. J Cell Biol 174:915–921. https://doi.org/10.1083/jcb.200604016
Kornmann B, Currie E, Collins SR, Schuldiner M, Nunnari J, Weissman JS, Walter P (2009) An ER-mitochondria tethering complex revealed by a synthetic biology screen. Science 325:477–481. https://doi.org/10.1126/science.1175088
Herrera-Cruz MS, Simmen T (2017) Over six decades of discovery and characterization of the architecture at mitochondria-associated membranes (MAMs). Adv Exp Med Biol 997:13–31. https://doi.org/10.1007/978-981-10-4567-7_2
Hirabayashi Y, Kwon SK, Paek H, Pernice WM, Paul MA, Lee J, Erfani P, Raczkowski A, Petrey DS, Pon LA et al (2017) ER-mitochondria tethering by PDZD8 regulates Ca(2+) dynamics in mammalian neurons. Science 358:623–630. https://doi.org/10.1126/science.aan6009
de Brito OM, Scorrano L (2008) Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456:605–610. https://doi.org/10.1038/nature07534
Cosson P, Marchetti A, Ravazzola M, Orci L (2012) Mitofusin-2 independent juxtaposition of endoplasmic reticulum and mitochondria: an ultrastructural study. PLoS ONE 7:e46293. https://doi.org/10.1371/journal.pone.0046293
Leal NS, Schreiner B, Pinho CM, Filadi R, Wiehager B, Karlstrom H, Pizzo P, Ankarcrona M (2016) Mitofusin-2 knockdown increases ER-mitochondria contact and decreases amyloid beta-peptide production. J Cell Mol Med 20:1686–1695. https://doi.org/10.1111/jcmm.12863
Galmes R, Houcine A, van Vliet AR, Agostinis P, Jackson CL, Giordano F (2016) ORP5/ORP8 localize to endoplasmic reticulum-mitochondria contacts and are involved in mitochondrial function. EMBO Rep 17:800–810. https://doi.org/10.15252/embr.201541108
Hung V, Lam SS, Udeshi ND, Svinkina T, Guzman G, Mootha VK, Carr SA, Ting AY (2017) Proteomic map** of cytosol-facing outer mitochondrial and ER membranes in living human cells by proximity biotinylation. Elife. https://doi.org/10.7554/eLife.24463
De Vos KJ, Morotz GM, Stoica R, Tudor EL, Lau KF, Ackerley S, Warley A, Shaw CE, Miller CC (2012) VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis. Hum Mol Genet 21:1299–1311. https://doi.org/10.1093/hmg/ddr559
Stoica R, De Vos KJ, Paillusson S, Mueller S, Sancho RM, Lau KF, Vizcay-Barrena G, Lin WL, Xu YF, Lewis J et al (2014) ER-mitochondria associations are regulated by the VAPB-PTPIP51 interaction and are disrupted by ALS/FTD-associated TDP-43. Nat Commun 5:3996. https://doi.org/10.1038/ncomms4996
Giorgi C, Missiroli S, Patergnani S, Duszynski J, Wieckowski MR, Pinton P (2015) Mitochondria-associated membranes: composition, molecular mechanisms, and physiopathological implications. Antioxid Redox Signal 22:995–1019. https://doi.org/10.1089/ars.2014.6223
Rodriguez-Arribas M, Yakhine-Diop SMS, Pedro JMB, Gomez-Suaga P, Gomez-Sanchez R, Martinez-Chacon G, Fuentes JM, Gonzalez-Polo RA, Niso-Santano M (2017) Mitochondria-associated membranes (MAMs): overview and its role in Parkinson’s disease. Mol Neurobiol 54:6287–6303. https://doi.org/10.1007/s12035-016-0140-8
Cardenas C, Miller RA, Smith I, Bui T, Molgo J, Muller M, Vais H, Cheung KH, Yang J, Parker I et al (2010) Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 142:270–283. https://doi.org/10.1016/j.cell.2010.06.007
Gomez-Suaga P, Paillusson S, Stoica R, Noble W, Hanger DP, Miller CCJ (2017) The ER-mitochondria tethering complex VAPB-PTPIP51 regulates autophagy. Curr Biol 27:371–385. https://doi.org/10.1016/j.cub.2016.12.038
van Vliet AR, Verfaillie T, Agostinis P (2014) New functions of mitochondria associated membranes in cellular signaling. Biochim Biophys Acta 1843:2253–2262. https://doi.org/10.1016/j.bbamcr.2014.03.009
Vance JE (2014) MAM (mitochondria-associated membranes) in mammalian cells: lipids and beyond. Biochim Biophys Acta 1841:595–609. https://doi.org/10.1016/j.bbalip.2013.11.014
Acehan D, Malhotra A, Xu Y, Ren M, Stokes DL, Schlame M (2011) Cardiolipin affects the supramolecular organization of ATP synthase in mitochondria. Biophys J 100:2184–2192. https://doi.org/10.1016/j.bpj.2011.03.031
Gohil VM, Hayes P, Matsuyama S, Schagger H, Schlame M, Greenberg ML (2004) Cardiolipin biosynthesis and mitochondrial respiratory chain function are interdependent. J Biol Chem 279:42612–42618. https://doi.org/10.1074/jbc.M402545200
Pfeiffer K, Gohil V, Stuart RA, Hunte C, Brandt U, Greenberg ML, Schagger H (2003) Cardiolipin stabilizes respiratory chain supercomplexes. J Biol Chem 278:52873–52880. https://doi.org/10.1074/jbc.M308366200
Schlame M, Rua D, Greenberg ML (2000) The biosynthesis and functional role of cardiolipin. Prog Lipid Res 39:257–288. https://doi.org/10.1016/s0163-7827(00)00005-9
Paillusson S, Stoica R, Gomez-Suaga P, Lau DHW, Mueller S, Miller T, Miller CCJ (2016) There’s something wrong with my MAM; the ER-mitochondria Axis and neurodegenerative diseases. Trends Neurosci 39:146–157. https://doi.org/10.1016/j.tins.2016.01.008
Area-Gomez E, de Groof AJ, Boldogh I, Bird TD, Gibson GE, Koehler CM, Yu WH, Duff KE, Yaffe MP, Pon LA et al (2009) Presenilins are enriched in endoplasmic reticulum membranes associated with mitochondria. Am J Pathol 175:1810–1816. https://doi.org/10.2353/ajpath.2009.090219
Marchi S, Patergnani S, Missiroli S, Morciano G, Rimessi A, Wieckowski MR, Giorgi C, Pinton P (2018) Mitochondrial and endoplasmic reticulum calcium homeostasis and cell death. Cell Calcium 69:62–72. https://doi.org/10.1016/j.ceca.2017.05.003
Pera M, Larrea D, Guardia-Laguarta C, Montesinos J, Velasco KR, Agrawal RR, Xu Y, Chan RB, Di Paolo G, Mehler MF et al (2017) Increased localization of APP-C99 in mitochondria-associated ER membranes causes mitochondrial dysfunction in Alzheimer disease. EMBO J 36:3356–3371. https://doi.org/10.15252/embj.201796797
Kogot-Levin A, Saada A (2014) Ceramide and the mitochondrial respiratory chain. Biochimie 100:88–94. https://doi.org/10.1016/j.biochi.2013.07.027
Yu J, Novgorodov SA, Chudakova D, Zhu H, Bielawska A, Bielawski J, Obeid LM, Kindy MS, Gudz TI (2007) JNK3 signaling pathway activates ceramide synthase leading to mitochondrial dysfunction. J Biol Chem 282:25940–25949. https://doi.org/10.1074/jbc.M701812200
Gautier CA, Erpapazoglou Z, Mouton-Liger F, Muriel MP, Cormier F, Bigou S, Duffaure S, Girard M, Foret B, Iannielli A et al (2016) The endoplasmic reticulum-mitochondria interface is perturbed in PARK2 knockout mice and patients with PARK2 mutations. Hum Mol Genet 25:2972–2984. https://doi.org/10.1093/hmg/ddw148
Gelmetti V, De Rosa P, Torosantucci L, Marini ES, Romagnoli A, Di Rienzo M, Arena G, Vignone D, Fimia GM, Valente EM (2017) PINK1 and BECN1 relocalize at mitochondria-associated membranes during mitophagy and promote ER-mitochondria tethering and autophagosome formation. Autophagy 13:654–669. https://doi.org/10.1080/15548627.2016.1277309
Paillusson S, Gomez-Suaga P, Stoica R, Little D, Gissen P, Devine MJ, Noble W, Hanger DP, Miller CCJ (2017) alpha-Synuclein binds to the ER-mitochondria tethering protein VAPB to disrupt Ca(2+) homeostasis and mitochondrial ATP production. Acta Neuropathol 134:129–149. https://doi.org/10.1007/s00401-017-1704-z
Guardia-Laguarta C, Area-Gomez E, Rub C, Liu Y, Magrane J, Becker D, Voos W, Schon EA, Przedborski S (2014) alpha-Synuclein is localized to mitochondria-associated ER membranes. J Neurosci Off J Soc Neurosci 34:249–259. https://doi.org/10.1523/JNEUROSCI.2507-13.2014
Bonifati V (2012) Autosomal recessive parkinsonism. Parkinsonism Relat Disord 18(Suppl 1):S4-6. https://doi.org/10.1016/S1353-8020(11)70004-9
Ottolini D, Cali T, Negro A, Brini M (2013) The Parkinson disease-related protein DJ-1 counteracts mitochondrial impairment induced by the tumour suppressor protein p53 by enhancing endoplasmic reticulum-mitochondria tethering. Hum Mol Genet 22:2152–2168. https://doi.org/10.1093/hmg/ddt068
Liu Y, Ma X, Fujioka H, Liu J, Chen S, Zhu X (2019) DJ-1 regulates the integrity and function of ER-mitochondria association through interaction with IP3R3-Grp75-VDAC1. Proc Natl Acad Sci USA 116:25322–25328. https://doi.org/10.1073/pnas.1906565116
Yang JY, Yang WY (2013) Bit-by-bit autophagic removal of parkin-labelled mitochondria. Nat Commun 4:2428. https://doi.org/10.1038/ncomms3428
Khacho M, Slack RS (2018) Mitochondrial dynamics in the regulation of neurogenesis: from development to the adult brain. Dev Dyn 247:47–53. https://doi.org/10.1002/dvdy.24538
Gomes LC, Di Benedetto G, Scorrano L (2011) During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol 13:589–598. https://doi.org/10.1038/ncb2220
Gao AW, Canto C, Houtkooper RH (2014) Mitochondrial response to nutrient availability and its role in metabolic disease. EMBO Mol Med 6:580–589. https://doi.org/10.1002/emmm.201303782
Li J, Wang Y, Wang Y, Wen X, Ma XN, Chen W, Huang F, Kou J, Qi LW, Liu B et al (2015) Pharmacological activation of AMPK prevents Drp1-mediated mitochondrial fission and alleviates endoplasmic reticulum stress-associated endothelial dysfunction. J Mol Cell Cardiol 86:62–74. https://doi.org/10.1016/j.yjmcc.2015.07.010
Gao J, Wang L, Liu J, **e F, Su B, Wang X (2017) Abnormalities of mitochondrial dynamics in neurodegenerative diseases. Antioxidants (Basel). https://doi.org/10.3390/antiox6020025
Frank M, Duvezin-Caubet S, Koob S, Occhipinti A, Jagasia R, Petcherski A, Ruonala MO, Priault M, Salin B, Reichert AS (2012) Mitophagy is triggered by mild oxidative stress in a mitochondrial fission dependent manner. Biochim Biophys Acta 1823:2297–2310. https://doi.org/10.1016/j.bbamcr.2012.08.007
Hirai K, Aliev G, Nunomura A, Fujioka H, Russell RL, Atwood CS, Johnson AB, Kress Y, Vinters HV, Tabaton M et al (2001) Mitochondrial abnormalities in Alzheimer’s disease. J Neurosci Off J Soc Neurosci 21:3017–3023
Reddy PH, McWeeney S, Park BS, Manczak M, Gutala RV, Partovi D, Jung Y, Yau V, Searles R, Mori M et al (2004) Gene expression profiles of transcripts in amyloid precursor protein transgenic mice: up-regulation of mitochondrial metabolism and apoptotic genes is an early cellular change in Alzheimer’s disease. Hum Mol Genet 13:1225–1240. https://doi.org/10.1093/hmg/ddh140
Diana A, Simic G, Sinforiani E, Orru N, Pichiri G, Bono G (2008) Mitochondria morphology and DNA content upon sublethal exposure to beta-amyloid(1–42) peptide. Coll Antropol 32(Suppl 1):51–58
Chandrasekaran K, Giordano T, Brady DR, Stoll J, Martin LJ, Rapoport SI (1994) Impairment in mitochondrial cytochrome oxidase gene expression in Alzheimer disease. Brain Res Mol Brain Res 24:336–340. https://doi.org/10.1016/0169-328x(94)90147-3
de la Monte SM, Luong T, Neely TR, Robinson D, Wands JR (2000) Mitochondrial DNA damage as a mechanism of cell loss in Alzheimer’s disease. Lab Invest 80:1323–1335. https://doi.org/10.1038/labinvest.3780140
Liang WS, Reiman EM, Valla J, Dunckley T, Beach TG, Grover A, Niedzielko TL, Schneider LE, Mastroeni D, Caselli R et al (2008) Alzheimer’s disease is associated with reduced expression of energy metabolism genes in posterior cingulate neurons. Proc Natl Acad Sci USA 105:4441–4446. https://doi.org/10.1073/pnas.0709259105
Cho DH, Nakamura T, Fang J, Cieplak P, Godzik A, Gu Z, Lipton SA (2009) S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science 324:102–105. https://doi.org/10.1126/science.1171091
Lin MK, Farrer MJ (2014) Genetics and genomics of Parkinson’s disease. Genome Med 6:48. https://doi.org/10.1186/gm566
Tanaka A (2010) Parkin-mediated selective mitochondrial autophagy, mitophagy: Parkin purges damaged organelles from the vital mitochondrial network. FEBS Lett 584:1386–1392. https://doi.org/10.1016/j.febslet.2010.02.060
Tsai PI, Lin CH, Hsieh CH, Papakyrikos AM, Kim MJ, Napolioni V, Schoor C, Couthouis J, Wu RM, Wszolek ZK et al (2018) PINK1 Phosphorylates MIC60/mitofilin to control structural plasticity of mitochondrial crista junctions. Mol Cell 69(744–756):e746. https://doi.org/10.1016/j.molcel.2018.01.026
Yao D, Gu Z, Nakamura T, Shi ZQ, Ma Y, Gaston B, Palmer LA, Rockenstein EM, Zhang Z, Masliah E et al (2004) Nitrosative stress linked to sporadic Parkinson’s disease: S-nitrosylation of parkin regulates its E3 ubiquitin ligase activity. Proc Natl Acad Sci USA 101:10810–10814. https://doi.org/10.1073/pnas.0404161101
Zhang Z, Liu L, Jiang X, Zhai S, **ng D (2016) The essential role of Drp1 and its regulation by S-nitrosylation of Parkin in dopaminergic neurodegeneration: implications for Parkinson’s disease. Antioxid Redox Signal 25:609–622. https://doi.org/10.1089/ars.2016.6634
Oh CK, Sultan A, Platzer J, Dolatabadi N, Soldner F, McClatchy DB, Diedrich JK, Yates JR 3rd, Ambasudhan R, Nakamura T et al (2017) S-Nitrosylation of PINK1 attenuates PINK1/Parkin-dependent mitophagy in hiPSC-based Parkinson’s disease models. Cell Rep 21:2171–2182. https://doi.org/10.1016/j.celrep.2017.10.068
Cui L, Jeong H, Borovecki F, Parkhurst CN, Tanese N, Krainc D (2006) Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell 127:59–69. https://doi.org/10.1016/j.cell.2006.09.015
Haun F, Nakamura T, Shiu AD, Cho DH, Tsunemi T, Holland EA, La Spada AR, Lipton SA (2013) S-nitrosylation of dynamin-related protein 1 mediates mutant huntingtin-induced mitochondrial fragmentation and neuronal injury in Huntington’s disease. Antioxid Redox Signal 19:1173–1184. https://doi.org/10.1089/ars.2012.4928
Wang H, Lim PJ, Karbowski M, Monteiro MJ (2009) Effects of overexpression of huntingtin proteins on mitochondrial integrity. Hum Mol Genet 18:737–752. https://doi.org/10.1093/hmg/ddn404
Franco-Iborra S, Plaza-Zabala A, Montpeyo M, Sebastian D, Vila M, Martinez-Vicente M (2020) Mutant HTT (huntingtin) impairs mitophagy in a cellular model of Huntington disease. . , Autophagy. https://doi.org/10.1080/15548627.2020.1728096
Nakamura T, Lipton SA (2017) ’SNO’-storms compromise protein activity and mitochondrial metabolism in neurodegenerative disorders. Trends Endocrinol Metab 28:879–892. https://doi.org/10.1016/j.tem.2017.10.004
Cenini G, Lloret A, Cascella R (2019) Oxidative stress in neurodegenerative diseases: from a mitochondrial point of view. Oxid Med Cell Longev 2019:2105607. https://doi.org/10.1155/2019/2105607
Nakamura T, Tu S, Akhtar MW, Sunico CR, Okamoto S, Lipton SA (2013) Aberrant protein s-nitrosylation in neurodegenerative diseases. Neuron 78:596–614. https://doi.org/10.1016/j.neuron.2013.05.005
Ischiropoulos H (2009) Protein tyrosine nitration—an update. Arch Biochem Biophys 484:117–121. https://doi.org/10.1016/j.abb.2008.10.034
Chouchani ET, Hurd TR, Nadtochiy SM, Brookes PS, Fearnley IM, Lilley KS, Smith RA, Murphy MP (2010) Identification of S-nitrosated mitochondrial proteins by S-nitrosothiol difference in gel electrophoresis (SNO-DIGE): implications for the regulation of mitochondrial function by reversible S-nitrosation. Biochem J 430:49–59. https://doi.org/10.1042/BJ20100633
Doulias PT, Tenopoulou M, Greene JL, Raju K, Ischiropoulos H (2013) Nitric oxide regulates mitochondrial fatty acid metabolism through reversible protein S-nitrosylation. Sci Signal 6:rs1. https://doi.org/10.1126/scisignal.2003252
Bak DW, Pizzagalli MD, Weerapana E (2017) Identifying functional cysteine residues in the mitochondria. ACS Chem Biol 12:947–957. https://doi.org/10.1021/acschembio.6b01074
Gupta KJ, Shah JK, Brotman Y, Jahnke K, Willmitzer L, Kaiser WM, Bauwe H, Igamberdiev AU (2012) Inhibition of aconitase by nitric oxide leads to induction of the alternative oxidase and to a shift of metabolism towards biosynthesis of amino acids. J Exp Bot 63:1773–1784. https://doi.org/10.1093/jxb/ers053
Nimmo GA, Nimmo HG (1984) The regulatory properties of isocitrate dehydrogenase kinase and isocitrate dehydrogenase phosphatase from Escherichia coli ML308 and the roles of these activities in the control of isocitrate dehydrogenase. Eur J Biochem 141:409–414. https://doi.org/10.1111/j.1432-1033.1984.tb08206.x
Borutaite V, Budriunaite A, Brown GC (2000) Reversal of nitric oxide-, peroxynitrite- and S-nitrosothiol-induced inhibition of mitochondrial respiration or complex I activity by light and thiols. Biochim Biophys Acta 1459:405–412. https://doi.org/10.1016/s0005-2728(00)00178-x
Clementi E, Brown GC, Feelisch M, Moncada S (1998) Persistent inhibition of cell respiration by nitric oxide: crucial role of S-nitrosylation of mitochondrial complex I and protective action of glutathione. Proc Natl Acad Sci USA 95:7631–7636. https://doi.org/10.1073/pnas.95.13.7631
Galkin A, Moncada S (2007) S-nitrosation of mitochondrial complex I depends on its structural conformation. J Biol Chem 282:37448–37453. https://doi.org/10.1074/jbc.M707543200
Zhang J, ** B, Li L, Block ER, Patel JM (2005) Nitric oxide-induced persistent inhibition and nitrosylation of active site cysteine residues of mitochondrial cytochrome-c oxidase in lung endothelial cells. Am J Physiol Cell Physiol 288:C840-849. https://doi.org/10.1152/ajpcell.00325.2004
Sun J, Morgan M, Shen RF, Steenbergen C, Murphy E (2007) Preconditioning results in S-nitrosylation of proteins involved in regulation of mitochondrial energetics and calcium transport. Circ Res 101:1155–1163. https://doi.org/10.1161/CIRCRESAHA.107.155879
Cassina A, Radi R (1996) Differential inhibitory action of nitric oxide and peroxynitrite on mitochondrial electron transport. Arch Biochem Biophys 328:309–316. https://doi.org/10.1006/abbi.1996.0178
Castro L, Demicheli V, Tortora V, Radi R (2011) Mitochondrial protein tyrosine nitration. Free Radic Res 45:37–52. https://doi.org/10.3109/10715762.2010.516254
Ryan SD, Dolatabadi N, Chan SF, Zhang X, Akhtar MW, Parker J, Soldner F, Sunico CR, Nagar S, Talantova M et al (2013) Isogenic human iPSC Parkinson’s model shows nitrosative stress-induced dysfunction in MEF2-PGC1alpha transcription. Cell 155:1351–1364. https://doi.org/10.1016/j.cell.2013.11.009
Okamoto S, Nakamura T, Cieplak P, Chan SF, Kalashnikova E, Liao L, Saleem S, Han X, Clemente A, Nutter A et al (2014) S-nitrosylation-mediated redox transcriptional switch modulates neurogenesis and neuronal cell death. Cell Rep 8:217–228. https://doi.org/10.1016/j.celrep.2014.06.005
Rossetti ZL, Sotgiu A, Sharp DE, Hadjiconstantinou M, Neff NH (1988) 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and free radicals in vitro. Biochem Pharmacol 37:4573–4574. https://doi.org/10.1016/0006-2952(88)90674-0
Sipos I, Tretter L, Adam-Vizi V (2003) Quantitative relationship between inhibition of respiratory complexes and formation of reactive oxygen species in isolated nerve terminals. J Neurochem 84:112–118. https://doi.org/10.1046/j.1471-4159.2003.01513.x
Cassarino DS, Fall CP, Swerdlow RH, Smith TS, Halvorsen EM, Miller SW, Parks JP, Parker WD Jr, Bennett JP Jr (1997) Elevated reactive oxygen species and antioxidant enzyme activities in animal and cellular models of Parkinson’s disease. Biochim Biophys Acta 1362:77–86. https://doi.org/10.1016/s0925-4439(97)00070-7
Calabrese V, Mancuso C, Calvani M, Rizzarelli E, Butterfield DA, Stella AM (2007) Nitric oxide in the central nervous system: neuroprotection versus neurotoxicity. Nat Rev Neurosci 8:766–775. https://doi.org/10.1038/nrn2214
Li MD, Burns TC, Morgan AA, Khatri P (2014) Integrated multi-cohort transcriptional meta-analysis of neurodegenerative diseases. Acta Neuropathol Commun 2:93. https://doi.org/10.1186/s40478-014-0093-y
Mattson MP (2012) Energy intake and exercise as determinants of brain health and vulnerability to injury and disease. Cell Metab 16:706–722. https://doi.org/10.1016/j.cmet.2012.08.012
Amato S, Man HY (2011) Bioenergy sensing in the brain: the role of AMP-activated protein kinase in neuronal metabolism, development and neurological diseases. Cell Cycle 10:3452–3460. https://doi.org/10.4161/cc.10.20.17953
Onyango IG, Lu J, Rodova M, Lezi E, Crafter AB, Swerdlow RH (2010) Regulation of neuron mitochondrial biogenesis and relevance to brain health. Biochim Biophys Acta 1802:228–234. https://doi.org/10.1016/j.bbadis.2009.07.014
Palacios OM, Carmona JJ, Michan S, Chen KY, Manabe Y, Ward JL 3rd, Goodyear LJ, Tong Q (2009) Diet and exercise signals regulate SIRT3 and activate AMPK and PGC-1alpha in skeletal muscle. Aging (Albany NY) 1:771–783. https://doi.org/10.18632/aging.100075
Liu L, Li Y, Wang J, Zhang D, Wu H, Li W, Wei H, Ta N, Fan Y, Liu Y et al (2021) Mitophagy receptor FUNDC1 is regulated by PGC-1alpha/NRF1 to fine tune mitochondrial homeostasis. EMBO Rep. https://doi.org/10.15252/embr.202050629
Vainshtein A, Desjardins EM, Armani A, Sandri M, Hood DA (2015) PGC-1alpha modulates denervation-induced mitophagy in skeletal muscle. Skelet Muscle 5:9. https://doi.org/10.1186/s13395-015-0033-y
Sheng B, Wang X, Su B, Lee HG, Casadesus G, Perry G, Zhu X (2012) Impaired mitochondrial biogenesis contributes to mitochondrial dysfunction in Alzheimer’s disease. J Neurochem 120:419–429. https://doi.org/10.1111/j.1471-4159.2011.07581.x
Robinson A, Grosgen S, Mett J, Zimmer VC, Haupenthal VJ, Hundsdorfer B, Stahlmann CP, Slobodskoy Y, Muller UC, Hartmann T et al (2014) Upregulation of PGC-1alpha expression by Alzheimer’s disease-associated pathway: presenilin 1/amyloid precursor protein (APP)/intracellular domain of APP. Aging Cell 13:263–272. https://doi.org/10.1111/acel.12183
Qin W, Haroutunian V, Katsel P, Cardozo CP, Ho L, Buxbaum JD, Pasinetti GM (2009) PGC-1alpha expression decreases in the Alzheimer disease brain as a function of dementia. Arch Neurol 66:352–361. https://doi.org/10.1001/archneurol.2008.588
Zheng B, Liao Z, Locascio JJ, Lesniak KA, Roderick SS, Watt ML, Eklund AC, Zhang-James Y, Kim PD, Hauser MA et al (2010) PGC-1alpha, a potential therapeutic target for early intervention in Parkinson’s disease. Sci Transl Med 2:52-a73. https://doi.org/10.1126/scitranslmed.3001059
Ciron C, Zheng L, Bobela W, Knott GW, Leone TC, Kelly DP, Schneider BL (2015) PGC-1alpha activity in nigral dopamine neurons determines vulnerability to alpha-synuclein. Acta Neuropathol Commun 3:16. https://doi.org/10.1186/s40478-015-0200-8
St-Pierre J, Drori S, Uldry M, Silvaggi JM, Rhee J, Jager S, Handschin C, Zheng K, Lin J, Yang W et al (2006) Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 127:397–408. https://doi.org/10.1016/j.cell.2006.09.024
Wareski P, Vaarmann A, Choubey V, Safiulina D, Liiv J, Kuum M, Kaasik A (2009) PGC-1{alpha} and PGC-1{beta} regulate mitochondrial density in neurons. J Biol Chem 284:21379–21385. https://doi.org/10.1074/jbc.M109.018911
Shin JH, Ko HS, Kang H, Lee Y, Lee YI, Pletinkova O, Troconso JC, Dawson VL, Dawson TM (2011) PARIS (ZNF746) repression of PGC-1alpha contributes to neurodegeneration in Parkinson’s disease. Cell 144:689–702. https://doi.org/10.1016/j.cell.2011.02.010
Leone TC, Lehman JJ, Finck BN, Schaeffer PJ, Wende AR, Boudina S, Courtois M, Wozniak DF, Sambandam N, Bernal-Mizrachi C et al (2005) PGC-1alpha deficiency causes multi-system energy metabolic derangements: muscle dysfunction, abnormal weight control and hepatic steatosis. PLoS Biol 3:e101. https://doi.org/10.1371/journal.pbio.0030101
Lin J, Wu PH, Tarr PT, Lindenberg KS, St-Pierre J, Zhang CY, Mootha VK, Jager S, Vianna CR, Reznick RM et al (2004) Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice. Cell 119:121–135. https://doi.org/10.1016/j.cell.2004.09.013
Tsunemi T, Ashe TD, Morrison BE, Soriano KR, Au J, Roque RA, Lazarowski ER, Damian VA, Masliah E, La Spada AR (2012) PGC-1alpha rescues Huntington’s disease proteotoxicity by preventing oxidative stress and promoting TFEB function. Sci Transl Med 4:142–197. https://doi.org/10.1126/scitranslmed.3003799
Chaturvedi RK, Calingasan NY, Yang L, Hennessey T, Johri A, Beal MF (2010) Impairment of PGC-1alpha expression, neuropathology and hepatic steatosis in a transgenic mouse model of Huntington’s disease following chronic energy deprivation. Hum Mol Genet 19:3190–3205. https://doi.org/10.1093/hmg/ddq229
Banerjee K, Munshi S, Frank DE, Gibson GE (2015) Abnormal glucose metabolism in Alzheimer’s disease: relation to autophagy/mitophagy and therapeutic approaches. Neurochem Res 40:2557–2569. https://doi.org/10.1007/s11064-015-1631-0
Arvanitakis Z, Wilson RS, Bienias JL, Evans DA, Bennett DA (2004) Diabetes mellitus and risk of Alzheimer disease and decline in cognitive function. Arch Neurol 61:661–666. https://doi.org/10.1001/archneur.61.5.661
Hu G, Jousilahti P, Bidel S, Antikainen R, Tuomilehto J (2007) Type 2 diabetes and the risk of Parkinson’s disease. Diabetes Care 30:842–847. https://doi.org/10.2337/dc06-2011
Santiago JA, Potashkin JA (2013) Shared dysregulated pathways lead to Parkinson’s disease and diabetes. Trends Mol Med 19:176–186. https://doi.org/10.1016/j.molmed.2013.01.002
Mattson MP (2015) Lifelong brain health is a lifelong challenge: from evolutionary principles to empirical evidence. Ageing Res Rev 20:37–45. https://doi.org/10.1016/j.arr.2014.12.011
**romerisiou G, Hadjigeorgiou GM, Papadimitriou A, Katsarogiannis E, Gourbali V, Singleton AB (2008) Association between AKT1 gene and Parkinson’s disease: a protective haplotype. Neurosci Lett 436:232–234. https://doi.org/10.1016/j.neulet.2008.03.026
Kwok JB, Hallupp M, Loy CT, Chan DK, Woo J, Mellick GD, Buchanan DD, Silburn PA, Halliday GM, Schofield PR (2005) GSK3B polymorphisms alter transcription and splicing in Parkinson’s disease. Ann Neurol 58:829–839. https://doi.org/10.1002/ana.20691
Harr SD, Simonian NA, Hyman BT (1995) Functional alterations in Alzheimer’s disease: decreased glucose transporter 3 immunoreactivity in the perforant pathway terminal zone. J Neuropathol Exp Neurol 54:38–41
Simpson IA, Vannucci SJ, Maher F (1994) Glucose transporters in mammalian brain. Biochem Soc Trans 22:671–675. https://doi.org/10.1042/bst0220671
Landau SM, Harvey D, Madison CM, Reiman EM, Foster NL, Aisen PS, Petersen RC, Shaw LM, Trojanowski JQ, Jack CR, Jr.et al, (2010) Comparing predictors of conversion and decline in mild cognitive impairment. Neurology 75:230–238. https://doi.org/10.1212/WNL.0b013e3181e8e8b8
Winkler EA, Nishida Y, Sagare AP, Rege SV, Bell RD, Perlmutter D, Sengillo JD, Hillman S, Kong P, Nelson AR et al (2015) GLUT1 reductions exacerbate Alzheimer’s disease vasculo-neuronal dysfunction and degeneration. Nat Neurosci 18:521–530. https://doi.org/10.1038/nn.3966
Ruegsegger GN, Manjunatha S, Summer P, Gopala S, Zabeilski P, Dasari S, Vanderboom PM, Lanza IR, Klaus KA, Nair KS (2019) Insulin deficiency and intranasal insulin alter brain mitochondrial function: a potential factor for dementia in diabetes. FASEB J 33:4458–4472. https://doi.org/10.1096/fj.201802043R
Palombo E, Porrino LJ, Bankiewicz KS, Crane AM, Sokoloff L, Kopin IJ (1990) Local cerebral glucose utilization in monkeys with hemiparkinsonism induced by intracarotid infusion of the neurotoxin MPTP. J Neurosci Off J Soc Neurosci 10:860–869
Schwartzman RJ, Alexander GM (1985) Changes in the local cerebral metabolic rate for glucose in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) primate model of Parkinson’s disease. Brain Res 358:137–143. https://doi.org/10.1016/0006-8993(85)90957-6
Schwartzman RJ, Alexander GM (1987) Changes in the local cerebral metabolic rate for glucose in the MPTP primate model of Parkinson’s disease. Adv Neurol 45:171–173
Malagelada C, ** ZH, Greene LA (2008) RTP801 is induced in Parkinson’s disease and mediates neuron death by inhibiting Akt phosphorylation/activation. J Neurosci: Off J Soc Neurosci 28:14363–14371. https://doi.org/10.1523/JNEUROSCI.3928-08.2008
Murata H, Sakaguchi M, ** Y, Sakaguchi Y, Futami J, Yamada H, Kataoka K, Huh NH (2011) A new cytosolic pathway from a Parkinson disease-associated kinase, BRPK/PINK1: activation of AKT via mTORC2. J Biol Chem 286:7182–7189. https://doi.org/10.1074/jbc.M110.179390
Tasaki Y, Omura T, Yamada T, Ohkubo T, Suno M, Iida S, Sakaguchi T, Asari M, Shimizu K, Matsubara K (2010) Meloxicam protects cell damage from 1-methyl-4-phenyl pyridinium toxicity via the phosphatidylinositol 3-kinase/Akt pathway in human dopaminergic neuroblastoma SH-SY5Y cells. Brain Res 1344:25–33. https://doi.org/10.1016/j.brainres.2010.04.085
Timmons S, Coakley MF, Moloney AM, C ON (2009) Akt signal transduction dysfunction in Parkinson’s disease. Neurosci Lett 467:30–35. https://doi.org/10.1016/j.neulet.2009.09.055
Greene LA, Levy O, Malagelada C (2011) Akt as a victim, villain and potential hero in Parkinson’s disease pathophysiology and treatment. Cell Mol Neurobiol 31:969–978. https://doi.org/10.1007/s10571-011-9671-8
Isaev NK, Stel’mashuk EV, Zorov DB (2007) Cellular mechanisms of brain hypoglycemia. Biochemistry (Mosc) 72:471–478. https://doi.org/10.1134/s0006297907050021
Guatteo E, Marinelli S, Geracitano R, Tozzi A, Federici M, Bernardi G, Mercuri NB (2005) Dopamine-containing neurons are silenced by energy deprivation: a defensive response or beginning of cell death? Neurotoxicology 26:857–868. https://doi.org/10.1016/j.neuro.2005.01.013
Bellucci A, Collo G, Sarnico I, Battistin L, Missale C, Spano P (2008) Alpha-synuclein aggregation and cell death triggered by energy deprivation and dopamine overload are counteracted by D2/D3 receptor activation. J Neurochem 106:560–577. https://doi.org/10.1111/j.1471-4159.2008.05406.x
Ciarmiello A, Cannella M, Lastoria S, Simonelli M, Frati L, Rubinsztein DC, Squitieri F (2006) Brain white-matter volume loss and glucose hypometabolism precede the clinical symptoms of Huntington’s disease. J Nucl Med 47:215–222
Gamberino WC, Brennan WA Jr (1994) Glucose transporter isoform expression in Huntington’s disease brain. J Neurochem 63:1392–1397. https://doi.org/10.1046/j.1471-4159.1994.63041392.x
McClory H, Williams D, Sapp E, Gatune LW, Wang P, DiFiglia M, Li X (2014) Glucose transporter 3 is a rab11-dependent trafficking cargo and its transport to the cell surface is reduced in neurons of CAG140 Huntington’s disease mice. Acta Neuropathol Commun 2:179. https://doi.org/10.1186/s40478-014-0178-7
Antonini A, Leenders KL, Spiegel R, Meier D, Vontobel P, Weigell-Weber M, Sanchez-Pernaute R, de Yebenez JG, Boesiger P, Weindl A et al (1996) Striatal glucose metabolism and dopamine D2 receptor binding in asymptomatic gene carriers and patients with Huntington’s disease. Brain 119(Pt 6):2085–2095. https://doi.org/10.1093/brain/119.6.2085
Grafton ST, Mazziotta JC, Pahl JJ, St George-Hyslop P, Haines JL, Gusella J, Hoffman JM, Baxter LR, Phelps ME (1992) Serial changes of cerebral glucose metabolism and caudate size in persons at risk for Huntington’s disease. Arch Neurol 49:1161–1167. https://doi.org/10.1001/archneur.1992.00530350075022
Mazziotta JC, Phelps ME, Pahl JJ, Huang SC, Baxter LR, Riege WH, Hoffman JM, Kuhl DE, Lanto AB, Wapenski JA et al (1987) Reduced cerebral glucose metabolism in asymptomatic subjects at risk for Huntington’s disease. N Engl J Med 316:357–362. https://doi.org/10.1056/NEJM198702123160701
Besson MT, Alegria K, Garrido-Gerter P, Barros LF, Lievens JC (2015) Enhanced neuronal glucose transporter expression reveals metabolic choice in a HD Drosophila model. PLoS ONE 10:e0118765. https://doi.org/10.1371/journal.pone.0118765
Vittori A, Breda C, Repici M, Orth M, Roos RA, Outeiro TF, Giorgini F, Hollox EJ, RiotEHsD N (2014) Copy-number variation of the neuronal glucose transporter gene SLC2A3 and age of onset in Huntington’s disease. Hum Mol Genet 23:3129–3137. https://doi.org/10.1093/hmg/ddu022
Liu YJ, Chern Y (2015) AMPK-mediated regulation of neuronal metabolism and function in brain diseases. J Neurogenet 29:50–58. https://doi.org/10.3109/01677063.2015.1067203
Ronnett GV, Ramamurthy S, Kleman AM, Landree LE, Aja S (2009) AMPK in the brain: its roles in energy balance and neuroprotection. J Neurochem 109(Suppl 1):17–23. https://doi.org/10.1111/j.1471-4159.2009.05916.x
Canto C, Auwerx J (2010) AMP-activated protein kinase and its downstream transcriptional pathways. Cell Mol Life Sci: CMLS 67:3407–3423. https://doi.org/10.1007/s00018-010-0454-z
Jager S, Handschin C, St-Pierre J, Spiegelman BM (2007) AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl Acad Sci USA 104:12017–12022. https://doi.org/10.1073/pnas.0705070104
Yu L, Yang SJ (2010) AMP-activated protein kinase mediates activity-dependent regulation of peroxisome proliferator-activated receptor gamma coactivator-1alpha and nuclear respiratory factor 1 expression in rat visual cortical neurons. Neuroscience 169:23–38. https://doi.org/10.1016/j.neuroscience.2010.04.063
Zong H, Ren JM, Young LH, Pypaert M, Mu J, Birnbaum MJ, Shulman GI (2002) AMP kinase is required for mitochondrial biogenesis in skeletal muscle in response to chronic energy deprivation. Proc Natl Acad Sci USA 99:15983–15987. https://doi.org/10.1073/pnas.252625599
Chaturvedi RK, Flint Beal M (2013) Mitochondrial diseases of the brain. Free Radic Biol Med 63:1–29. https://doi.org/10.1016/j.freeradbiomed.2013.03.018
Federico A, Cardaioli E, Da Pozzo P, Formichi P, Gallus GN, Radi E (2012) Mitochondria, oxidative stress and neurodegeneration. J Neurol Sci 322:254–262. https://doi.org/10.1016/j.jns.2012.05.030
Ju TC, Chen HM, Chen YC, Chang CP, Chang C, Chern Y (2014) AMPK-alpha1 functions downstream of oxidative stress to mediate neuronal atrophy in Huntington’s disease. Biochim Biophys Acta 1842:1668–1680. https://doi.org/10.1016/j.bbadis.2014.06.012
Mairet-Coello G, Courchet J, Pieraut S, Courchet V, Maximov A, Polleux F (2013) The CAMKK2-AMPK kinase pathway mediates the synaptotoxic effects of Abeta oligomers through Tau phosphorylation. Neuron 78:94–108. https://doi.org/10.1016/j.neuron.2013.02.003
Son SM, Jung ES, Shin HJ, Byun J, Mook-Jung I (2012) Abeta-induced formation of autophagosomes is mediated by RAGE-CaMKKbeta-AMPK signaling. Neurobiol Aging 33(1006):e1011-1023. https://doi.org/10.1016/j.neurobiolaging.2011.09.039
Du H, Guo L, Yan S, Sosunov AA, McKhann GM, Yan SS (2010) Early deficits in synaptic mitochondria in an Alzheimer’s disease mouse model. Proc Natl Acad Sci USA 107:18670–18675. https://doi.org/10.1073/pnas.1006586107
Manczak M, Anekonda TS, Henson E, Park BS, Quinn J, Reddy PH (2006) Mitochondria are a direct site of A beta accumulation in Alzheimer’s disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum Mol Genet 15:1437–1449. https://doi.org/10.1093/hmg/ddl066
Calkins MJ, Reddy PH (2011) Assessment of newly synthesized mitochondrial DNA using BrdU labeling in primary neurons from Alzheimer’s disease mice: Implications for impaired mitochondrial biogenesis and synaptic damage. Biochim Biophys Acta 1812:1182–1189. https://doi.org/10.1016/j.bbadis.2011.04.006
Thornton C, Bright NJ, Sastre M, Muckett PJ, Carling D (2011) AMP-activated protein kinase (AMPK) is a tau kinase, activated in response to amyloid beta-peptide exposure. Biochem J 434:503–512. https://doi.org/10.1042/BJ20101485
Abramov AY, Canevari L, Duchen MR (2004) Calcium signals induced by amyloid beta peptide and their consequences in neurons and astrocytes in culture. Biochim Biophys Acta 1742:81–87. https://doi.org/10.1016/j.bbamcr.2004.09.006
Shimohama S, Tanino H, Kawakami N, Okamura N, Kodama H, Yamaguchi T, Hayakawa T, Nunomura A, Chiba S, Perry G et al (2000) Activation of NADPH oxidase in Alzheimer’s disease brains. Biochem Biophys Res Commun 273:5–9. https://doi.org/10.1006/bbrc.2000.2897
Vingtdeux V, Davies P, Dickson DW, Marambaud P (2011) AMPK is abnormally activated in tangle- and pre-tangle-bearing neurons in Alzheimer’s disease and other tauopathies. Acta Neuropathol 121:337–349. https://doi.org/10.1007/s00401-010-0759-x
Ma T, Chen Y, Vingtdeux V, Zhao H, Viollet B, Marambaud P, Klann E (2014) Inhibition of AMP-activated protein kinase signaling alleviates impairments in hippocampal synaptic plasticity induced by amyloid beta. J Neurosci Off J Soc Neurosci 34:12230–12238. https://doi.org/10.1523/JNEUROSCI.1694-14.2014
Chen Y, Zhou K, Wang R, Liu Y, Kwak YD, Ma T, Thompson RC, Zhao Y, Smith L, Gasparini L et al (2009) Antidiabetic drug metformin (GlucophageR) increases biogenesis of Alzheimer’s amyloid peptides via up-regulating BACE1 transcription. Proc Natl Acad Sci USA 106:3907–3912. https://doi.org/10.1073/pnas.0807991106
DiTacchio KA, Heinemann SF, Dziewczapolski G (2015) Metformin treatment alters memory function in a mouse model of Alzheimer’s disease. J Alzheimers Dis 44:43–48. https://doi.org/10.3233/JAD-141332
Park H, Kam TI, Kim Y, Choi H, Gwon Y, Kim C, Koh JY, Jung YK (2012) Neuropathogenic role of adenylate kinase-1 in Abeta-mediated tau phosphorylation via AMPK and GSK3beta. Hum Mol Genet 21:2725–2737. https://doi.org/10.1093/hmg/dds100
Won JS, Im YB, Kim J, Singh AK, Singh I (2010) Involvement of AMP-activated-protein-kinase (AMPK) in neuronal amyloidogenesis. Biochem Biophys Res Commun 399:487–491. https://doi.org/10.1016/j.bbrc.2010.07.081
Greco SJ, Hamzelou A, Johnston JM, Smith MA, Ashford JW, Tezapsidis N (2011) Leptin boosts cellular metabolism by activating AMPK and the sirtuins to reduce tau phosphorylation and beta-amyloid in neurons. Biochem Biophys Res Commun 414:170–174. https://doi.org/10.1016/j.bbrc.2011.09.050
Greco SJ, Sarkar S, Johnston JM, Tezapsidis N (2009) Leptin regulates tau phosphorylation and amyloid through AMPK in neuronal cells. Biochem Biophys Res Commun 380:98–104. https://doi.org/10.1016/j.bbrc.2009.01.041
Chiang MC, Cheng YC, Chen SJ, Yen CH, Huang RN (2016) Metformin activation of AMPK-dependent pathways is neuroprotective in human neural stem cells against Amyloid-beta-induced mitochondrial dysfunction. Exp Cell Res 347:322–331. https://doi.org/10.1016/j.yexcr.2016.08.013
Vingtdeux V, Giliberto L, Zhao H, Chandakkar P, Wu Q, Simon JE, Janle EM, Lobo J, Ferruzzi MG, Davies P et al (2010) AMP-activated protein kinase signaling activation by resveratrol modulates amyloid-beta peptide metabolism. J Biol Chem 285:9100–9113. https://doi.org/10.1074/jbc.M109.060061
Wang ZG, Yang C, Zhu B, Hua F (2015) AMPK-dependent autophagic activation is probably involved in the mechanism of resveratrol exerting therapeutic effects for Alzheimer’s disease. Rejuvenation Res 18:101–102. https://doi.org/10.1089/rej.2014.1652
Hang L, Thundyil J, Lim KL (2015) Mitochondrial dysfunction and Parkinson disease: a Parkin-AMPK alliance in neuroprotection. Ann NY Acad Sci 1350:37–47. https://doi.org/10.1111/nyas.12820
Choi JS, Park C, Jeong JW (2010) AMP-activated protein kinase is activated in Parkinson’s disease models mediated by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Biochem Biophys Res Commun 391:147–151. https://doi.org/10.1016/j.bbrc.2009.11.022
Horvath TL, Erion DM, Elsworth JD, Roth RH, Shulman GI, Andrews ZB (2011) GPA protects the nigrostriatal dopamine system by enhancing mitochondrial function. Neurobiol Dis 43:152–162. https://doi.org/10.1016/j.nbd.2011.03.005
Kim TW, Cho HM, Choi SY, Suguira Y, Hayasaka T, Setou M, Koh HC, Hwang EM, Park JY, Kang SJ et al (2013) (ADP-ribose) polymerase 1 and AMP-activated protein kinase mediate progressive dopaminergic neuronal degeneration in a mouse model of Parkinson’s disease. Cell Death Dis 4:e919. https://doi.org/10.1038/cddis.2013.447
Chou SY, Lee YC, Chen HM, Chiang MC, Lai HL, Chang HH, Wu YC, Sun CN, Chien CL, Lin YS et al (2005) CGS21680 attenuates symptoms of Huntington’s disease in a transgenic mouse model. J Neurochem 93:310–320. https://doi.org/10.1111/j.1471-4159.2005.03029.x
Ju TC, Chen HM, Lin JT, Chang CP, Chang WC, Kang JJ, Sun CP, Tao MH, Tu PH, Chang C et al (2011) Nuclear translocation of AMPK-alpha1 potentiates striatal neurodegeneration in Huntington’s disease. J Cell Biol 194:209–227. https://doi.org/10.1083/jcb.201105010
Zheng J, Winderickx J, Franssens V, Liu B (2018) A mitochondria-associated oxidative stress perspective on Huntington’s disease. Front Mol Neurosci 11:329. https://doi.org/10.3389/fnmol.2018.00329
Choo YS, Johnson GV, MacDonald M, Detloff PJ, Lesort M (2004) Mutant huntingtin directly increases susceptibility of mitochondria to the calcium-induced permeability transition and cytochrome c release. Hum Mol Genet 13:1407–1420. https://doi.org/10.1093/hmg/ddh162
Lim D, Fedrizzi L, Tartari M, Zuccato C, Cattaneo E, Brini M, Carafoli E (2008) Calcium homeostasis and mitochondrial dysfunction in striatal neurons of Huntington disease. J Biol Chem 283:5780–5789. https://doi.org/10.1074/jbc.M704704200
Panov AV, Lund S, Greenamyre JT (2005) Ca2+-induced permeability transition in human lymphoblastoid cell mitochondria from normal and Huntington’s disease individuals. Mol Cell Biochem 269:143–152. https://doi.org/10.1007/s11010-005-3454-9
Solans A, Zambrano A, Rodriguez M, Barrientos A (2006) Cytotoxicity of a mutant huntingtin fragment in yeast involves early alterations in mitochondrial OXPHOS complexes II and III. Hum Mol Genet 15:3063–3081. https://doi.org/10.1093/hmg/ddl248
Chang DT, Rintoul GL, Pandipati S, Reynolds IJ (2006) Mutant huntingtin aggregates impair mitochondrial movement and trafficking in cortical neurons. Neurobiol Dis 22:388–400. https://doi.org/10.1016/j.nbd.2005.12.007
Subhramanyam CS, Wang C, Hu Q, Dheen ST (2019) Microglia-mediated neuroinflammation in neurodegenerative diseases. Semin Cell Dev Biol 94:112–120. https://doi.org/10.1016/j.semcdb.2019.05.004
van Horssen J, van Schaik P, Witte M (2019) Inflammation and mitochondrial dysfunction: a vicious circle in neurodegenerative disorders? Neurosci Lett 710:132931. https://doi.org/10.1016/j.neulet.2017.06.050
Hughes V (2012) Microglia: the constant gardeners. Nature 485:570–572. https://doi.org/10.1038/485570a
Stence N, Waite M, Dailey ME (2001) Dynamics of microglial activation: a confocal time-lapse analysis in hippocampal slices. Glia 33:256–266
Brown MR, Sullivan PG, Geddes JW (2006) Synaptic mitochondria are more susceptible to Ca2+overload than nonsynaptic mitochondria. J Biol Chem 281:11658–11668. https://doi.org/10.1074/jbc.M510303200
Kostuk EW, Cai J, Iacovitti L (2018) Regional microglia are transcriptionally distinct but similarly exacerbate neurodegeneration in a culture model of Parkinson’s disease. J Neuroinflammation 15:139. https://doi.org/10.1186/s12974-018-1181-x
Liemburg-Apers DC, Willems PH, Koopman WJ, Grefte S (2015) Interactions between mitochondrial reactive oxygen species and cellular glucose metabolism. Arch Toxicol 89:1209–1226. https://doi.org/10.1007/s00204-015-1520-y
Wada J (2017) Reprogramming of metabolism in immune-mediated cells. Diabetol Int 8:244–247. https://doi.org/10.1007/s13340-017-0321-3
Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N et al (2014) An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci Off J Soc Neurosci 34:11929–11947. https://doi.org/10.1523/JNEUROSCI.1860-14.2014
Ghosh S, Castillo E, Frias ES, Swanson RA (2018) Bioenergetic regulation of microglia. Glia 66:1200–1212. https://doi.org/10.1002/glia.23271
Gimeno-Bayon J, Lopez-Lopez A, Rodriguez MJ, Mahy N (2014) Glucose pathways adaptation supports acquisition of activated microglia phenotype. J Neurosci Res 92:723–731. https://doi.org/10.1002/jnr.23356
Voloboueva LA, Emery JF, Sun X, Giffard RG (2013) Inflammatory response of microglial BV-2 cells includes a glycolytic shift and is modulated by mitochondrial glucose-regulated protein 75/mortalin. FEBS Lett 587:756–762. https://doi.org/10.1016/j.febslet.2013.01.067
Zell R, Geck P, Werdan K, Boekstegers P (1997) TNF-alpha and IL-1 alpha inhibit both pyruvate dehydrogenase activity and mitochondrial function in cardiomyocytes: evidence for primary impairment of mitochondrial function. Mol Cell Biochem 177:61–67. https://doi.org/10.1023/a:1006896832582
Reynolds WF, Rhees J, Maciejewski D, Paladino T, Sieburg H, Maki RA, Masliah E (1999) Myeloperoxidase polymorphism is associated with gender specific risk for Alzheimer’s disease. Exp Neurol 155:31–41. https://doi.org/10.1006/exnr.1998.6977
Palomer X, Alvarez-Guardia D, Rodriguez-Calvo R, Coll T, Laguna JC, Davidson MM, Chan TO, Feldman AM, Vazquez-Carrera M (2009) TNF-alpha reduces PGC-1alpha expression through NF-kappaB and p38 MAPK leading to increased glucose oxidation in a human cardiac cell model. Cardiovasc Res 81:703–712. https://doi.org/10.1093/cvr/cvn327
Islinger M, Voelkl A, Fahimi HD, Schrader M (2018) The peroxisome: an update on mysteries 2.0. Histochem Cell Biol 150:443–471. https://doi.org/10.1007/s00418-018-1722-5
Ginsberg L, Rafique S, Xuereb JH, Rapoport SI, Gershfeld NL (1995) Disease and anatomic specificity of ethanolamine plasmalogen deficiency in Alzheimer’s disease brain. Brain Res 698:223–226. https://doi.org/10.1016/0006-8993(95)00931-f
Kou J, Kovacs GG, Hoftberger R, Kulik W, Brodde A, Forss-Petter S, Honigschnabl S, Gleiss A, Brugger B, Wanders R et al (2011) Peroxisomal alterations in Alzheimer’s disease. Acta Neuropathol 122:271–283. https://doi.org/10.1007/s00401-011-0836-9
Grimm MO, Grimm HS, Tomic I, Beyreuther K, Hartmann T, Bergmann C (2008) Independent inhibition of Alzheimer disease beta- and gamma-secretase cleavage by lowered cholesterol levels. J Biol Chem 283:11302–11311. https://doi.org/10.1074/jbc.M801520200
Jo DS, Park NY, Cho DH (2020) Peroxisome quality control and dysregulated lipid metabolism in neurodegenerative diseases. Exp Mol Med 52:1486–1495. https://doi.org/10.1038/s12276-020-00503-9
Fabelo N, Martin V, Santpere G, Marin R, Torrent L, Ferrer I, Diaz M (2011) Severe alterations in lipid composition of frontal cortex lipid rafts from Parkinson’s disease and incidental Parkinson’s disease. Mol Med 17:1107–1118. https://doi.org/10.2119/molmed.2011.00119
Brodde A, Teigler A, Brugger B, Lehmann WD, Wieland F, Berger J, Just WW (2012) Impaired neurotransmission in ether lipid-deficient nerve terminals. Hum Mol Genet 21:2713–2724. https://doi.org/10.1093/hmg/dds097
Fransen M, Lismont C, Walton P (2017) The peroxisome-mitochondria connection: how and why? Int J Mol Sci. https://doi.org/10.3390/ijms18061126
Lautenschager J, Kaminski Schierle GS (2019) Mitochondrial degradation of amyloidogenic proteins—a new perspective for neurodegenerative diseases. Prog Neurobiol 181:101660. https://doi.org/10.1016/j.pneurobio.2019.101660
Lu B, Guo S (2020) Mechanisms linking mitochondrial dysfunction and proteostasis failure. Trends Cell Biol 30:317–328. https://doi.org/10.1016/j.tcb.2020.01.008
Lambert JP, Luongo TS, Tomar D, Jadiya P, Gao E, Zhang X, Lucchese AM, Kolmetzky DW, Shah NS, Elrod JW (2019) MCUB regulates the molecular composition of the mitochondrial calcium uniporter channel to limit mitochondrial calcium overload during stress. Circulation 140:1720–1733. https://doi.org/10.1161/CIRCULATIONAHA.118.037968
Csordas G, Golenar T, Seifert EL, Kamer KJ, Sancak Y, Perocchi F, Moffat C, Weaver D, Perez SF, Bogorad R et al (2013) MICU1 controls both the threshold and cooperative activation of the mitochondrial Ca(2)(+) uniporter. Cell Metab 17:976–987. https://doi.org/10.1016/j.cmet.2013.04.020
Mallilankaraman K, Doonan P, Cardenas C, Chandramoorthy HC, Muller M, Miller R, Hoffman NE, Gandhirajan RK, Molgo J, Birnbaum MJ et al (2012) MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca(2+) uptake that regulates cell survival. Cell 151:630–644. https://doi.org/10.1016/j.cell.2012.10.011
Gottschalk B, Klec C, Leitinger G, Bernhart E, Rost R, Bischof H, Madreiter-Sokolowski CT, Radulovic S, Eroglu E, Sattler W et al (2019) MICU1 controls cristae junction and spatially anchors mitochondrial Ca(2+) uniporter complex. Nat Commun 10:3732. https://doi.org/10.1038/s41467-019-11692-x
Patron M, Granatiero V, Espino J, Rizzuto R, De Stefani D (2019) MICU3 is a tissue-specific enhancer of mitochondrial calcium uptake. Cell Death Differ 26:179–195. https://doi.org/10.1038/s41418-018-0113-8
Sancak Y, Markhard AL, Kitami T, Kovacs-Bogdan E, Kamer KJ, Udeshi ND, Carr SA, Chaudhuri D, Clapham DE, Li AA et al (2013) EMRE is an essential component of the mitochondrial calcium uniporter complex. Science 342:1379–1382. https://doi.org/10.1126/science.1242993
van der Laan M, Horvath SE, Pfanner N (2016) Mitochondrial contact site and cristae organizing system. Curr Opin Cell Biol 41:33–42. https://doi.org/10.1016/j.ceb.2016.03.013
Gottschalk B, Klec C, Waldeck-Weiermair M, Malli R, Graier WF (2018) Intracellular Ca(2+) release decelerates mitochondrial cristae dynamics within the junctions to the endoplasmic reticulum. Pflugers Arch 470:1193–1203. https://doi.org/10.1007/s00424-018-2133-0
Konig T, Troder SE, Bakka K, Korwitz A, Richter-Dennerlein R, Lampe PA, Patron M, Muhlmeister M, Guerrero-Castillo S, Brandt U et al (2016) The m-AAA protease associated with neurodegeneration limits MCU activity in mitochondria. Mol Cell 64:148–162. https://doi.org/10.1016/j.molcel.2016.08.020
Patron M, Sprenger HG, Langer T (2018) m-AAA proteases, mitochondrial calcium homeostasis and neurodegeneration. Cell Res 28:296–306. https://doi.org/10.1038/cr.2018.17
Acknowledgements
This work was supported by NIHR01HL136954, R01HL142271, P01HL147841 and P01HL134608 to J.W.E., NIH K99AG065445 to P.J., and NIH F32HL151146 to J.F.G.
Author information
Authors and Affiliations
Contributions
PJ, JFG wrote the manuscript and PJ designed and generated the figures. JWE conceived the review, and wrote and edited the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interests
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Jadiya, P., Garbincius, J.F. & Elrod, J.W. Reappraisal of metabolic dysfunction in neurodegeneration: Focus on mitochondrial function and calcium signaling. acta neuropathol commun 9, 124 (2021). https://doi.org/10.1186/s40478-021-01224-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40478-021-01224-4