
Abstract
Background
It is now realized that Parkinson’s disease (PD) pathology extends beyond the substantia nigra, affecting both central and peripheral nervous systems, and exhibits a variety of non-motor symptoms often preceding motor features. Neuroinflammation induced by activated microglia and astrocytes is thought to underlie these manifestations. α-Synuclein aggregation has been linked with sustained neuroinflammation in PD, aggravating neuronal degeneration; however, there is still a lack of critical information about the structural identity of the α-synuclein conformers that activate microglia and/or astrocytes and the molecular pathways involved.
Methods
To investigate the role of α-synuclein conformers in the development and maintenance of neuroinflammation, we used primary quiescent microglia and astrocytes, post-mortem brain tissues from PD patients and A53T α-synuclein transgenic mice that recapitulate key features of PD-related inflammatory responses in the absence of cell death, i.e., increased levels of pro-inflammatory cytokines and complement proteins. Biochemical and -omics techniques including RNAseq and secretomic analyses, combined with 3D reconstruction of individual astrocytes and live calcium imaging, were used to uncover the molecular mechanisms underlying glial responses in the presence of α-synuclein oligomers in vivo and in vitro.
Results
We found that the presence of SDS-resistant hyper-phosphorylated α-synuclein oligomers, but not monomers, was correlated with sustained inflammatory responses, such as elevated levels of endogenous antibodies and cytokines and microglial activation. Similar oligomeric α-synuclein species were found in post-mortem human brain samples of PD patients but not control individuals. Detailed analysis revealed a decrease in Iba1Low/CD68Low microglia and robust alterations in astrocyte number and morphology including process retraction. Our data indicated an activation of the p38/ATF2 signaling pathway mostly in microglia and a sustained induction of the NF-κB pathway in astrocytes of A53T mice. The sustained NF-κB activity triggered the upregulation of astrocytic T-type Cav3.2 Ca2+ channels, altering the astrocytic secretome and promoting the secretion of IGFBPL1, an IGF-1 binding protein with anti-inflammatory and neuroprotective potential.
Conclusions
Our work supports a causative link between the neuron-produced α-synuclein oligomers and sustained neuroinflammation in vivo and maps the signaling pathways that are stimulated in microglia and astrocytes. It also highlights the recruitment of astrocytic Cav3.2 channels as a potential neuroprotective mediator against the α-synuclein-induced neuroinflammation.
Graphical Abstract

Similar content being viewed by others


Introduction
Parkinson’s disease (PD) is a multifactorial movement disorder characterized by progressive neurodegeneration in certain brain areas and deposits of aggregated protein materials at the soma and axons of neurons, termed Lewy bodies (LBs) and Lewy neurites [1]. Despite the pivotal role of neuronal cell death in the central nervous system (CNS), PD is now recognized as a multi-system disorder characterized by notable neuroinflammation and immune dysfunction underlying many of the non-motor PD deficits such as sleep and gastrointestinal abnormalities, which in most cases, precede the onset of motor symptoms [2]. In this context, neuroinflammation and glial cell activation are not considered a response to cell death but rather a significant contributor to the pathogenic microenvironment [2, 3]. Post-mortem analysis of brain tissues of PD patients has shown widespread inflammatory manifestations as indicated by the sustained activation of CNS resident immune cells (microglia and astrocytes) and the infiltration of peripherally derived immune cells such as mononuclear phagocytes, neutrophils and lymphocytes. These features possibly initiate from intestinal dysbiosis and gut inflammation that lead to an increase in the circulating pro-inflammatory cytokines [2]. Both the innate and the adaptive peripheral systems are reported to be activated by cytokines and contribute to the generation and maintenance of immune responses in the PD brain. As a result of the extensive gliosis, the levels of pro-inflammatory cytokines are found to be elevated in PD brain and cerebrospinal fluid samples as well as in animal models of PD [4, 5].
At the cellular level, under pathological conditions in PD, damage-associated molecular patterns (DAMPs) are released from neurons and activate microglia and astrocytes through distinct molecular pathways including toll-like receptor (TLR)-mediated pathways and endocytosis. Activation of microglia dysregulates their phagocytic activity and induces activation of the inflammasome and complement proteins, secretion of pro-inflammatory cytokines and ROS production. The microglia-released cytokines induce a neurotoxic phenotype in astrocytes, which stop providing neuronal support, secrete neurotoxins and further amplify neuroinflammation. However, the exact trigger of glial reactivity and its contribution to neuronal loss and disease progression in the PD brain remain unclear.
Being the major constituent of LBs, aggregated α-synuclein has been extensively studied as a causative factor in PD etiology and pathogenesis [6]. Under normal conditions, α-synuclein is predominantly localized at the presynaptic nerve terminals where it possibly regulates synaptic plasticity, membrane remodeling and neurotransmitter release [7, 8]. Apart from acting in the cytoplasm of cells, neuron-derived α-synuclein species are secreted into the extracellular space where they can be taken up by nearby neurons, thereby promoting disease propagation along interconnected neuronal networks [9]. In addition, extracellular α-synuclein can trigger the NOD-, LRR- and pyrin domain-containing 3 (NLRP3) inflammasome signaling in microglia in a conformation-specific manner [10]. As a result of this activation, the phagocytic capacity of microglia is increased, ultimately leading to α-synuclein degradation through an autophagy-like process that requires the receptors TLR2 and p62 [11, 12]. Studies using pre-formed fibrils (PFFs) have confirmed that PFF treatment induces microglial activation towards the M1 phenotype and triggers the release of pro-inflammatory cytokines [13]. Such microglial activation is thought to subsequently induce a neurotoxic astrocytic phenotype, the A1 neurotoxic reactive astrocytes, further promoting neuroinflammation [14]. In the same line, exogenously added PFFs can be internalized by human or rodent astrocytes, promoting neurotoxic astrocyte activation [15]. Although the resulting neuroinflammation can initially act as a dynamic mechanism to eliminate the potential toxic insult, the sustained inflammation can gradually induce glial-dependent synaptic loss and neuronal death. However, the exact cell-produced α-synuclein conformers that contribute to glial activation in vivo and the molecular pathways they stimulate to establish an immune response are still unknown.
In the current work, we set out to address the roles of α-synuclein conformers in the development, maintenance and progression of non-motor symptoms that appear early in the disease and are mostly related to glial activation and neuroinflammation prior to neurodegeneration. We focused on the striatum, a brain area that has a pivotal role early in the disease and exhibits neuroinflammatory responses in the absence of cell death. The significance of the striatum in PD is also underlined by experimental and clinical evidence supporting a retrograde degeneration of dopaminergic neurons starting from the striatal axon terminals [16, 17]. In fact, dopaminergic denervation is greater in the striatum than in the SNpc in early disease, suggesting that the neocortex can be the area of initial neuronal dysfunction in some PD cases [18, 19]. In support of this mechanism, automatic and habitual performance that depends on the caudal putamen is greatly impaired in PD patients, often preceding PD diagnosis for several years [20].
To clarify the specific α-synuclein species that serve as the inflammatory signals and elucidate the molecular mechanisms through which these signals are transduced to immune and neuronal responses, we performed a series of genetic and biochemical analyses in the striatum of adult A53T α-synuclein transgenic mice (A53T Tg), a PD model with neuron-specific moderate expression of the human A53T α-synuclein variant under the control of the prion promoter [21, 22]. In these mice, the onset of α-synuclein expression starts during development, reaching a 2–3-fold increased expression in early adulthood (2–3 months of age). This pattern of increase of α-synuclein monomer allows the gradual oligomerization and accumulation of various conformers in vivo in a time-dependent manner.
We show here that these mice are characterized by sustained inflammation and intense astrocyte activation that is correlated with the presence of aberrant α-synuclein oligomeric conformers also found in post-mortem brain tissue samples from PD patients. Our data indicate that such α-synuclein oligomers, but not monomers, can activate microglia through induction of the p38MAPK/ATF2/7 pathway, which in turn results in an unconstrained activation of the NF-κB pathway in astrocytes. These cellular responses potently drive an up-regulation of astrocytic low-threshold T-type Cav3.2 voltage gated Ca2+ channels (VGCCs) leading to the Ca2+-dependent release of the neuroprotective protein, insulin growth factor (IGF) binding protein like 1 (IGFBPL1). Our work suggests that such Cav3.2 induction and the subsequent IGFBPL1 secretion from astrocytes could act as a compensatory mechanism against the damaging immune responses triggered by α-synuclein oligomers, thus highlighting the role of astrocytes as neuroprotective mediators in neurodegenerative disorders.
Materials and methods
Study design
This study was designed to determine the role of α-synuclein in neuroinflammation in mouse and human brains. We performed experiments to address: (i) the specific α-synuclein species associated with neuroinflammatory responses, (ii) the signaling pathways involved in α-synuclein-induced activation of microglia and astrocytes, and (iii) the changes in astrocyte calcium signaling and secretion upon neuroinflammation. For in vivo studies, age-matched homozygous A53T Tg mice and their wild-type (WT) littermates were randomly used for all experiments. To establish the numbers of mice required for each technique used, three basic parameters were taken into consideration, ethical limitations (homozygous A53T mice exhibit low fertility and are reproduced through crossbreeding of heterozygous A53T mice), method accuracy and reproducibility, and potential requirement for age-grou**. Animal numbers were determined by the investigators based on previous experience. Mice were humanely euthanized, and all experimental procedures were carried out in accordance with local institutional animal ethics approvals. Whenever possible, a brain tissue was used for multiple techniques. Cell culture experiments were performed using at least three biological replicates and each experimental condition was assessed in triplicate in addition to pilot optimization studies for dose and time-point determination.
Human brain samples
The use of human brain material was approved under the protocol number 46/07-01-2020 by the Bioethics Committee of Biomedical Research Foundation Academy of Athens. Post-mortem tissues from 8 PD patients and 8 non-PD control individuals (Additional file 1: Table S1) corresponding to the putamen and the caudate nucleus were obtained from the PD UK Brain Bank. Human samples were stored at − 80 °C until further use for protein and RNA extraction.
Mice
Adult homozygous A53T Tg C57BI/C3H mice (line M83-RRID: IMSR_JAX:004479) and WT littermates were used at 4–11 months of age. The generation and phenotypes of these mice have been described previously [22]. The release of α-synuclein does not show sex difference as indicated by our previous in vivo study using microdialysis to monitor α-synuclein secretion in living mice of either sex [23]. Considering this, we have used mice of mixed sex (61% and 50% females in WT and A53T Tg mice, respectively) throughout our study. The characteristics of the mouse groups are described in Additional file 1: Table S2. Animals were housed in the animal facility of the Biomedical Research Foundation of the Academy of Athens in a room with a controlled light–dark cycle (12 h light–12 h dark) with continuous access to food and water. All animal procedures were approved by the National Ethics Committee for Animal Welfare (Protocol Numbers 2143/14-05-18 and 656899/03-08-21).
Cell lines
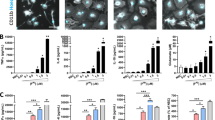
Human neuroblastoma SH-SY5Y cells were cultured in the RPMI 1640 medium and maintained at 37 °C in a humidified 5% CO2 environment. The medium was supplemented with 10% (v/v) heat-inactivated fetal bovine serum (FBS), 1% antibiotic/antimycotic (10,000 units/ml of penicillin, 10,000 µg/ml of streptomycin, and 25 µg/ml of amphotericin B) and 1% L-glutamine. Stable tet-off SH-SY5Y cells inducibly over-expressing the WT human α-synuclein were generated as previously described [3l, n and Additional file 1: Fig. S2g, i). Taken together, our data suggested that α-synuclein expression in A53T Tg mice induces astrocyte proliferation and activation and can compromise the arbor complexity of astrocytes, leading to a partial retraction of their processes.
High levels of α-synuclein motivate neuropeptide-, mitogen-activated protein (MAP) kinase (MAPK)- and Ca2+-dependent signaling pathways
Our results suggest that α-synuclein oligomers are related to consistent neuroinflammation characterized by increased cytokine release and distinct biochemical and morphological alterations in microglia and astrocytes. To dissect the molecular pathways underlying these changes, we performed RNA sequencing (RNAseq) analysis in WT and A53T Tg mice. Striatum was collected in duplicate for each mouse genotype at 6 months of age and processed for RNA extraction and sequencing using the Illumina HiSeq platform. We assessed the quality and purity of our RNAseq profiles by map** quality (> 70% of reads for all our samples were mapped). Our analysis revealed 526 DEGs in A53T Tg compared with WT mice (≥ 1.5 fold) (Additional file 2), including 304 up-regulated and 222 down-regulated DEGs. Prnp and SNCA genes were found to be up-regulated with the highest significance, as expected. Six basic clusters were identified in the heat map (Additional file 1: Fig. S3a). Enrichr-based KEGG pathway enrichment analysis (https://maayanlab.cloud/Enrichr/) highlighted the “neuroactive ligand-receptor interaction”, the “MAPK signaling”, the “calcium signaling” and the “cAMP signaling” pathways as the most affected in the presence of high α-synuclein levels (Additional file 1: Fig. S3b). Cholinergic, dopaminergic and GABAergic synaptic pathways were also significantly altered, indicating changes at the level of neuronal circuits and distorted neurotransmitter release (Additional file 1: Fig. S3b). Of the DEGs that were most significantly altered in the A53T Tg striatum (Additional file 1: Fig. S3c), Mylk3, encoding for myocin light chain kinase (MYLK), Shox2, encoding for a transcription homeobox regulator, and Pappa-2, encoding for pappalysin-2 protease implicated in IGF-1 signaling, were most up-regulated. Nine of these DEGs were selected for qPCR validation. Assessment of the Adcyap gene encoding PACAP verified its up-regulation in the A53T mice (Additional file 1: Fig. S3d). PACAP is up-regulated in neurons and astrocytes in response to inflammation and can exert potent anti-inflammatory actions that are protective for neurons [47]. Its mechanism of action is mediated by cAMP signaling, which could probably explain why this pathway was underscored in the RNAseq. Two other DEGs that belong to the “neuroactive ligand-receptor interaction” pathway, NpY and Npsr1, were also up-regulated in the A53T Tg mice, further supporting our RNAseq data (Additional file 1: Fig. S3e, f). Like PACAP, NpY signaling is suggested to inhibit microglial activation and has a neuroprotective role against neurodegeneration in PD [8h). We propose that the Cav3.2-mediated release of IGBPL1 by astrocytes could act as a compensatory mechanism to resolve the α-synuclein-induced inflammation via promoting IGF1 signaling.
α-Synuclein oligomers but not monomers trigger immune responses in the brain
The generation of aberrant oligomeric and/or fibrillar forms of α-synuclein stems from the intrinsic propensity of α-synuclein to self-aggregate and bind to membranous structures. The process of aggregation is thought to initiate from a local accumulation of monomers, possibly structurally distorted, that gradually oligomerize to generate a repertoire of β-sheet-rich fibrillar and oligomeric multimers of high molecular weight. In this sense, the selective removal of oligomeric α-synuclein in vivo, in the presence of high amounts of monomers, as happens in the case of transgenic mouse models, is technically challenging and remains a limitation of the current study. Such an idea has been put forward by El Agnaf and colleagues who administered oligomer-specific antibodies produced in-house to transgenic mice that over-express WT α-synuclein [72]. Indeed, in this study, a decrease in α-synuclein oligomers/fibrils was associated with a lower level of phosphorylated, oligomeric and total α-synuclein and alleviated both neurodegeneration and neuroinflammation.
Our results from several brain areas including the striatum, cortex, hippocampus and midbrain showed that the generation and accumulation of α-synuclein oligomers in A53T Tg mice were correlated with active immune responses in vivo. Such responses were absent from young A53T Tg mice that express high levels of monomeric but lack oligomeric α-synuclein. To further provide a causative link between α-synuclein oligomers and microglial activation, we used primary mouse microglia and a neuroblastoma cell line inducibly expressing α-synuclein, which readily secrete both oligomeric and monomeric α-synuclein. This in vitro approach verified that the cell-secreted α-synuclein oligomers, but not monomers, have the potential to activate primary quiescent microglia, stimulating the NF-κB pathway and inducing TNFα release. Even though previous studies showed that microglia can recognize and interact with exogenously supplied recombinant monomeric α-synuclein or PFFs [12, 73, 77]. Our data show that such a loss in Iba1 and CD68 proteins is also evident in the A53T model of synucleinopathy, suggesting a common theme in microglial responses in neurodegeneration.
We found that the p38 microglial activation is combined with a profound up-regulation of the NF-κB signaling pathway in astrocytes. Such reactivity has been associated with the cytokine-induced transformation of astrocytes to the A1 neurotoxic reactive phenotype as a means to amplify neuroinflammation [14] and α-synuclein propagation [11, 15]. Interventions that reverse the A1 astrocyte reactivity can be neuroprotective in PD models [73]. Due to its fundamental importance in the regulation of immune cell responses, the NF-κB activation status is tightly controlled. In contrast, we found that in A53T Tg mice, increased activity of NF-κB is maintained through downregulation of its transcriptional repressor EGR-1 and the NF-κΒ cascade terminator A20/TNFAIP3. Under these conditions, the activation of NF-κB is not resolved and further contributes to a neurotoxic environment.
Cav3.2-mediated IGFBPL1 secretion as a means to counterbalance the α-synuclein-induced neuroinflammation
Unexpectedly, we found that the NF-κB activity in astrocytes promotes the up-regulation of astrocytic T-type Cav3.2 Ca2+ channels in vitro and in vivo. Despite the documented expression of almost all the VGCC subtypes in cultured astrocytes [78], we found that at least in striatum, only the L-type Cav1.2 and the T-type Cav3 VGCCs are expressed in astrocytes in vivo. From these subtypes, the expression and function of T-type Cav3.2 Ca2+ channels are modulated by inflammatory mediators [61, 62]. The functional regulation of Cav3.2 depends on the coordinated action of specific ubiquitinases and de-ubiquitinases that regulate its proteasomal degradation. In neurons, IL-1β-mediated enhancement of the interaction between Cav3.2 and the deubiquitinase USP5 inhibits the proteasome degradation of Cav3.2, thereby promoting its accumulation in the plasma membrane, a mechanism that contributes to the maintenance of chronic pain [61, 62, 79]. Here, using quiescent primary astrocytes, we showed that when TNFα and IL-1β trigger the NF-κB pathway, Cav3.2 can accumulate in the membranes of astrocytes within minutes, indicating a rapid blockage of its proteasomal degradation. This leads to an enhancement of the Cav3.2 VGCC-mediated Ca2+ influx in astrocytes as demonstrated using the selective Cav3.2 channel blocker, NiCl2. Cav3.2 induction was dependent on NF-κB activity as confirmed by using the specific NF-κB inhibitor, BAY-11-708, that reversed the IL-1β-induced Cav3.2 up-regulation. Respectively, we can assume that Cav3.2 upregulation in A53T astrocytes could be due to the chronic NF-κB activation.
Neuronal Cav3.2 channels are established contributors to the development of seizures and neuropathic and inflammatory pain [80]. However, the role of Cav3.2 channels in astrocytes has not been studied. Our work highlights a novel function of astrocytic Cav3.2 channels to mediate the secretion of the chemokine CXCL10 and the IGF-1 binding protein IGFBPL1. Even though we did not find a significant elevation of CXCL10 in vivo at the time point of our analysis, our results showed that IGFBPL1 was highly up-regulated in the striatal astrocytes of A53T Tg mice, in which the sustained NF-κB activation results in Cav3.2 induction. IGFBPL1 has recently emerged as a molecular switch, turning inflammatory microglia to their homeostatic state to limit neuroinflammation [68]. The induction of IGFBPL1 via operation of astrocytic Cav3.2 channels reveals a novel neuroprotective mechanism through which astrocytes safeguard neuronal integrity under conditions of chronic inflammation. Since we found a significant induction of pappalysin-2 (encoded by PAPP-A2) expression, a protease that specifically cleaves the IGF/IGFBP complex to potentiate IGF signaling [81], it is tempting to speculate that pappalysin-2 and IGFBPL1 synergistically promote IGF-1 signaling to protect from α-synuclein-produced inflammatory damage. Further experimental work is required to test whether IGFBPL1 targets neurons, microglia or astrocytes in our model.
Relevance to human disease
Several genetic mouse models expressing the human WT or the mutant variant of α-synuclein have been generated, in which α-synuclein is expressed under the control of various promoters. Most of them can recapitulate, albeit to different extents, some characteristic biochemical and pathological features of PD, and have proven useful for identifying potential neuroprotective strategies for human disease [34]. The A53T Tg mice used in this study exhibit a moderate increase in α-synuclein expression, reflecting changes that can be found in human patients, i.e., when duplication or triplication of SNCA gene locus occurs. In this model, inflammation is documented by elevated levels of cytokines such as TNFα and IL-1β, which are also consistently increased in the PD brain. Even though we and others show no evidence of progressive neurodegeneration in the striatum and SNpc of A53T Tg mice, we found activation of the synapse-tagging mechanism, suggesting that defects related to synaptic maturation and function could be present in these areas as was recently shown for A53T-BAC-SNCA mice using high-resolution electron microscopy [82].
Even though our work was mainly focused on the striatum, the presence of similar α-synuclein oligomers and the profound increase of astrocytic Cav3.2 VGCCs in the cortex and SNpc relate our findings also in the context of PD pathology. Striatum is a brain region severely affected in PD, mostly due to the loss of its dopaminergic innervation caused by neurodegeneration in the SNpc and to some extent from the denervation of striatal axon terminals. Our investigation was focused on neuroinflammation that is established early in the diseased brain, and is related with the development, maintenance and progression of the non-motor symptoms that precede cell death. As such, we selected an area in which neuronal viability and synaptic integrity are maintained and a transgenic mouse model (A53T Tg) that exhibits a moderate increase in α-synuclein expression (up to three-fold). In this model, the spontaneously generated oligomeric hyperphosphorylated α-synuclein conformers gradually accumulate in the striatum and are associated with elevated C3 complement, pro-inflammatory cytokines and increased levels of endogenous IgG antibodies. Similar species were found in other brain areas in mouse brain such as the cortex, midbrain and hippocampus, and also in human PD brain tissue, where they were also associated with C3 elevation.
Our data suggest that the α-synuclein-induced neuroinflammation in A53T Tg mice is linked with an up-regulation of Cav3.2 VGCCs in astrocytes. Even though we could not directly assess astrocyte-specific Cav3.2 expression in the human tissue, we found that Cav3.2 mRNA was significantly increased in the putamen of PD patients compared to control individuals. The T-type Ca2+ channels have emerged as therapeutic targets for PD, particularly to attenuate the burst discharges in subthalamic neurons and improve the parkinsonian locomotor symptoms [55]. In fact, due to the growing evidence linking Cav3.2 channels with neurological conditions, the T-type channels are now considered one of the most highly regarded druggable targets of the past decade, with > 40 patents (since 2012) for new small organic blockers of T-type channels [83, 84]. Our study adds to the functional properties of the T-type Cav3.2 channels and underscores their involvement in the resolution of neuroinflammation in neurodegenerative disorders.
The finding that astrocytic Cav1.2 channels are expressed in astrocytes in vivo is also important based on the use of isradipine, an L-type Ca2+ channel (LTCC) blocker currently approved as a drug for the treatment of high blood pressure, as a potential therapeutic approach for PD. However, despite the encouraging findings in pre-clinical models showing alleviation of the LTCC-mediated Ca2+ load in the dopaminergic neurons, isradipine failed to confer neuroprotection in a phase III clinical trial on early PD patients [85]. Our study revealed that Cav1.2 channels are significantly decreased in the A53T Tg mouse model of synucleinopathy. In this paradigm, the use of selective LTCC agonists to restore Cav1.2 activity will allow us to investigate the involvement of this channel in astrocyte function in future studies.
Conclusions
We have defined the molecular mechanisms by which aberrant α-synuclein oligomers prolong neuroinflammation in vivo by sequentially activating specific signaling pathways in microglia and astrocytes. We have shown that these species are also present in the human PD brain. Further, we present a novel function of astrocytic T-type Cav3.2 channels to counterbalance the α-synuclein-produced inflammation by mediating the secretion of the neuroprotective protein IGFBPL1. Even though the neuroprotective potential of IGFBPL1 has to be further verified in synucleinopathy models, this protein could represent a novel molecular target against α-synuclein-induced neuroinflammation.
Targeting this molecular mechanism could provide an alternative anti-inflammatory strategy in diseases associated with unconstrained activation of the NF-κB pathway such as synucleinopathies. However, the direct targeting of NF-κB signaling for therapy is challenging due to the vast cell-type heterogeneity of the brain tissue and the wide distribution of pleiotropic NF-κB activity in all cell types. In this context, our work highlights Cav3.2 as a novel druggable molecular target to alleviate the damaging effects of microglial and astrocytic activation. Cav3.2 regulation has been extensively studied in the context of inflammatory and neuropathic pain. Considering the ubiquitous expression of Cav3.2 VGCCs and the structural similarities among T-type Ca2+ channels, the design of astrocyte-selective agonists or the indirect targeting of the ubiquitinases and de-ubiquitinases that regulate the turnover of Cav3.2 channels could represent new neuroprotective approaches for synucleinopathies.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its supplementary information files. The datasets produced during the current study can be also available from the corresponding author on reasonable request.
Abbreviations
- PD:
-
Parkinson’s disease
- GFAP:
-
Glial fibrillary acidic protein
- JNK:
-
C-Jun N-terminal kinase
- NF-κB:
-
Nuclear factor-κB
- IGFBPL1:
-
Insulin like growth factor binding protein like 1
- IGF1:
-
Insulin-like growth factor 1
- LB:
-
Lewy bodies
- CNS:
-
Central nervous system
- NLRP3:
-
NOD-, LRR- and pyrin domain-containing 3
- TLR2:
-
Toll-like receptor 2
- A53T Tg:
-
A53T α-Synuclein transgenic mice
- ATF2/7:
-
Activating transcription factor 2/7
- VGCC:
-
Voltage-gated Ca2+ channel
- WT:
-
Wild type
- Dox:
-
Doxycycline
- FGF2:
-
Fibroblast growth factor-2
- EGF:
-
Epidermal growth factor
- TNFα:
-
Tumor necrosis factor α
- IL-1β:
-
Interleukin 1β
- IFNγ:
-
Interferon γ
- PFFs:
-
Preformed fibrils
- Hprt1:
-
Hypoxanthine–guanine Phosphoribosyltransferase 1
- RPL13A:
-
60S ribosomal protein L13a
- NGS:
-
Normal goat serum
- PFA:
-
Paraformaldehyde
- Iba1:
-
Ionized calcium-binding adapter molecule 1
- CHAPS:
-
3-[(3-Cholamidopropyl) dimethylammonio]-1-propane-sulphonate
- ECL:
-
Electrogenerated chemiluminescence
- CM:
-
Conditioned medium
- FASP:
-
Filter aided sample preparation
- FDR:
-
False discovery rate
- TH:
-
Tyrosine hydroxylase
- SYB2:
-
Synaptobrevin-2
- NFL:
-
Neurofilament
- NE:
-
Norepinephrine
- DBH:
-
Dopamine β-hydroxylase
- PACAP:
-
Pituitary adenylyl cyclase-activating peptide
- VIP:
-
Vasoactive intestinal peptide
- DEG:
-
Differentially expressed gene
- MYLK:
-
Myocin light chain kinase
- IFNα:
-
Interferon-α
- LPS:
-
Lipopolysaccharides
- NAB2:
-
NGFI-A-binding protein 2
- REST:
-
RE1 silencing transcription factor
- Egr-1:
-
Early growth response 1
- IκΒα:
-
Nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha
- CXCL10:
-
C-X-C motif chemokine 10
- PAPP-A:
-
Pappalysin-1
References
Goedert M, Jakes R, Spillantini MG. The synucleinopathies: twenty years on. J Parkinsons Dis. 2017;7(s1):S51-69.
Tansey MG, Wallings RL, Houser MC, Herrick MK, Keating CE, Joers V. Inflammation and immune dysfunction in Parkinson disease. Nat Rev Immunol. 2022;22(11):657–73.
Lim S, Chun Y, Lee JS, Lee S-J. Neuroinflammation in synucleinopathies. Brain Pathol. 2016;26(3):404–9.
Weiss F, Labrador-Garrido A, Dzamko N, Halliday G. Immune responses in the Parkinson’s disease brain. Neurobiol Dis. 2022;168:105700.
Whitton PS. Inflammation as a causative factor in the aetiology of Parkinson’s disease. Br J Pharmacol. 2007;150(8):963–76.
Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M. α-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. Proc Natl Acad Sci. 1998;95(11):6469–73.
Vargas KJ, Makani S, Davis T, Westphal CH, Castillo PE, Chandra SS. Synucleins regulate the kinetics of synaptic vesicle endocytosis. J Neurosci Off J Soc Neurosci. 2014;34(28):9364–76.
Logan T, Bendor J, Toupin C, Thorn K, Edwards RH. α-Synuclein promotes dilation of the exocytotic fusion pore. Nat Neurosci. 2017;20(5):681–9.
Luk KC, Kehm V, Carroll J, Zhang B, O’Brien P, Trojanowski JQ, et al. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science. 2012;338(6109):949–53.
Scheiblich H, Bousset L, Schwartz S, Griep A, Latz E, Melki R, et al. Microglial NLRP3 inflammasome activation upon TLR2 and TLR5 ligation by distinct α-synuclein assemblies. J Immunol. 2021;207(8):2143–54.
Dutta D, Jana M, Majumder M, Mondal S, Roy A, Pahan K. Selective targeting of the TLR2/MyD88/NF-κB pathway reduces α-synuclein spreading in vitro and in vivo. Nat Commun. 2021;12(1):5382.
Choi I, Zhang Y, Seegobin SP, Pruvost M, Wang Q, Purtell K, et al. Microglia clear neuron-released α-synuclein via selective autophagy and prevent neurodegeneration. Nat Commun. 2020;11(1):1386.
Izco M, Blesa J, Verona G, Cooper JM, Alvarez-Erviti L. Glial activation precedes alpha-synuclein pathology in a mouse model of Parkinson’s disease. Neurosci Res. 2021;170:330–40.
Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541(7638):481–7.
Chou T-W, Chang NP, Krishnagiri M, Patel AP, Lindman M, Angel JP, et al. Fibrillar α-synuclein induces neurotoxic astrocyte activation via RIP kinase signaling and NF-κB. Cell Death Dis. 2021;12(8):756.
Volpicelli-Daley LA. Effects of α-synuclein on axonal transport. Neurobiol Dis. 2017;105:321–7.
Uchihara T, Giasson BI. Propagation of alpha-synuclein pathology: hypotheses, discoveries, and yet unresolved questions from experimental and human brain studies. Acta Neuropathol. 2016;131(1):49–73.
Kordower JH, Olanow CW, Dodiya HB, Chu Y, Beach TG, Adler CH, et al. Disease duration and the integrity of the nigrostriatal system in Parkinson’s disease. Brain. 2013;136(Pt 8):2419–31.
Foffani G, Obeso JA. A cortical pathogenic theory of Parkinson’s disease. Neuron. 2018;99(6):1116–28.
Darweesh SKL, Verlinden VJA, Stricker BH, Hofman A, Koudstaal PJ, Ikram MA. Trajectories of prediagnostic functioning in Parkinson’s disease. Brain. 2017;140(2):429–41.
Maskri L, Zhu X, Fritzen S, Kühn K, Ullmer C, Engels P, et al. Influence of different promoters on the expression pattern of mutated human alpha-synuclein in transgenic mice. Neurodegener Dis. 2004;1(6):255–65.
Giasson BI, Duda JE, Quinn SM, Zhang B, Trojanowski JQ, Lee VM-Y. Neuronal alpha-synucleinopathy with severe movement disorder in mice expressing A53T human alpha-synuclein. Neuron. 2002;34(4):521–33.
Emmanouilidou E, Minakaki G, Keramioti MV, Xylaki M, Balafas E, Chrysanthou-Piterou M, et al. GABA transmission via ATP-dependent K+ channels regulates α-synuclein secretion in mouse striatum. Brain. 2016;139(Pt 3):871–90.
Vekrellis K, **louri M, Emmanouilidou E, Stefanis L. Inducible over-expression of wild type alpha-synuclein in human neuronal cells leads to caspase-dependent non-apoptotic death. J Neurochem. 2009;109(5):1348–62.
Emmanouilidou E, Melachroinou K, Roumeliotis T, Garbis SD, Ntzouni M, Margaritis LH, et al. Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J Neurosci Off J Soc Neurosci. 2010;30(20):6838–51.
Prah J, Winters A, Chaudhari K, Hersh J, Liu R, Yang S-H. A novel serum free primary astrocyte culture method that mimic quiescent astrocyte phenotype. J Neurosci Methods. 2019;320:50–63.
Pantazopoulou M, Lamprokostopoulou A, Karampela DS, Alexaki A, Delis A, Coens A, et al. Differential intracellular trafficking of extracellular vesicles in microglia and astrocytes. Cell Mol Life Sci. 2023;80(7):193.
Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9(7):676–82.
Sternberg SR. Biomedical image processing. Computer (Long Beach Calif). 1983;16(1):22–34.
Wiśniewski JR, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nat Methods. 2009;6(5):359–62.
Stroggilos R, Mokou M, Latosinska A, Makridakis M, Lygirou V, Mavrogeorgis E, et al. Proteome-based classification of nonmuscle invasive bladder cancer. Int J Cancer. 2020;146(1):281–94.
Taguchi T, Ikuno M, Hondo M, Parajuli LK, Taguchi K, Ueda J, et al. α-Synuclein BAC transgenic mice exhibit RBD-like behaviour and hyposmia: a prodromal Parkinson’s disease model. Brain. 2019;143(1):249–65.
Aniszewska A, Bergström J, Ingelsson M, Ekmark-Lewén S. Modeling Parkinson’s disease-related symptoms in alpha-synuclein overexpressing mice. Brain Behav. 2022;12(7):e2628.
Chesselet M-F, Richter F. Modelling of Parkinson’s disease in mice. Lancet Neurol. 2011;10(12):1108–18.
Luchena C, Zuazo-Ibarra J, Alberdi E, Matute C, Capetillo-Zarate E. Contribution of neurons and glial cells to complement-mediated synapse removal during development, aging and in Alzheimer’s disease. Mediators Inflamm. 2018;2018:2530414.
Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, et al. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131(6):1164–78.
Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. 2016;352(6286):712–6.
Ding B, Lin C, Liu Q, He Y, Ruganzu JB, ** H, et al. Tanshinone IIA attenuates neuroinflammation via inhibiting RAGE/NF-κB signaling pathway in vivo and in vitro. J Neuroinflammation. 2020;17(1):1–17.
Lier J, Streit WJ, Bechmann I. Beyond activation: characterizing microglial functional phenotypes. Cells. 2021;10(9):2236.
Su X, Federoff HJ, Maguire-Zeiss KA. Mutant alpha-synuclein overexpression mediates early proinflammatory activity. Neurotox Res. 2009;16(3):238–54.
Gyoneva S, Traynelis SF. Norepinephrine modulates the motility of resting and activated microglia via different adrenergic receptors. J Biol Chem. 2013;288(21):15291–302.
Kim S, Park J-M, Moon J, Choi HJ. Alpha-synuclein interferes with cAMP/PKA-dependent upregulation of dopamine β-hydroxylase and is associated with abnormal adaptive responses to immobilization stress. Exp Neurol. 2014;252:63–74.
Liddelow SA, Barres BA. Reactive astrocytes: production, function, and therapeutic potential. Immunity. 2017;46(6):957–67.
Braak H, Sastre M, Del Tredici K. Development of alpha-synuclein immunoreactive astrocytes in the forebrain parallels stages of intraneuronal pathology in sporadic Parkinson’s disease. Acta Neuropathol. 2007;114(3):231–41.
Nanclares C, Poynter J, Martell-Martinez HA, Vermilyea S, Araque A, Kofuji P, et al. Dysregulation of astrocytic Ca(2+) signaling and gliotransmitter release in mouse models of α-synucleinopathies. Acta Neuropathol. 2023;145(5):597–610.
Sholl DA. Dendritic organization in the neurons of the visual and motor cortices of the cat. J Anat. 1953;87(4):387–406.
Waschek JA. VIP and PACAP: neuropeptide modulators of CNS inflammation, injury, and repair. Br J Pharmacol. 2013;169(3):512–23.
Zheng Y, Zhang L, **e J, Shi L. The emerging role of neuropeptides in Parkinson’s disease. Front Aging Neurosci. 2021;13:646726.
Figiel M, Engele J. Pituitary adenylate cyclase-activating polypeptide (PACAP), a neuron- derived peptide regulating glial glutamate transport and metabolism. J Neurosci. 2000;20(10):3596–605.
Yu T, Li YJ, Bian AH, Bin ZH, Zhu TW, Ji SX, et al. The regulatory role of activating transcription factor 2 in inflammation. Mediators Inflamm. 2014;2014:950472.
Bachstetter AD, **ng B, de Almeida L, Dimayuga ER, Watterson DM, Van Eldik LJ. Microglial p38α MAPK is a key regulator of proinflammatory cytokine up-regulation induced by toll-like receptor (TLR) ligands or beta-amyloid (Aβ). J Neuroinflammation. 2011;8:79.
Liu B, Teschemacher AG, Kasparov S. Astroglia as a cellular target for neuroprotection and treatment of neuro-psychiatric disorders. Glia. 2017;65(8):1205–26.
Chapman NR, Perkins ND. Inhibition of the RelA(p65) NF-kappaB subunit by Egr-1. J Biol Chem. 2000;275(7):4719–25.
Das T, Chen Z, Hendriks RW, Kool M. A20/tumor necrosis factor α-induced protein 3 in immune cells controls development of autoinflammation and autoimmunity: lessons from mouse models. Front Immunol. 2018;9:104.
Yang Y-C, Tai C-H, Pan M-K, Kuo C-C. The T-type calcium channel as a new therapeutic target for Parkinson’s disease. Pflugers Arch. 2014;466(4):747–55.
Harding EK, Zamponi GW. The calcium channel terminator: hasta la vista pain. Trends Pharmacol Sci. 2022;43(10):801–3.
Magistri M, Khoury N, Mazza EMC, Velmeshev D, Lee JK, Bicciato S, et al. A comparative transcriptomic analysis of astrocytes differentiation from human neural progenitor cells. Eur J Neurosci. 2016;44(10):2858–70.
Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, Giffard RG, et al. Genomic analysis of reactive astrogliosis. J Neurosci [Internet]. 2012;32(18):6391–410.
Schlessinger J. Common and distinct elements in cellular signaling via EGF and FGF receptors. Science. 2004;306(5701):1506–7.
van Loo KMJ, Schaub C, Pernhorst K, Yaari Y, Beck H, Schoch S, et al. Transcriptional regulation of T-type calcium channel CaV3.2: bi-directionality by early growth response 1 (Egr1) and repressor element 1 (RE-1) protein-silencing transcription factor (REST). J Biol Chem. 2012;287(19):15489–501.
Stemkowski PL, Garcia-Caballero A, Gadotti VM, M’Dahoma S, Chen L, Souza IA, et al. Identification of interleukin-1 beta as a key mediator in the upregulation of Cav3.2-USP5 interactions in the pain pathway. Mol Pain. 2017;13:1744806917724698.
García-Caballero A, Gadotti VM, Stemkowski P, Weiss N, Souza IA, Hodgkinson V, et al. The deubiquitinating enzyme USP5 modulates neuropathic and inflammatory pain by enhancing Cav3.2 channel activity. Neuron. 2014;83(5):1144–58.
van Weering HRJ, de Jong APH, de Haas AH, Biber KPH, Boddeke HWGM. CCL21-induced calcium transients and proliferation in primary mouse astrocytes: CXCR3-dependent and independent responses. Brain Behav Immun. 2010;24(5):768–75.
Verkhratsky A, Matteoli M, Parpura V, Mothet J-P, Zorec R. Astrocytes as secretory cells of the central nervous system: idiosyncrasies of vesicular secretion. EMBO J. 2016;35(3):239–57.
Weiss N, Zamponi GW. Control of low-threshold exocytosis by T-type calcium channels. Biochim Biophys Acta. 2013;1828(7):1579–86.
Labandeira-Garcia JL, Costa-Besada MA, Labandeira CM, Villar-Cheda B, Rodríguez-Perez AI. Insulin-like growth factor-1 and neuroinflammation. Front Aging Neurosci. 2017;9:365.
Farina C, Aloisi F, Meinl E. Astrocytes are active players in cerebral innate immunity. Trends Immunol. 2007;28(3):138–45.
Pan L, Cho KS, Wei X, Xu F, Lennikov A, Hu G, et al. IGFBPL1 is a master driver of microglia homeostasis and resolution of neuroinflammation in glaucoma and brain tauopathy. Cell Rep. 2023;42(8):112889.
Guo C, Cho K-S, Li Y, Tchedre K, Antolik C, Ma J, et al. IGFBPL1 regulates axon growth through IGF-1-mediated signaling cascades. Sci Rep. 2018;8(1):2054.
Carter SL, Müller M, Manders PM, Campbell IL. Induction of the genes for Cxcl9 and Cxcl10 is dependent on IFN-γ but shows differential cellular expression in experimental autoimmune encephalomyelitis and by astrocytes and microglia in vitro. Glia. 2007;55(16):1728–39.
Ichikawa A, Kuba K, Morita M, Chida S, Tezuka H, Hara H, et al. CXCL10-CXCR3 enhances the development of neutrophil-mediated fulminant lung injury of viral and nonviral origin. Am J Respir Crit Care Med. 2013;187(1):65–77.
El-Agnaf O, Overk C, Rockenstein E, Mante M, Florio J, Adame A, et al. Differential effects of immunotherapy with antibodies targeting α-synuclein oligomers and fibrils in a transgenic model of synucleinopathy. Neurobiol Dis. 2017;104:85–96.
Yun SP, Kam TI, Panicker N, Kim S, Oh Y, Park JS, et al. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat Med. 2018;24(7):931–8.
**a Y, Zhang G, Kou L, Yin S, Han C, Hu J, et al. Reactive microglia enhance the transmission of exosomal α-synuclein via toll-like receptor 2. Brain. 2021;144(7):2024–37.
Long H, Zhang S, Zeng S, Tong Y, Liu J, Liu C, et al. Interaction of RAGE with α-synuclein fibrils mediates inflammatory response of microglia. Cell Rep. 2022;40(12):111401.
Swanson MEV, Scotter EL, Smyth LCD, Murray HC, Ryan B, Turner C, et al. Identification of a dysfunctional microglial population in human Alzheimer’s disease cortex using novel single-cell histology image analysis. Acta Neuropathol Commun. 2020;8(1):170.
Hendrickx DAE, van Eden CG, Schuurman KG, Hamann J, Huitinga I. Staining of HLA-DR, Iba1 and CD68 in human microglia reveals partially overlap** expression depending on cellular morphology and pathology. J Neuroimmunol. 2017;309:12–22.
Latour I, Hamid J, Beedle AM, Zamponi GW, Macvicar BA. Expression of voltage-gated Ca2+ channel subtypes in cultured astrocytes. Glia. 2003;41(4):347–53.
Gadotti VM, Caballero AG, Berger ND, Gladding CM, Chen L, Pfeifer TA, et al. Small organic molecule disruptors of Cav3.2 - USP5 interactions reverse inflammatory and neuropathic pain. Mol Pain. 2015;11:12.
Harding EK, Zamponi GW. Central and peripheral contributions of T-type calcium channels in pain. Mol Brain. 2022;15(1):39.
Allard JB, Duan C. IGF-binding proteins: why do they exist and why are there so many? Front Endocrinol (Lausanne). 2018;9:117.
Parajuli LK, Wako K, Maruo S, Kakuta S, Taguchi T, Ikuno M, et al. Developmental changes in dendritic spine morphology in the striatum and their alteration in an A53T α-synuclein transgenic mouse model of Parkinson’s disease. eNeuro. 2020. https://doi.org/10.1523/ENEURO.0072-20.2020.
Weiss N, Zamponi GW. T-type calcium channels: from molecule to therapeutic opportunities. Int J Biochem Cell Biol. 2019;108:34–9.
Nam G. T-type calcium channel blockers: a patent review (2012–2018). Expert Opin Ther Pat. 2018;28(12):883–901.
Maiti B, Perlmutter JS. A clinical trial of isradipine: What went wrong? Ann Intern Med. 2020;172(9):625–6.
Acknowledgements
We would like to acknowledge Dr. S. Pagakis and Dr. A. Delis (Biological Imaging Unit, BRFAA) for their contribution to image acquisition and analysis. We would also like to thank P. Alexakos (Laboratory Animal Facility, BRFAA) for providing the mouse pups. We also thank Dr. Yannis Dalezios and Ms. Katerina Andreatidi (School of Medicine, University of Crete) for the immunoreactions and electron microscopic analysis, and Vasilis Galanopoulos (Laboratory of Electron Microscopy, University of Crete) for the use of electron microscope. We acknowledge Dr. G. Vatsellas (Greek Genome Center, BRFAA) for library preparation and RNA sequencing analysis. We also thank Dr. E. Stratikos (Department of Chemistry, NKUA) for useful discussions.
Funding
This study was funded by a Michael J. Fox Foundation grant, Target Advancement Program 2018 and a Hellenic Foundation for Research and Innovation (HFRI) Grant (581) to EE. Partial financial support was received from Special Account for Research Grants of NKUA (18638) to EE and a PhD scholarship grant to D. A. (2022-050-0502-52576) from the Greek State Scholarships Foundation (I.K.Y.) through the action “Scholarships Programs for post-graduate studies” in the framework of the Operational Program “Human Resources Development Program, Education and Lifelong Learning” of the National Strategic Reference Framework (NSRF 2014–2020).
Author information
Authors and Affiliations
Contributions
EE, conceptualized, designed and supervised research, analyzed data, and wrote the paper. EL, IC designed and performed research experiments, analyzed data and contributed to the writing of the original manuscript. DA, CM and MS performed research experiments. VF and EA performed the RNAseq bioinformatic analysis. MM and AV performed the secretomic analysis. KV edited the original draft. All authors were involved in revising the manuscript for intellectual content. All authors read and approved the submitted manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Post-mortem human brain tissues were obtained from the PD UK Brain Bank. The use of human brain material was approved under the Protocol Number 46/07-01-2020 by the Bioethics Committee of Biomedical Research Foundation Academy of Athens. All animal procedures were approved by the National Ethics Committee for Animal Welfare (Protocol Numbers 2143/14-05-18 and 656899/03-08-21).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Supplementary Information
Additional file 1
: Supplementary Materials and Methods. Figure S1. Moderate overexpression of α-synuclein in A53T transgenic mice results in α-synuclein oligomerization and complement neuronal tagging in the absence of neuronal death. Figure S2. Morphological and biochemical alterations in A53T microglia and astrocytes in vivo. Figure S3. α-Synuclein expression motivates neuropeptide-, MAPK- and Ca2+ -dependent signaling pathways. Figure S4. PACAP activity is not induced in A53T transgenic mice. Figure S5. Investigation of the signalling pathways potentially involved in inflammatory responses in A53T transgenic mice. Figure S6. The p38, ATF-2 and NF-κB pathways are selectively activated in A53T glial cells. Figure S7. Localization of L- and T-type VGCCs in striatal neurons. Figure S8. Cav2.1, Cav2.2 and Cav1.3 VGCCs are not expressed in mouse astrocytes in vivo. Figure S9. Primary quiescent astrocytes recapitulate biochemical and functional characteristics of mature astrocytes including responsiveness to cytokines. Figure S10. Nab2 and Rest mRNA levels are not altered in A53T Tg mice. Figure S11. Neuroinflammation and astrocytic Cav3.2 upregulation is not restricted to the striatum of A53T mice. Figure S12. p38/NF-κB pathway and Cav3.2 levels are not induced in microglia and astrocytes upon PFFs treatment. Table S1. Demographic information of non-PD and PD individuals. Table S2. Characteristics of mice groups used in the study. Table S3. Primer sequences used in qPCR analysis of mouse and human tissue. Table S4. List of antibodies used in the study.
Additional file 2
: DEGs up- and down-regulated in WT and A53T Tg mice. Analysis of RNAseq data. Differentially up- and down-regulated genes in the striatum of WT and A53T Tg mice.
Additional file 3
: Proteins detected in the CM of a1H- and mock-transfected astrocytes. Detection of all the proteins that are secreted from astrocytes 48 hours following transfection. Proteins identified in 3 independent experiments are listed.
Additional file 4
: Sorting of the proteins detected in the CM of a1H- and mock-transfected astrocytes using the SignalP and SecretomeP databases. List of differentially secreted proteins from mock- and a1H-transfected astrocytes identified in three independent experiments.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Leandrou, E., Chalatsa, I., Anagnostou, D. et al. α-Synuclein oligomers potentiate neuroinflammatory NF-κB activity and induce Cav3.2 calcium signaling in astrocytes. Transl Neurodegener 13, 11 (2024). https://doi.org/10.1186/s40035-024-00401-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40035-024-00401-4