
Abstract
Background
Germline mutations in the BRCA1 and BRCA2 genes account for 20–25 % of inherited breast cancers and about 10 % of all breast cancer cases. Detection of BRCA mutation carriers can lead to therapeutic interventions such as mastectomy, oophorectomy, hormonal prevention therapy, improved screening, and targeted therapies such as PARP-inhibition. We estimate that African Americans and Hispanics are 4–5 times less likely to receive BRCA screening, despite having similar mutation frequencies as non-Jewish Caucasians, who have higher breast cancer mortality. To begin addressing this health disparity, we initiated a nationwide trial of BRCA testing of Latin American women with breast cancer. Patients were recruited through community organizations, clinics, public events, and by mail and Internet. Subjects completed the consent process and questionnaire, and provided a saliva sample by mail or in person. DNA from 120 subjects was used to sequence the entirety of BRCA1 and BRCA2 coding regions and splice sites, and validate pathogenic mutations, with a total material cost of $85/subject. Subjects ranged in age from 23 to 81 years (mean age, 51 years), 6 % had bilateral disease, 57 % were ER/PR+, 23 % HER2+, and 17 % had triple-negative disease.
Results
A total of seven different predicted deleterious mutations were identified, one newly described and the rest rare. In addition, four variants of unknown effect were found.
Conclusions
Application of this strategy on a larger scale could lead to improved cancer care of minority and underserved populations.
Similar content being viewed by others

Background
Mutations in the BRCA1 and BRCA2 genes result in predisposition to breast and ovarian cancers [1, 2]. In addition, there is increasing evidence that BRCA mutations confer risk for cancers of the prostate, pancreas, stomach and skin [3–5]; there is also suggestive evidence for their involvement in esophageal and gastric cancers [6]. In Caucasian and Asian ethnicities, BRCA mutations are associated with basal-type and/or –triple-negative disease (in which the estrogen, progesterone, and HER2 receptors are absent from the tumor); however, the nature of this relationship in other ethnicities is understudied [7–10].
The identification of BRCA1/2 carriers is a critical component of breast and ovarian cancer prevention as there are multiple screening, surgical, and chemoprevention strategies that can be employed. Magnetic resonance imaging (MRI) screening is effective in detecting early cancers in BRCA1/2 carriers; mastectomy and oophorectomy can reduce ovarian cancers; and estrogen inhibition can reduce both type of malignancy [11]. However, relatively few minorities have participated in these prevention studies.
Although African American and Hispanic women have a lower incidence of breast cancer, they have a higher mortality. Triple-negative breast cancer is more common in African Americans and Hispanic women, accounting, in part, for this health disparity [12–16]. A study of over 45,000 women referred for BRCA1 and BRCA2 mutation screening from 2006 to 2008, found that 13–16 % of African Americans, Native Americans and Hispanics possess disease-causing mutations and a high rate of variants of unknown significance [17]. An independent study of 389 African Americans and 425 Hispanic women with incomplete gene sequencing, found that 8–10 % of high-risk subjects have a BRCA1 or BRCA2 gene mutation, and a small Puerto Rican study reported a 52 % mutation rate [18, 19]. Common recurrent mutations, including large deletions in BRCA1 and BRCA2, exist in Hispanic/Latin American communities, which accounted for 35–45 % of the mutation carriers [17–20].
The BRCA1 and BRCA2 proteins play a role in the repair of double-stranded breaks in DNA. A synthetic lethal strategy for cancer therapy has been developed using DNA damaging chemotherapy agents to cause single-stranded breaks, combined with poly-ADP ribose polymerase (PARP) inhibitors, to inhibit single-stranded DNA repair. This approach may be particularly effective in BRCA mutation carriers, as the tumor will be unable to repair the double-stranded breaks [21, 22]. Clinical trials of PARP inhibitors demonstrate partial response or stable disease in breast, ovarian and prostate cancer subjects [23–27].
Mutations in BRCA1 and BRCA2 have been detected with a variety of techniques including multiple mutation scanning methods and Sanger sequencing (reviewed in [28]). Next-generation sequencing (NGS) has the benefit of high-throughput, automated sequence analysis, and single strand reads. DNA capture, droplet PCR and multiplex PCR methods of template preparation, and sequencing on 454, Illumina and Ion Torrent platforms have all been employed [29–32]. A clinical diagnostic laboratory validation of the Ion Torrent platform demonstrated an absence of false negatives and a 10 % false positive rate [33]. With the availability of a three-tube multiplex for the complete BRCA1 and BRCA2 genes we sought to apply this approach to a cohort of Hispanic/Latin American breast cancer patients.
Data description
The data involve variants in the coding and flanking intron sequences of the human BRCA1 and BRCA2 genes in Hispanic subjects with breast cancer. The sequence was identified through amplification, library preparation and semi-conductor sequencing on an Ion Torrent Personal Genome Machine (PGM) Sequencer (Thermo Fisher Scientific) to an average (for all samples and amplicons of 293-307X) coverage in runs with 92 samples/chip and 466X with 46 samples/chip. Excluding samples giving fewer than 20,000 total reads, 1 amplicon of BRCA1 was below 100x average reads (beginning of exon 2, containing 32 bp of the 5’UTR and 1 splice acceptor site); and 4 amplicons of BRCA2 had <100× average, covering 261 bp of coding region and 5 splice sites. Therefore, the coverage of the coding regions is 100 % for BRCA1 and 98 % for BRCA2.
The data consist of raw sequence reads mapped to the human genome, and the resulting BAM files. These files were used to predict sequence variants using the Torrent Suite Variant Caller (TSVC) and a modified Genome Analysis Tool Kit (GATK) variant caller, optimized for PGM data. For SNPs, the two variant callers (VCs) give virtually identical results, each calling one intronic SNP the other missed (both present on manual inspection). GATK is known to be ineffective at calling indels on the Ion Torrent platform. Parameter files for TSVC are given, as well as the raw and annotated variant files. Variants were manually examined in the Integrated Genome Viewer (IGV) and selected screen shots are provided. Rare variants were annotated to be Deleterious, Probably Benign, or Benign through inspection of appropriate databases (see Methods). Deleterious variants were validated by Sanger sequencing and displayed in Mutation Surveyor (SoftGenetics). Information on variants has been deposited in the LOVD ID #0000058963 [34].
Clinical data consists of information on the pathology of the tumor extracted from pathology reports, and results of a questionnaire administered by study personnel. Data are managed in a FileMaker relational database, and information on mutation carriers was double checked for accuracy. The composite information is displayed in Table 2, as well as the age-of-onset, pathology, hormone receptor status, and family history of cancer status of those subjects with mutations.
Analyses
Study design and patient population
To estimate the participation rate of minorities in BRCA1 and BRCA2 testing we used data from Hall et al. [35], on 64,717 non-Ashkenazi women receiving testing at Myriad Genetic Laboratories between the years 2006 and 2008, in order to calculate participation. Women of Western European descent made up 78 % of the subjects receiving testing during this time period. Latin American and African American women each made up only 4 % of the samples, despite representing 16 and 13 % of the US population, respectively. This represents 18.4 European Americans screened/100,000 as compared to 3.8/100,000 for Hispanics and 4.7/100,000 for African Americans (Table 1). Therefore, Hispanic and African American women are 4–5 times less likely to receive BRCA genetic testing than Western European women (Fig. 1). Economic factors, education, concern about genetic testing, and insurance coverage are likely to play roles in this deficit.

We designed a clinical trial to address some of these issues, with recruitment of Latin American women through community organizations, clinics with large Hispanic populations, public events, and the Internet. Study materials were available in Spanish and English, and the patients were protected by a certificate of confidentiality [36]. We recruited a total of 135 subjects from 10 different states, and had an 88 % success rate in terms of completion and collection of consent forms, questionnaires, saliva samples, and pathology reports. The use of a saliva collection device that can be sent by regular mail allowed the materials cost of collection, ship**, and DNA extraction to be less than US$25.
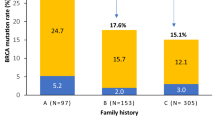
The subjects had an age range at diagnosis of 23–81 years (mean 50.6 years); 60 % had a household income below $25,000; and 81 % were educated to high school level or less (Table 2). A total of 85 % of the subjects had invasive ductal carcinoma, 6 % bilateral disease, and 57 % ER/PR+, 23 % HER2+, and 17 % triple-negative disease. A family history of either a first or second-degree relative with breast cancer was identified in 35 % of cases.
DNA sequencing of BRCA1 and BRCA2
An aliquot of each DNA sample was stripped of all identifiers in order to comply with the requirements of the protocol. A previously validated panel of primers (Ion AmpliSeg BRCA1 and BRCA2 Community Panel) was used to amplify all coding exons and splice sites (24,143 bp) with an average coverage of 313–466X and 100 % coverage (>100X average) of the BRCA1 and 98 % of the BRCA2 coding sequence. Variants were predicted using the Torrent Variant Caller; all predicted frameshift and premature stop codon alleles, and all other variants represented at less than 5 % in the 1000genomes database [37] and with quality scores greater than 40 were manually examined in IGV, and predicted deleterious variants were further confirmed by manual Sanger sequencing. A total of seven clearly deleterious alleles were identified, including a newly described allele: a single nucleotide deletion in BRCA1 (6005delT, c.5777delT). The other six alleles were unique and, except for 189del11 in BRCA1 and E1308X in BRCA2, all represent mutations uncommonly seen in these genes (Fig. 2, Table 3). Six of the seven mutations are frameshift or termination codons. The one missense variant is a compound allele C1787S and G1788D. These alleles have been reported five times in BIC [38] and have been proposed to be in cis on the same allele. NGS confirms this (Fig. 2).

Selected BRCA2 mutations. The Ion Torrent data displayed in IGV [51] is shown for the newly described 6005delT mutation in the left panel, a. The display shows individual forward (F) sequence reads in red and reverse (R) reads in blue. The 6005delT mutation can be seen as a gap in the sequence (arrow) in approximately half of the F and R reads – this is consistent with a heterozygous mutation. Sanger sequencing (not shown) confirmed this mutation. b. Both the Ion Torrent (above) and Sanger sequence (displayed in Mutation Explorer, SoftGenetics, below) for the C1787S and G1788D mutations are displayed in the right panel. The co-occurrence of the C1787S and G1788D variants on the same allele can be clearly seen in the Ion Torrent reads, whereas phase cannot be determined from the Sanger traces. Note: BRCA1 is in reverse orientation in the genome, and so IGV display is of the reverse complement

Screenshots of selected variants. a, A region with multiple homopolymers in BRCA1 is shown, sequenced by the two enzyme formulations. b. A missense variant (benign) that was not called with the standard enzyme, but was by Hi-Q
Several rare missense alleles were found, but by using data in BIC and ClinVar, all but four could be excluded as: known; likely non-pathogenic variants; or those found in a sample with an existing mutation (Table 3). Two of these variants of unknown significance (VUS) are not present in BIC (BRCA1 E577Q and BRCA2 F266L). To further interrogate the variants of unknown significance, the Align GVGD site for evolutionary conservation [39] and LOVD [34] databases were examined. One of the VUS (BRCA1 T826K) is listed as neutral in LOVD and the other three (F266L, E577Q and D1781N) are ranked as C0 (not conserved) in GVGD. It has been shown that an alignment of only primate sequences provides a potentially more appropriate model for human genetic variants [40]. Therefore, an alignment of all available primate BRCA1 and BRCA2 amino acid sequences was generated and the conservation of the residues determined. This analysis ranked all but one of the VUS (E577Q) as likely benign or benign (Table 3). E577Q is found in a patient with triple-negative disease and one second-degree relative with breast cancer.
Women with deleterious mutations were younger (43 versus 51 years of age, P = 0.029). Two of the seven patients with deleterious mutations had triple-negative disease (29 %); this is not significantly different from those patients without identified mutations. Only two of the patients with identified mutations had a family history of breast cancer, with one second-degree relative in each case. This frequency of a family history, 29 %, was not significantly different to that of the cohort as a whole.
Testing of a highly accurate sequencing enzyme
Most sequencing technologies have a higher error rate at mononucleotide stretches of DNA. We previously documented this in a comparison of Ion Torrent, Illumina and Complete Genomics NGS machines [41]. While the latest version of TVC has eliminated most apparent 1 bp deletion artifacts, we identified a substantial number of apparent erroneous coding 1 bp insertions and deletions in mononucleotide regions, especially poly-A or poly-T repeats (Table 4).
A recently developed enzyme, Ion Hi-Q Sequencing Chemistry, has been designed by Life Technologies for higher accuracy with respect to insertions and deletions (indels) and homopolymers (Ion Hi-Q Sequencing Chemistry Technology Access Program Information). We sequenced the same library of 91 DNA samples using both the standard and the Hi-Q enzyme, and analyzed the results according to the manufacturer’s instructions. With the Hi-Q enzyme the number of false positives was lower especially for 1 bp deletion alleles (Table 4, Fig. 3). The use of Hi-Q could greatly streamline variant prediction by reducing the number of variants requiring manual review.
Discussion
Rates of mortality, triple-negative disease, and BRCA1 and BRCA2 mutational and allelic diversity are all higher in African American and Hispanic populations. Although these populations could benefit significantly from genetic testing and screening, the combination of lower average income; lower insurance coverage; reduced knowledge of the benefits of testing; and mistrust of medical and government agencies, have led to a large disparity in participation [42–44]. From 64,717 women in the Myriad database, this under-participation is 3.9–4.8 times lower in these two populations, which together account for 29 % of the US population [35]. A similarly low rate of participation from the first 10,000 women tested at Myriad was noted by Forman and Hall [43].
To begin to address these issues in Hispanic populations, we designed a study with a number of potential advantages:
-
a fully de-linked sample not requiring extensive counseling regarding BRCA testing;
-
collection of saliva that can be completed in the home, community clinics or public events;
-
bilingual study materials; and
-
full protection of confidentiality.
Our recruitment success was very modest from online publicity (Facebook, clinicaltrials.gov), and few minority patients participate in public fund-raising events, such as Avon fundraising walks. However, partnerships with community groups such as Nueva Vida (Baltimore, Richmond) and the Latino Community Development Agency (Oklahoma City) were successful in recruiting a number of Hispanic women of diverse backgrounds. The remainder of the population samples were collected at the Texas Tech University Health Science Hospitals in Lubbock and El Paso, Texas. By sending and receiving saliva kits through the US mail we were able to keep the cost of the collection/ship** and DNA preparation materials to under $20 per subject. Targeted sequencing, performed in batch sizes of 90 samples, has a materials cost of approximately $50, making the total reagent cost under $100, including Sanger validation but excluding labor. This is an important factor if sample sizes in the order of thousands of subjects are to be eventually attained.
We did identify amplicons in BRCA2 that performed poorly in most samples, having <100X average coverage, comprising 261 bp of coding region and 4 splice sites, and 1 splice site of BRCA1 poorly covered. The initial run of 92 samples had three that underperformed and required repetition (3 %); however, these samples performed well on a second run. Our second run of 46 samples allowed the average coverage to increase from 313–361X to 466X. Running 46 samples on a single chip would raise the cost/sample by $5, and may be advisable for clinical testing.
The BRCA1 gene contains multiple mononucleotide repeats, for which it is challenging to accurately detect 1 bp indels; these regions are the most problematic to detect for all current methods [36, 40, 41]. Early versions of the TVC demonstrated relatively higher error rates on homopolymer sequences [40, 41]. This performance has improved with subsequent version of TVC and three studies demonstrate that 70-130X coverage across homopolymer regions is sufficient to get accurate mutation calls in the BRCA1 and BRCA2 genes (Table 5, Additional file 1) [42, 43]. Using the 70X standard of Dacheva et al., besides the poor performing exons mentioned above, there are only 2 A5 homopolymer repeats in coding regions (BRCA2, exon 12, 65X average coverage) that are below this threshold in our data. In a small sample set, we show here that the Hi-Q formulation and current TVC analysis settings result in a substantial reduction in 1 bp indel calls, especially in false positive mononucleotide regions. Manual analysis of BAM files in IGV allows most of these variants to be excluded from consideration, although the difference in background between the two enzymes visible in IGV displays are not dramatic. The addition of overlap** amplicons in the most difficult regions could improve this result. Although we chose an amplicon-based method on Ion Torrent, the samples could also be run on multiple platforms or used in a capture-based method [29]. Color Genomics (https://getcolor.com/) now offers a clinical test of 19 breast cancer related genes for $249.
While this initial phase of the project was designed to maximize participation and diversity at a modest cost, it has the disadvantage that subjects with mutations could not be retrospectively identified in order to benefit from the testing. A second phase, in which identifiers will be retained and patients counseled about testing, will now be initiated. We expect recruitment to increase as several individuals and groups declined to participate in the unlinked arm of the study. Population growth; advancement in age of the US Hispanic populations; and large family size make it imperative that innovative means be employed to increase participation in clinical and genetic studies. The increasingly recognized involvement of germline mutations in BRCA1 and BRCA2 in diverse cancers, as well as the active design of targeted therapies, further adds to the need to recruit minority subjects.
To date there have been no large nationwide surveys of Latin American with breast cancer. In the report by Hall et al., only the identities of mutations reaching 4 % or greater were made available [35]. The Clinical Cancer Genetics Community Research Network collected samples and data from 746 patients from 14 clinics concentrated in Southwestern USA, and individual data from centers in Texas, California, and Puerto Rico [46]. Our study adds to the diversity of Hispanic BRCA mutations, and – interestingly – we did not find the most common allele in all mainland US Hispanic studies to date, the BRCA1 185delAG mutation. Of the six mutations we identified, only one, BRCA2 E1308X, has been reported in multiple studies. A much larger study, incorporating all the regional and ethnic diversities of Hispanic populations, will have to be carried out to fully understand mutational diversity, and to aid in the classification of VUS.
Clearly, a comprehensive characterization of our samples will require copy number analysis to identify large insertion/deletion mutations. Recurrent large deletions in BRCA1 have been found in both Mexican and Puerto Rican breast cancer patients [19, 20]. Rare germline mutations in other genes have been identified in familial breast cancer, and our samples could be used to scan these genes [32] or complete exomes or genomes.
While we and others have documented that the major next-generation sequencers do an excellent job in identifying single nucleotide variants, they can be deficient in the prediction of insertion and deletion variants, especially in mononucleotide repeats [41]. The BRCA1 and BRCA2 genes have numerous mononucleotide repeat regions, and these areas are rich in known mutations. Thus, a method to increase accuracy of sequencing in repeats would be welcome. We documented that the Hi-Q enzyme can achieve a significantly higher accuracy in sequencing through mononucleotide repeats. When combined with methods or specific assays for the most prevalent large deletions, a high percentage of germline mutations can be identified. A recent study with the same BRCA1/2 panel was tested in a diagnostics laboratory with high accuracy [47].
In summary, we have succeeded in recruiting and anonymously testing diverse groups of Latin American women with breast cancer in the US, all at a materials cost of less than $100 for samples collection, shipment and sequencing, and confirmation. Using DNA sequencing, we found almost exclusively rare mutations, most of which have been observed in other studies. Expansion of this approach could be a component of a larger effort to improve the application of the benefits of genetic testing to Hispanic American women.
Potential implications
The benefits of BRCA1 and BRCA2 testing for women with breast and/or ovarian cancers, women with a family history and/or elevated risk, or even all women, are evident [48]. A range of options for women at risk is currently available, including increased and more effective screening, risk reduction through hormone reduction therapy, and surgical intervention. As cancers in BRCA1 and BRCA2 mutation carrier subjects occur at an earlier age, identification, education and implementation of risk reduction has a high cost-to-benefit ratio in favor of benefit. By reducing the cost of testing, simplifying sample collection, and working with organizations and clinics focusing on Hispanic communities, we address some of the barriers to utilizing this technology. Extending this approach to larger populations, employing counseling and analysis in approved clinical genetics laboratories, could contribute to reducing the higher mortality from these cancers in minority populations.
Methods
Study design and patients
Hispanic patients with breast cancer were recruited through community organizations, dedicated clinics, public events (Avon Walk) and through online contacts. The study was approved by the National Cancer Institute (NCI) Institutional Review Board (IRB) as well as the IRBs of Texas Tech Health Science Center Hospitals at Lubbock and El Paso, and included a confidentiality agreement from the National Institutes for Health (NIH) [36]. Subjects were newly diagnosed or previously treated, and gave consent in both English and Spanish, with the protocol and questionnaire available in both languages. A validated questionnaire was used to capture data on reproductive health, education, income, and family history, and a pathology report was collected to capture data on pathology and estrogen, progesterone receptor status, as well as HER2 status.
DNA collection and extraction
Saliva (~5 ml) was collected in Oragene collection devices (DNA-Genotek, Ontario, Canada), stored at room temperature, and shipped at ambient temperature through the US mail. A 0.5 ml aliquot was extracted according to manufacturer’s instructions and quantified by a NanoDrop (ND-1000) spectrophotometer (Thermo-Fisher, Wilmington, DE). Per the IRB protocol, DNA samples and selected clinical data were given new numbers unlinked to patient identifiers.
DNA sequencing
Starting with 30 ng of genomic DNA, samples were processed according to the standard protocol for Ampliseq target amplification and library preparation using the targeted, multiplex Ion AmpliSeq BRCA1 and BRCA2 Community Panel. The panel contains 167 amplicons, covers 16.3 kb and provides 98–100 % coverage of the coding regions of the BRCA1 and BRCA2 genes [49]. The libraries were prepared following the manufacturer’s Ion AmpliSeq Library Preparation protocol (Life Technologies, Carlsbad, CA, USA) and individual samples were barcoded, pooled together for the template emulsion preparation, and then sequenced on a P1 chip and Ion Torrent PGM Sequencer (Thermo Fisher Scientific). Each run produced over 10 Gb of sequence data, and each sample had an average depth of coverage surpassing 500X. Raw sequencing reads generated by the Ion Torrent sequencer were quality and adapter-trimmed by Ion Torrent Suite, then aligned to the hg19 reference sequence by TMAP [50] using default parameters (parameter file provided). Resulting BAM files were merged according to sample names and processed through an in-house quality control (QC) and coverage analysis pipeline, which generated coverage summary plots and per sample per amplicon read count heatmaps (heatmaps provided). Aligned BAM files were left-aligned using the GATK LeftAlignIndels module. Amplicon primers were trimmed from aligned reads. Variant calls and filtering was made by Torrent Variant Caller 4.0 (TSVC). Two slightly difference parameter settings were used for standard sequencing enzyme and Hi-Q enzyme. In the Hi-Q enzyme parameter set, variant recalibration was enabled. All other parameters, such as minimum coverage, minimum alternative allele frequency, and strand bias were the same between the two settings (parameter files provided). Filtered variants were annotated by the Glu Genetics annotation pipeline [51].
Sequence analysis
Predicted variants were manually reviewed in IGV [51] and manually confirmed variants examined for data in the Breast Cancer Information Core (BIC) [38], and the ClinVar database [52]. Further analysis of variants was performed using the Leiden Open Variation Database (LOVD) and ALIGN-GVGD [34, 39, 54, 55]. Selected sites for common mutations were manually examined across all samples to ensure that the false positive rate was low, and no additional variants detected. Selected mutations were repeated by Sanger sequencing and gave identical results to the PGM sequence. The newly described mutation, BRCA2 6005delT has been submitted to LOVD (Variant ID 0000058963).
Data analysis
Data was compiled in a relational database (Filemaker) and statistical analysis performed in STATA (StataCorp LP, College Station, TX, USA).
Availability of supporting data
Supporting data including BAM files of standard and Hi-Q enzyme libraries and ABI Trace files of validated mutations are available from the GigaScience GigaDB [56]. The newly described BRCA2 6005delT mutation has been submitted to LOVD (Variant ID 0000058963).
Abbreviations
- BIC:
-
Breast Information Core
- ER:
-
Estogen receptor
- PR:
-
Progesterone receptor
- GATK:
-
Genome Analysis Tool Kit
- IGV:
-
Integrated Genome Viewer
- IRB:
-
Institutional Review Board
- LOVD:
-
Leiden Open Variation Database
- MRI:
-
Magnetic resonance imaging
- NCI:
-
National Cancer Institute
- NGS:
-
Next-generation sequencing
- NIH:
-
National Institutes for Health
- PARP:
-
poly-ADP ribose polymerase
- PGM:
-
Personal Genome Machine
- TSVC:
-
Torrent Suite Variant Caller
- VC:
-
Variant caller
- VUS:
-
Variants of unknown significance
References
Lynch HT, Casey MJ, Snyder CL, Bewtra C, Lynch JF, Butts M, et al. Hereditary ovarian carcinoma: heterogeneity, molecular genetics, pathology, and management. Mol Oncol. 2009;3:97–137.
Fackenthal JD, Olopade OI. Breast cancer risk associated with BRCA1 and BRCA2 in diverse populations. Nat Rev. 2007;7:937–48.
Agalliu I, Gern R, Leanza S, Burk RD. Associations of high-grade prostate cancer with BRCA1 and BRCA2 founder mutations. Clin Cancer Res. 2009;15:1112–20.
Friedenson B. BRCA1 and BRCA2 pathways and the risk of cancers other than breast or ovarian. MedGenMed. 2005;7:60.
Lorenzo Bermejo J, Hemminki K. Risk of cancer at sites other than the breast in Swedish families eligible for BRCA1 or BRCA2 mutation testing. Ann Oncol. 2004;15:1834–41.
Moran A, O'Hara C, Khan S, Shack L, Woodward E, Maher ER, et al. Risk of cancer other than breast or ovarian in individuals with BRCA1 and BRCA2 mutations. Fam Cancer. 2012;11:235–42.
Young SR, Pilarski RT, Donenberg T, Shapiro C, Hammond LS, Miller J, et al. The prevalence of BRCA1 mutations among young women with triple-negative breast cancer. BMC Cancer. 2009;9:86.
Li WF, Hu Z, Rao NY, Song CG, Zhang B, Cao MZ, et al. The prevalence of BRCA1 and BRCA2 germline mutations in high-risk breast cancer patients of Chinese Han nationality: two recurrent mutations were identified. Breast Cancer Res Treat. 2008;110:99–109.
Turner NC, Reis-Filho JS. Basal-like breast cancer and the BRCA1 phenotype. Oncogene. 2006;25:5846–53.
Tun NM, Villani G, Ong K, Yoe L, Bo ZM. Risk of having BRCA1 mutation in high-risk women with triple-negative breast cancer: a meta-analysis. Clin Genet. 2014;85:43–8.
Couch FJ, Nathanson KL, Offit K. Two decades after BRCA: setting paradigms in personalized cancer care and prevention. Science. 2014;343:1466–70.
Parise CA, Bauer KR, Caggiano V. Variation in breast cancer subtypes with age and race/ethnicity. Crit Rev Oncol Hematol. 2010;76:44–52.
Morris GJ, Naidu S, Topham AK, Guiles F, Xu Y, McCue P, et al. Differences in breast carcinoma characteristics in newly diagnosed African-American and Caucasian patients: a single-institution compilation compared with the National Cancer Institute's Surveillance, Epidemiology, and End Results database. Cancer. 2007;110:876–84.
Stead LA, Lash TL, Sobieraj JE, Chi DD, Westrup JL, Charlot M, et al. Triple-negative breast cancers are increased in black women regardless of age or body mass index. Breast Cancer Res. 2009;11:R18.
Li CI, Malone KE, Daling JR. Differences in breast cancer stage, treatment, and survival by race and ethnicity. Arch Intern Med. 2003;163:49–56.
Lara-Medina F, Perez-Sanchez V, Saavedra-Perez D, Blake-Cerda M, Arce C, Motola-Kuba D, et al. Triple-negative breast cancer in Hispanic patients: high prevalence, poor prognosis, and association with menopausal status, body mass index, and parity. Cancer. 2011;117:3658–69.
Hall MJ, Reid JE, Burbidge LA, Pruss D, Deffenbaugh AM, Frye C, et al. BRCA1 and BRCA2 mutations in women of different ethnicities undergoing testing for hereditary breast-ovarian cancer. Cancer. 2009;115:2222–33.
Kurian AW, Gong GD, John EM, Miron A, Felberg A, Phipps AI, et al. Performance of prediction models for BRCA mutation carriage in three racial/ethnic groups: findings from the Northern California Breast Cancer Family Registry. Cancer Epidemiol Biomarkers Prev. 2009;18:1084–91.
Dutil J, Colon-Colon JL, Matta JL, Sutphen R, Echenique M. Identification of the prevalent BRCA1 and BRCA2 mutations in the female population of Puerto Rico. Cancer Genet. 2012;205:242–8.
Weitzel JN, Lagos VI, Herzog JS, Judkins T, Hendrickson B, Ho JS, et al. Evidence for common ancestral origin of a recurring BRCA1 genomic rearrangement identified in high-risk Hispanic families. Cancer Epidemiology Biomarkers Prev. 2007;16:1615–20.
Rottenberg S, Jaspers JE, Kersbergen A, van der Burg E, Nygren AO, Zander SA, et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc Natl Acad Sci U S A. 2008;105:17079–84.
Rodon J, Iniesta MD, Papadopoulos K. Development of PARP inhibitors in oncology. Expert Opin Investig Drugs. 2009;18:31–43.
Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361:123–34.
Fong PC, Yap TA, Boss DS, Carden CP, Mergui-Roelvink M, Gourley C, et al. Poly(ADP)-ribose polymerase inhibition: frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J Clin Oncol. 2010;28:2512–9.
Tutt A, Robson M, Garber JE, Domchek SM, Audeh MW, Weitzel JN, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet. 2010;376:235–44.
Audeh MW, Carmichael J, Penson RT, Friedlander M, Powell B, Bell-McGuinn KM, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet. 2010;376:245–51.
Sandhu SK, Schelman WR, Wilding G, Moreno V, Baird RD, Miranda S, et al. The poly(ADP-ribose) polymerase inhibitor niraparib (MK4827) in BRCA mutation carriers and patients with sporadic cancer: a phase 1 dose-escalation trial. Lancet Oncol. 2013;14:882–92.
Gerhardus A, Schleberger H, Schlegelberger B, Gadzicki D. Diagnostic accuracy of methods for the detection of BRCA1 and BRCA2 mutations: a systematic review. Eur J Hum Genet. 2007;15:619–27.
De Leeneer K, Hellemans J, De Schrijver J, Baetens M, Poppe B, Van Criekinge W, et al. Massive parallel amplicon sequencing of the breast cancer genes BRCA1 and BRCA2: opportunities, challenges, and limitations. Hum Mutat. 2011;32:335–44.
Morgan JE, Carr IM, Sheridan E, Chu CE, Hayward B, Camm N, et al. Genetic diagnosis of familial breast cancer using clonal sequencing. Hum Mutat. 2010;31:484–91.
Ozcelik H, Shi X, Chang MC, Tram E, Vlasschaert M, Di Nicola N, et al. Long-range PCR and next-generation sequencing of BRCA1 and BRCA2 in breast cancer. J Mol Diagn. 2012;14:467–75.
Walsh T, Casadei S, Lee MK, Pennil CC, Nord AS, Thornton AM, et al. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc Natl Acad Sci U S A. 2011;108:18032–7.
Costa JL, Sousa S, Justino A, Kay T, Fernandes S, Cirnes L, et al. Nonoptical massive parallel DNA sequencing of BRCA1 and BRCA2 genes in a diagnostic setting. Hum Mutat. 2013;34:629–35.
LOVD BRCA database. http://brca.iarc.fr/LOVD. Accessed 13 August 2015.
Hall MJ, Reid JE, Wenstrup RJ. Prevalence of BRCA1 and BRCA2 mutations in women with breast carcinoma In Situ and referred for genetic testing. Cancer Prev Res. 2010;3:1579–85.
BRCA1 and BRCA2 Mutations and Triple Negative Disease in Hispanic/Latino Breast Cancer Subjects. http://clinicaltrials.gov/show/NCT01251900. Accessed 13 August 2015.
1000 genomes website. http://www.1000genomes.org. Accessed 13 August 2015.
Breast Cancer Information Core. http://research.nhgri.nih.gov/projects/bic. Accessed 13 August 2015.
Align-GVGD website. http://agvgd.iarc.fr. Accessed 13 August 2015.
Perelman P, Johnson WE, Roos C, Seuanez HN, Horvath JE, Moreira MA, et al. A molecular phylogeny of living primates. PLoS Genet. 2011;7:e1001342.
Boland JF, Chung CC, Roberson D, Mitchell J, Zhang X, Im KM, et al. The new sequencer on the block: comparison of Life Technology's Proton sequencer to an Illumina HiSeq for whole-exome sequencing. Hum Genet. 2013;132:1153–63.
Armstrong K, Micco E, Carney A, Stopfer J, Putt M. Racial differences in the use of BRCA1/2 testing among women with a family history of breast or ovarian cancer. JAMA. 2005;293:1729–36.
Forman AD, Hall MJ. Influence of race/ethnicity on genetic counseling and testing for hereditary breast and ovarian cancer. Breast J. 2009;15 Suppl 1:S56–62.
Levy DE, Byfield SD, Comstock CB, Garber JE, Syngal S, Crown WH, et al. Underutilization of BRCA1/2 testing to guide breast cancer treatment: black and Hispanic women particularly at risk. Genet Med. 2011;13:349–55.
GetColor [https://getcolor.com]
Weitzel JN, Clague J, Martir-Negron A, Ogaz R, Herzog J, Ricker C, et al. Prevalence and type of BRCA mutations in Hispanics undergoing genetic cancer risk assessment in the southwestern United States: a report from the Clinical Cancer Genetics Community Research Network. J Clin Oncol. 2013;31:210–6.
Trujillano D, Weiss ME, Schneider J, Koster J, Papachristos EB, Saviouk V, et al. Next-Generation Sequencing of the BRCA1 and BRCA2 Genes for the Genetic Diagnostics of Hereditary Breast and/or Ovarian Cancer. J Mol Diagn. 2015;17:162–70.
Gabai-Kapara E, Lahad A, Kaufman B, Friedman E, Segev S, Renbaum P, et al. Population-based screening for breast and ovarian cancer risk due to BRCA1 and BRCA2. Proc Natl Acad Sci U S A. 2014;111:14205–10.
Ion Ampliseq Designer website. http://www.ampliseq.com. Accessed 13 August 2015.
TMAP - torrent map** alignment program GitHub page. http://github.com/iontorrent/TS/tree/master/Analysis/TMAP. Accessed 13 August 2015.
GLU: Genotype Library and Utilities google code papge. http://code.google.com/p/glu-genetics. Accessed 13 August 2015.
Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013;14:178–92.
ClinVar website. http://www.ncbi.nlm.nih.gov/clinvar. Accessed 13 August 2015.
Tavtigian SV, Deffenbaugh AM, Yin L, Judkins T, Scholl T, Samollow PB, et al. Comprehensive statistical study of 452 BRCA1 missense substitutions with classification of eight recurrent substitutions as neutral. J Med Genet. 2006;43:295–305.
Mathe E, Olivier M, Kato S, Ishioka C, Hainaut P, Tavtigian SV. Computational approaches for predicting the biological effect of p53 missense mutations: a comparison of three sequence analysis based methods. Nucleic Acids Res.2006;34:1317–25.
Dean M, Boland J, Yeager M, Im KM, Garland L, Rodriguez-Herrera M, et al. Supporting data for “Addressing health disparities in Hispanic breast cancer: Accurate and inexpensive sequencing of BRCA1 and BRCA2”. GigaScience Database. 2015. http://dx.doi.org/10.5524/100154
Tarabeux J, Zeitouni B, Moncoutier V, Tenreiro H, Abidallah K, Lair S, et al: Streamlined ion torrent PGM-based diagnostics: BRCA1 and BRCA2 genes as a model. European journal of human genetics : EJHG 2014;22:535–41.
Dacheva D, Dodova R, Popov I, Goranova T, Mitkova A, Mitev V, et al. Validation of an NGS Approach for Diagnostic BRCA1/BRCA2 Mutation Testing. Molecular diagnosis & therapy 2015;19:119–30
Kluska A, Balabas A, Paziewska A, Kulecka M, Nowakowska D, Mikula M, et al. New recurrent BRCA1/2 mutations in Polish patients with familial breast/ovarian cancer detected by next generation sequencing. BMC medical genomics 2015;8:19
Yeo ZX, Wong JC, Rozen SG, Lee AS: Evaluation and optimisation of indel detection workflows for ion torrent sequencing of the BRCA1 and BRCA2 genes. BMC genomics 2014;15:516
Chan M, Ji SM, Yeo ZX, Gan L, Yap E, Yap YS, et al. Development of a next-generation sequencing method for BRCA mutation screening: a comparison between a high-throughput and a benchtop platform. The Journal of molecular diagnostics : JMD 2012;14:602–12
Bosdet IE, Docking TR, Butterfield YS, Mungall AJ, Zeng T, Coope RJ, et al. A clinically validated diagnostic second-generation sequencing assay for detection of hereditary BRCA1 and BRCA2 mutations. The Journal of molecular diagnostics : JMD 2013;15:796–809
Acknowledgements
The authors would like to thank the staff and health professionals from Nueva Vida (Baltimore, MD and Richmond, VA), the Latino Community Development Agency (Oklahoma City, OK), and the Texas Tech University Health Sciences Centers, in Lubbock and El Paso, TX; Mark Hurlbert (Avon Foundation); Dr Jennifer Loud for advice on approval of the study; Russ Hanson for managing study documents; and the BSP-CCR Genetics Core for technical support. The work was supported in part by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research, and by Leidos Biomedical Research, Inc., under contract # HHSN261200800001E.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
JB has received travel reimbursements from Life Technologies. The authors declare that they have no other competing interests.
Authors’ contributions
JB, LG, MR-H, JH, DR, HJL, and RE carried out the molecular genetic studies. KMI, KJ, JS, SB, and XZ participated in the sequence analysis, bioinformatics and data analysis. VR, CH, CB, ER, CA JAF, DDN, and ZN carried out patient recruitment, consent and collection of clinical data. MD conceived of the study, participated in its design and coordination, and helped to draft the manuscript. All authors read and approved the final manuscript.
Additional file
Additional file 1: Table S1.
Mutation details for variants in Table 5. (DOCX 27 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Dean, M., Boland, J., Yeager, M. et al. Addressing health disparities in Hispanic breast cancer: accurate and inexpensive sequencing of BRCA1 and BRCA2 . GigaSci 4, 50 (2015). https://doi.org/10.1186/s13742-015-0088-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13742-015-0088-z