
Abstract
The CCCTC-binding factor (CTCF) protein and its modified forms regulate gene expression and genome organization. However, information on CTCF acetylation and its biological function is still lacking. Here, we show that CTCF can be acetylated at lysine 20 (CTCF-K20) by CREB-binding protein (CBP) and deacetylated by histone deacetylase 6 (HDAC6). CTCF-K20 is required for the CTCF interaction with CBP. A CTCF point mutation at lysine 20 had no effect on self-renewal but blocked the mesoderm differentiation of mouse embryonic stem cells (mESCs). The CTCF-K20 mutation reduced CTCF binding to the promoters and enhancers of genes associated with early cardiac mesoderm differentiation, resulting in diminished chromatin accessibility and decreased enhancer-promoter interactions, impairing gene expression. In summary, this study reveals the important roles of CTCF-K20 in regulating CTCF genomic functions and mESC differentiation into mesoderm.
Similar content being viewed by others


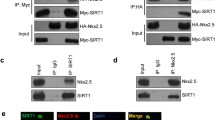
Background
Interphase chromosomes in eukaryotic cells are partitioned into discrete megabase-sized topologically associated domains (TADs), and the boundaries of TADs are enriched for the binding of the architectural protein CCCTC-binding factor (CTCF) (Dixon et al. 2012, Nora et al. 2012, Ong and Corces 2014). CTCF, which is ubiquitously expressed and highly conserved in eukaryotes (Filippova et al. 1996, Klenova et al. 1993), is composed of an N-terminal domain, a C-terminal domain, and a DNA-binding domain with 11 zinc fingers (Ghirlando and Felsenfeld 2016). CTCF is implicated in diverse regulatory functions, including transcriptional activation/repression, gene insulation, imprinting, X chromosome inactivation, and long-range DNA–DNA contacts (Phillips and Corces 2009). A number of CTCF cobinding partners have been identified and might promote CTCF loop formation (Hu et al. 2020). CTCF regulates loop extrusion of the cohesin complex, and cohesin progressively forms DNA loops but stalls at convergent CTCF sites (Fudenberg et al. 2016). The N-terminus of CTCF contains a YDF motif that interacts with a pocket formed by the SA2-SCC1 subunits of cohesin and is required for cohesin-mediated extrusion (Li et al. 2020). We recently identified a short CTCF isoform lacking the N-terminus and 2.5 zinc fingers of CTCF which is encoded by an alternatively spliced transcript skip** exons 3 and 4 (Li et al. 2019). CTCF genomic sites bound by the CTCF short isoform might fail to stop cohesin-mediated loop extrusion in the genome. These observations suggest that the N-terminus of CTCF plays a critical role in genome organization.
In addition to the important roles of CTCF in regulating gene expression and chromatin organization, posttranslational modifications of the CTCF protein also mediate its biological functions. Phosphorylated CTCF is present during interphase and mitosis, suggesting that CTCF phosphorylation may play different roles in different stages of the cell cycle (** (Li et al. 2020, Pugacheva et al. 2020), suggesting that the N-terminal domain of CTCF plays an essential role in genome organization. Indeed, although the CTCF short isoform (CTCF-s), which lacks the N-terminus and 2.5 zinc fingers, competes with canonical CTCF for genomic DNA binding and disrupts the CTCF/cohesin-mediated long-range chromatin loops (Li et al. 2019), CTCF-s could not mediate long-range DNA–DNA interactions and might not regulate cohesin-mediated loop extrusion.
Protein acetylation may mediate the protein interaction, and the CTCF-K20R mutation reduced the interaction between CTCF and CBP, suggesting that lysine 20 acetylation of CTCF might regulate dimer formation and subsequently modulate the organization of CTCF-mediated long-distance interactions. Lysine 20 is present in the N-terminus of CTCF, and additional experiments are needed to determine whether lysine 20 acetylation in CTCF regulates cohesin-mediated loop extrusion.
Conclusions
In summary, we found that CTCF could be acetylated at K20 by CBP and deacetylated by HDAC6. CTCF-K20R mutation had no effect on mESC self-renewal but hindered cardiac mesoderm differentiation. Mechanism studies revealed that CTCF-K20R mutation resulted in the decrease of a subset of CTCF binding sites during differentiation, further leading to the down-regulation of chromatin accessibility and EP interactions, which both might be harmful to the activation of genes related to cardiac mesoderm differentiation.
Methods
Cell culture and differentiation
HEK293T and HeLa cells were cultured in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% FBS and 1% penicillin and streptomycin. The mESC line E14Tg2A was cultured on mitomycin C-inactivated mouse embryo fibroblasts in DMEM containing 15% fetal bovine serum (FBS; Gibco), 1 mM sodium pyruvate (Gibco), 1 mM nonessential amino acids (Gibco), 1 mM GlutaMAX (Gibco), 0.1 mM 2-mercaptoethanol (Gibco), 1000 U/mL leukemia inhibitory factor, and 2i inhibitors (3 μM CHIR99021 and 1 μM PD0325901). All mammalian cell lines were grown in a 37 °C incubator with 5% CO2. For ESC differentiation, ESCs were differentiated into EBs using the hanging-drop method (with 1,000 cells/drop) in DMEM supplemented with 15% serum without LIF.
Plasmid construction
shRNA oligos were designed and cloned into the pLKO.1 plasmid. The sequences of shRNA oligos are listed in Supplementary Table 1. The CTCF or CTCF-K20R cDNAs were cloned into pSin-FLAG and pGEX4T-2 vectors, and the sequences of their primers are listed in Supplementary Table 2.
Lentivirus-mediated shRNA knockdown
The recombinant constructs (pLKO.1 empty, pLKO.1-CTCF, pLKO.1-HDAC6, and pLKO.1-MOF) were transfected into HEK293T cells together with lentiviral helper plasmids (pMD2.G and psPAX.2). Viral supernatants were collected after 48 h, filtered through a 0.45 μm filter and then mixed with 8 μg/mL polybrene to infect the target cells. Stable cell lines were selected with 2 μg/mL puromycin 2 days after infection.
Generation of CTCF-K20R mESCs
A point mutation in CTCF in which lysine 20 was replaced with arginine (K20R) was generated in mouse ESCs using the CRISPR/Cas9 system. The sgRNA target sequences were designed using crispr.mit.edu and inserted into the pX330 plasmid. Then, pX330, along with the linearized donor vector, were electroporated into mESCs for gene editing. The correctly targeted colonies were chosen through drug selection, genomic DNA sequencing, and Western blotting. The oligos used for generating mESCs expressing the CTCF-K20R point mutation are shown in Supplementary Table 3.
Antibodies
The following antibodies were used in this study: rabbit anti-CTCF (active motif, 61,311), mouse anti-Flag (Sigma, F1804), rabbit anti-acetylated lysine (Cell Signaling Technology, 9441S), mouse anti-β-ACTIN (Abcam, ab8226), rabbit anti-HDAC6 (Proteintech, 12,834–1-AP), rabbit anti-CBP (Cell Signaling Technology, 7389S), mouse anti-MOF (Boster, A02757), rabbit anti-H3K27ac (Active Motif, 39,133), anti-OCT4 (Santa Cruz Biotechnology, sc-5279), anti-SOX2 (Abcam, ab79351), mouse anti-cTnT (Thermo, MA5-12,960), rabbit IgG (Santa Cruz Biotechnology, sc-2027), and anti-6 × HIS (Abcam, ab18184).
Immunoprecipitation and Western blotting
Cells were harvested and lysed in cell lysis buffer (50 mM Tris–HCl (pH 7.6), 1% Triton X-100, 1 mM EDTA, 10% glycerol, 1 mM DTT, 1 mM PMSF, and protease inhibitor cocktail). Proteins were resolved on an SDS–PAGE gel and then transferred to a PVDF membrane. Then, the membrane was washed with TBS-T buffer, and immunoblotting was performed with the indicated antibodies.
For immunoprecipitation, cells were lysed in lysis buffer (20 mM Tris–HCl (pH 7.5), 1% Triton X-100, 150 mM NaCl, and 10% glycerol) supplemented with a complete protease inhibitor cocktail. Then, immunoprecipitation was performed using the indicated antibodies. Generally, 2 μg of antibody was added to Protein A/G beads and incubated at 4 °C for 6 h before the supernatant was discarded. After adding the protein extract, the incubation was continued for 12 h, and then immune complexes were washed four times with lysis buffer, resolved on SDS–PAGE gels, and analyzed using immunoblotting. Western blot results were quantified with ImageJ software.
Dot blot
CTCF-K20 acetylated and unmodified polypeptides were synthesized by Guangzhou IGE Biotechnology. Equal amounts of polypeptides containing K20 acetylated polypeptide and unmodified polypeptide were incubated with an equal amount of diluted anti-CTCF-K20Ac antibody at 37 °C. The immunoblot analysis was performed with anti-CTCF-K20Ac antibodies.
Quantitative RT–PCR analysis
Total RNA was isolated from samples with TRIzol reagent (Invitrogen). One milligram of total RNA was then reverse transcribed with a reverse transcription system (Promega). The cDNAs of interest were then quantified using real-time qPCR. The primers used in the RT–qPCR assays are listed in Supplementary Table 4. All experiments were repeated three times.
RNA sequencing and bioinformatics analysis
Total RNA was extracted as described above. RNA sequencing libraries were constructed using the VAHTS mRNA-seq V3 Library Prep Kit (Vazyme Biotech, NR611). Two rounds of mRNA purification were performed to guarantee the removal of rRNA. Briefly, 50 μL of mRNA capture beads were incubated with 1.5 μg of total RNA at 65 °C for 5 min and then at 25 °C for 5 min. The supernatant was discarded, and 200 μL of bead wash buffer were added to clean the beads. Fifty microliters of Tris buffer were added to resuspend the beads, and the sample was incubated at 80 °C for 5 min to release mRNA. Then, 50 μL of bead binding buffer were added to facilitate the binding of mRNA to the beads. Furthermore, ribosome-depleted mRNAs were fragmented at 85 °C for 6 min, and cDNAs were synthesized. The cDNAs were purified with AMPure XP beads (Beckman Coulter, A63882), followed by end repair, adaptor ligation, size selection of the library, and library amplification. The libraries from two biological replicates were purified using AMPure XP beads and then sequenced with an illumina NovaSeq instrument (Annoroad Gene Technology Co., Ltd.).
Adaptors were trimmed from raw reads using Trim_Galore (v0.4.4) and then quasi-mapped and quantified to the mouse mm10 genome with salmon (v0.9.1) (Patro et al. 2017). Transcript-level counts were collapsed to gene-level counts using tximport (v1.20.0) (Soneson et al. 2015) and analyzed with DESeq2 (v1.10.1) (Love et al. 2014). Genes with a fold change greater than 2 and a q-value less than 0.05 were considered differentially expressed genes (DEGs). The DEGs are listed in Supplementary Table 5. GO analysis and GSEA were conducted with clusterProfiler (v4.0.0) (Yu et al. 2012).
GST pull-down assay
BL21 E. coli were treated with 0.1 mM IPTG at 37 °C to induce protein expression, harvested, and resuspended in PBS containing 0.5% Triton X-100, 2 mM EDTA, and 1 mM PMSF, followed by sonication. The protein was then purified using GST antibody-conjugated beads. Beads were subsequently added to HEK293T cell lysate transfected with the HDAC6-Flag construct and mixed at 4 °C overnight. Next, beads were harvested through centrifugation and washed four times with 0.3% Nonidet P40 buffer before boiling with 1 × SDS–PAGE loading buffer and subjected to Western blotting.
Cellular immunofluorescence staining
Cells were fixed with 4% paraformaldehyde and permeabilized with 0.2% Triton X-100 containing 10% FBS (Invitrogen)/1% BSA in PBS at room temperature for 15 min. Samples were then incubated with primary antibodies at 4 °C overnight. The antibody used for cellular immunofluorescence staining was raised against cTnT. The cells were then washed four times, and 0.1 μg/mL DAPI (Sigma) was included in the final wash to stain nuclei. Images were captured with an inverted microscope (DMI4000, Leica Microsystems).
Flow cytometry
mESCs were dissociated with trypsin, and cells were fixed with 4% paraformaldehyde and permeabilized with 0.2% Triton X-100 containing 10% FBS (Invitrogen)/1% BSA in PBS at room temperature for 15 min. Samples were then incubated with primary antibodies at 37 °C for 30 min. Cells were washed twice and incubated with a secondary antibody in a cassette at room temperature for 30 min. The cTnT antibody was used in this experiment. The analysis was performed using FlowJo software.
Chromatin immunoprecipitation and sequencing
ChIP experiments were performed as previously described (Li et al. 2019). Briefly, 1 × 107 cells were crosslinked with 1% formaldehyde at room temperature for 10 min. Then, the crosslinking reaction was stopped by adding glycine (final concentration, 0.125 M). The cells were sonicated in SDS lysis buffer containing a 1 × protease inhibitor cocktail and 1 mM PMSF to achieve a chromatin size of 100–300 bp. The sonicated chromatin was incubated with the indicated antibodies coupled with Dynabeads conjugated to Protein A and G (1:1 mixed) at 4 °C overnight with rotation. Immune complexes were washed with the following buffers: low-salt wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris–HCl (pH 8.0), and 150 mM NaCl), high-salt wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris–HCl (pH 8.0), and 500 mM NaCl), LiCl wash buffer (0.25 M LiCl, 1% IGEPAL CA-630, 1% deoxycholic acid (sodium salt), 1 mM EDTA, and 10 mM Tris–HCl (pH 8.0)) and TE buffer (10 mM Tris–HCl (pH 8.0) and 1 mM EDTA). After reversing the crosslinks, the ChIPed DNA samples were purified, and libraries were constructed according to the Illumina ChIP-seq library generation protocol.
ATAC sequencing
ATAC-seq experiments were performed as previously described (Buenrostro et al. 2015). Briefly, 50,000 cells were harvested and washed once with 50 μL of cold PBS. Then, the cells were resuspended in 50 μL of lysis buffer (10 mM Tris–HCl (pH 7.4), 10 mM NaCl, 3 mM MgCl2, and 0.2% (v/v) IGEPAL CA-630). Then, the suspension of nuclei was centrifuged at 500 × g at 4 °C for 10 min. The pellet was resuspended in 50 μL of transposition reaction mix (10 μL of TD buffer, 5 μL of Tn5 transposase, and 35 μL of nuclease-free H2O) and incubated at 37 °C for 30 min. Finally, DNA was extracted using a MinElute PCR Purification Kit (QIAGEN). ATAC-seq libraries were constructed and purified with AMPure XP beads (Beckman Coulter). The libraries were denatured and diluted and then sequenced with the HiSeq X-Ten platform (Annoroad Gene Technology Co., Ltd).
ATAC-seq and ChIP-seq data analysis
All ATAC-seq and CTCF ChIP-seq experiments were performed using two biological replicates. Raw reads were trimmed with Trim_Galore and aligned to the mouse mm10 genome using bowtie2 (v2.2.5) (Langmead and Salzberg 2012) with the parameter “–very-sensitive –end-to-end –no-unal”. Reads with a map** quality lower than 30 were discarded. Duplicate reads were removed with sambamba (v0.6.7). Reads overlap** with mouse mm10 blacklist regions (http://mitra.stanford.edu/kundaje/akundaje/release/blacklists) were excluded. For the CTCF ChIP-seq experiment, 10 million reads were randomly subsampled. Peaks were called using MACS2 (v2.1.0) (Zhang et al. 2008) with default parameters for CTCF ChIP-seq and ATAC-seq experiments. Broad peaks were called for histone H3K27ac ChIP-seq using MACS2 with the parameter “–broad”. CTCF peak summits were submitted to the Homer findMotifsGenome.pl tool (Heinz et al. 2010) for the motif analysis with the parameters “-len 8,10,12,20”. bamCoverage from deepTools (v2.2.4) (Ramirez et al. 2016) was used to generate a normalized bigwig file with the parameter “-of bigwig -bs 1 –normalizeUsing RPGC”. Histone H3K27ac bigwig files were normalized such that the total enrichment of all H3K27ac peaks was similar. Regions that were enriched with H3K27ac and overlapped with chromatin-accessible peaks but were not transcription start sites (TSSs) or transcription termination sites (TTSs) were identified as enhancers. Heatmaps were drawn with deepTools (v2.2.4). Differential sites were identified using DiffBind (v3.2.4) (Ross-Innes et al. 2012) with default parameters, and peaks with q values less than 0.05 and log2(fold change) values greater than 1 were considered differential binding sites. The analysis of differential CTCF binding in day 4 differentiated cells is provided in Supplementary Table 6.
BL-Hi-C experiments
The BL-Hi-C libraries were constructed as previously described (Liang et al. 2017). Briefly, the cells were treated with 1% formaldehyde at room temperature for 10 min, and the crosslinking reaction was quenched by adding 2.5 M glycine to a final concentration of 0.2 M. Then, the cells were resuspended with BL-Hi-C lysis buffer 1 and incubated on ice for 15 min. After centrifugation, the cell pellet was resuspended in BL-Hi-C lysis buffer 2 and rotated at 4 °C for 15 min. The cell pellet was washed once with BL-Hi-C lysis buffer 1, resuspended with 0.5% SDS and incubated at 62 °C. At the end of the incubation, SDS was quenched by adding 10% Triton X-100 and ddH2O, and the sample was incubated at 37 °C for 10 min. Afterward, the genomic DNA was digested with HaeIII (NEB) at 37 °C for 2 h to generate blunt-end fragments. Chromatin was cleaved by adding HaeIII (NEB) and incubating the sample at 37 °C. Cleaved chromatin was A-tailed by adding a 10 mM dATP solution (Thermo) and Klenow Fragment (NEB) with rotation at 37 °C for 40 min. Then, chromatin was treated with adenine and ligated with biotinylated bridge linker S2 (annealed by /5Phos/CGCGATATC/iBIOdT/TATCTGACT and /5Phos/GTCAGATAAGATATCGCGT) at 16 °C for 4 h. The unligated DNA fragments were digested with DNA exonuclease (NEB) at 37 °C for 1 h. After centrifugation at 3500 g at 4 °C for 5 min and removal of the supernatant, the pellet was resuspended in ddH2O with lambda exonuclease buffer, lambda exonuclease, and exonuclease I and rocked at 900 rpm at 37 °C for 1 h in a ThermoMixer C. Next, the samples were treated with SDS and proteinase K at 55 °C overnight to digest the proteins, and the DNA was purified using phenol:chloroform:isoamyl alcohol (25:24:1) extraction followed by ethanol precipitation. Then, the DNA was fragmented into 300 bp fragments on average by sonication, and the biotin-labeled DNA fragments were pulled down with Dynabeads M-280 conjugated to streptavidin. The beads were washed twice with 2 × B&W buffer (10 mM Tris–HCl (pH 7.5), 1 mM EDTA, and 2 M NaCl) and blocked with 1 × I-Block buffer (2% I-block protein-based blocking reagent and 0.5% SDS) at room temperature for 45 min. Next, the beads were washed twice with 1 × B&W buffer and treated with 1 mg of preheated salmon sperm DNA with rotation at room temperature for 30 min. After washing with 1 × B&W buffer, the beads were resuspended with 2 × B&W buffer, combined with sonicated DNA and rotated at room temperature for 45 min. The beads were washed five times with 2 × SSC containing 0.5% SDS, twice with 1 × B&W buffer and once with Buffer EB (QIAGEN). DNA bound to beads was end-repaired using T4 DNA polymerase (NEB), T4 polynucleotide kinase (NEB) and large (Klenow) fragment (NEB) with shaking at 900 rpm at 37 °C for 30 min. After two washes with 1 × TWB (5 mM Tris–HCl (pH 7.5), 0.5 mM EDTA, 1 M NaCl, and 0.05% Tween 20) at 55 °C for 2 min, DNA on beads was A-tailed using Klenow fragment (3’/5’ exo-) (NEB) with shaking at 900 rpm at 37 °C for 30 min. Beads were washed twice with 1 × TWB at 55 °C for 2 min and once with 1 × Quick ligation buffer (NEB). DNA on beads was ligated with an adaptor using Quick ligase (NEB) and 20 mM Y-Adaptor (Annealed by /5Phos/GATCGGAAGAGCACACGTCTGAACTCCAGTCAC and TACACTCTTTCCCTACACGACGCTCTTCCGATCT) at room temperature for 45 min. Beads were washed twice with 1 × TWB at 55 °C for 2 min and once with EB buffer (QIAGEN).
The libraries were constructed using Q5 Hot Star DNA Polymerase (NEB) for PCR amplification. PCR products with sizes ranging from 300–700 bp were purified using Ampure XP beads (Beckman Coulter) and subjected to sequencing with the Illumina NovaSeq platform (Annoroad Gene Technology Co., Ltd.).
BL-Hi-C analysis
All BL-Hi-C experiments were performed using two biological replicates. Raw reads were first submitted to Trim_Galore to remove adaptors, and then linkers were trimmed with the parameters “-m 1 -k 2 -e 1 -l 15 -A ACGCGATATCTTATC -B AGTCAGATAAGATAT” using ChIA-PET2 (v0.9.3) (Li et al. 2017). The trimmed reads were handled using HiC-Pro (v2.11.1) (Servant et al. 2015). Quality control information was collected and is listed in Supplementary Table 7. The distance decay analysis was performed using hicPlotDistVsCounts from the hicexplorer suite (v3.6) at a 100 kb resolution (Wolff et al. 2020). The reproducibility of Hi-C data was assessed using HiCRep (v0.2.6) for all chromosomes except chrY and chrM at 50 kb resolution (Yang et al. 2017), and the average correlation score for all chromosomes was used. Hi-C data were normalized using HiCcompare (v1.14.0) (Stansfield et al. 2018). The normalized matrix was transformed to Hi-C format using juicer tools (v1.13) (Durand et al. 2016) and visualized in WashU Epigenome Browser (Li et al. 2019). Compartments were analyzed using juicer tools with default parameters at 100 kb resolution, and the insulation score and TAD boundaries were identified by FAN-C (v0.9.20) at 100 kb resolution with parameter “-w 400 kb” (Kruse et al. 2020). Significant loops were identified as previously described (Song et al. 2022) and are listed in Supplementary Table 8.
Quantification and statistical analysis
Two-tailed Student’s t tests were used for all comparisons, including the RT–qPCR analysis. All values included in the figures are presented as the means ± s.d. Error bars represent ± s.d. for triplicate experiments. The statistical significance is indicated with asterisks (*). A two-sided P value of < 0.05 was considered to be statistically significant (*P < 0.05, **P < 0.01, and ***P < 0.001).
Availability of data and materials
The RNA-seq, ATAC-seq, ChIP-seq, and BL-Hi-C data described in this paper are deposited in GEO under accession number GSE181705 and GSA under accession number CRA004748.
Abbreviations
- CTCF:
-
CCCTC-binding factor
- CTCF-K20:
-
Lysine 20 of CTCF
- mESCs:
-
Mouse embryonic stem cells
- TADs:
-
Topologically associated domains
- CKII:
-
Casein kinase II
- CBP:
-
CREB-binding protein
- ChIP-seq:
-
Chromatin immunoprecipitation sequencing
- EP interactions:
-
Enhancer-promoter interactions
- TSA:
-
Trichostatin A
- co-IP:
-
Co-immunoprecipitation
- NAM:
-
Nicotinamide
- shRNA:
-
Short hairpin RNA
- TBSA:
-
Tubastatin A
- CRISPR:
-
Clustered regularly interspaced short palindromic repeats
- EBs:
-
Embryoid bodies
- WT:
-
Wild-type
- GO:
-
Gene Ontology
- GSEA:
-
Gene set enrichment analysis
- APA:
-
Aggregate peak analysis
References
Alexanian M, Maric D, Jenkinson SP, Mina M, Friedman CE, Ting CC, et al. A transcribed enhancer dictates mesendoderm specification in pluripotency. Nat Commun. 2017;8(1):1806. https://doi.org/10.1038/s41467-017-01804-w.
Bitterman KJ, Anderson RM, Cohen HY, Latorre-Esteves M, Sinclair DA. Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast sir2 and human SIRT1. J Biol Chem. 2002;277(47):45099–107. https://doi.org/10.1074/jbc.M205670200.
Bonchuk A, Kamalyan S, Mariasina S, Boyko K, Popov V, Maksimenko O, et al. N-terminal domain of the architectural protein CTCF has similar structural organization and ability to self-association in bilaterian organisms. Sci Rep. 2020;10(1):2677. https://doi.org/10.1038/s41598-020-59459-5.
Bowers EM, Yan G, Mukherjee C, Orry A, Wang L, Holbert MA, et al. Virtual ligand screening of the p300/CBP histone acetyltransferase: identification of a selective small molecule inhibitor. Chem Biol. 2010;17(5):471–82. https://doi.org/10.1016/j.chembiol.2010.03.006.
Buenrostro JD, Wu B, Chang HY, Greenleaf WJ. ATAC-seq: a method for assaying chromatin accessibility genome-wide. Curr Protoc Mol Biol. 2015;109:21.29.1-21.29.9. https://doi.org/10.1002/0471142727.mb2129s109.
Butler KV, Kalin J, Brochier C, Vistoli G, Langley B, Kozikowski AP. Rational design and simple chemistry yield a superior, neuroprotective HDAC6 inhibitor, tubastatin A. J Am Chem Soc. 2010;132(31):10842–6. https://doi.org/10.1021/ja102758v.
Cunningham TJ, Yu MS, McKeithan WL, Spiering S, Carrette F, Huang CT, et al. Id genes are essential for early heart formation. Genes Dev. 2017;31(13):1325–38. https://doi.org/10.1101/gad.300400.117.
Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485(7398):376–80. https://doi.org/10.1038/nature11082.
Docquier F, Kita GX, Farrar D, Jat P, O’Hare M, Chernukhin I, et al. Decreased poly(ADP-ribosyl)ation of CTCF, a transcription factor, is associated with breast cancer phenotype and cell proliferation. Clin Cancer Res. 2009;15(18):5762–71. https://doi.org/10.1158/1078-0432.CCR-09-0329.
Durand NC, Shamim MS, Machol I, Rao SS, Huntley MH, Lander ES, et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 2016;3(1):95–8. https://doi.org/10.1016/j.cels.2016.07.002.
El-Kady A, Klenova E. Regulation of the transcription factor, CTCF, by phosphorylation with protein kinase CK2. FEBS Lett. 2005;579(6):1424–34. https://doi.org/10.1016/j.febslet.2005.01.044.
Evans MJ, Kaufman MH. Establishment in culture of pluripotential cells from mouse embryos. Nature. 1981;292(5819):154–6. https://doi.org/10.1038/292154a0.
Filippova GN, Fagerlie S, Klenova EM, Myers C, Dehner Y, Goodwin G, et al. An exceptionally conserved transcriptional repressor, CTCF, employs different combinations of zinc fingers to bind diverged promoter sequences of avian and mammalian c-myc oncogenes. Mol Cell Biol. 1996;16(6):2802–13. https://doi.org/10.1128/MCB.16.6.2802.
Fudenberg G, Imakaev M, Lu C, Goloborodko A, Abdennur N, Mirny LA. Formation of chromosomal domains by loop extrusion. Cell Rep. 2016;15(9):2038–49. https://doi.org/10.1016/j.celrep.2016.04.085.
Ghirlando R, Felsenfeld G. CTCF: making the right connections. Genes Dev. 2016;30(8):881–91. https://doi.org/10.1101/gad.277863.116.
Gomez-Velazquez M, Badia-Careaga C, Lechuga-Vieco AV, Nieto-Arellano R, Tena JJ, Rollan I, et al. CTCF counter-regulates cardiomyocyte development and maturation programs in the embryonic heart. PLoS Genet. 2017;13(8):e1006985. https://doi.org/10.1371/journal.pgen.1006985.
Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38(4):576–89. https://doi.org/10.1016/j.molcel.2010.05.004.
Herrmann BG, Labeit S, Poustka A, King TR, Lehrach H. Cloning of the T gene required in mesoderm formation in the mouse. Nature. 1990;343(6259):617–22. https://doi.org/10.1038/343617a0.
Hirayama T, Tarusawa E, Yoshimura Y, Galjart N, Yagi T. CTCF is required for neural development and stochastic expression of clustered Pcdh genes in neurons. Cell Rep. 2012;2(2):345–57. https://doi.org/10.1016/j.celrep.2012.06.014.
Hornbeck PV, Zhang B, Murray B, Kornhauser JM, Latham V, Skrzypek E. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 2015;43(Database issue):D512-20. https://doi.org/10.1093/nar/gku1267.
Hu G, Dong X, Gong S, Song Y, Hutchins AP, Yao H. Systematic screening of CTCF binding partners identifies that BHLHE40 regulates CTCF genome-wide distribution and long-range chromatin interactions. Nucleic Acids Res. 2020;48(17):9606–20. https://doi.org/10.1093/nar/gkaa705.
Klenova EM, Nicolas RH, Paterson HF, Carne AF, Heath CM, Goodwin GH, et al. CTCF, a conserved nuclear factor required for optimal transcriptional activity of the chicken c-myc gene, is an 11-Zn-finger protein differentially expressed in multiple forms. Mol Cell Biol. 1993;13(12):7612–24. https://doi.org/10.1128/mcb.13.12.7612-7624.1993.
Klenova EM, Chernukhin IV, El-Kady A, Lee RE, Pugacheva EM, Loukinov DI, et al. Functional phosphorylation sites in the C-terminal region of the multivalent multifunctional transcriptional factor CTCF. Mol Cell Biol. 2001;21(6):2221–34. https://doi.org/10.1128/MCB.21.6.2221-2234.2001.
Kruse K, Hug CB, Vaquerizas JM. FAN-C: a feature-rich framework for the analysis and visualisation of chromosome conformation capture data. Genome Biol. 2020;21(1):303. https://doi.org/10.1186/s13059-020-02215-9.
Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–9. https://doi.org/10.1038/nmeth.1923.
Li G, Chen Y, Snyder MP, Zhang MQ. ChIA-PET2: a versatile and flexible pipeline for ChIA-PET data analysis. Nucleic Acids Res. 2017;45(1):e4. https://doi.org/10.1093/nar/gkw809.
Li D, Hsu S, Purushotham D, Sears RL, Wang T. WashU Epigenome Browser update 2019. Nucleic Acids Res. 2019;47(W1):W158–65. https://doi.org/10.1093/nar/gkz348.
Li J, Huang K, Hu G, Babarinde IA, Li Y, Dong X, et al. An alternative CTCF isoform antagonizes canonical CTCF occupancy and changes chromatin architecture to promote apoptosis. Nat Commun. 2019;10(1):1535. https://doi.org/10.1038/s41467-019-08949-w.
Li Y, Haarhuis JHI, Sedeno Cacciatore A, Oldenkamp R, van Ruiten MS, Willems L, et al. The structural basis for cohesin-CTCF-anchored loops. Nature. 2020;578(7795):472–6. https://doi.org/10.1038/s41586-019-1910-z.
Liang Z, Li G, Wang Z, Djekidel MN, Li Y, Qian MP, et al. BL-Hi-C is an efficient and sensitive approach for capturing structural and regulatory chromatin interactions. Nat Commun. 2017;8(1):1622. https://doi.org/10.1038/s41467-017-01754-3.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. https://doi.org/10.1186/s13059-014-0550-8.
Luo H, Yu Q, Liu Y, Tang M, Liang M, Zhang D, et al. LATS kinase-mediated CTCF phosphorylation and selective loss of genomic binding. Sci Adv. 2020;6(8):eaaw4651. https://doi.org/10.1126/sciadv.aaw4651.
MacPherson MJ, Beatty LG, Zhou W, Du M, Sadowski PD. The CTCF insulator protein is posttranslationally modified by SUMO. Mol Cell Biol. 2009;29(3):714–25. https://doi.org/10.1128/MCB.00825-08.
Moore JM, Rabaia NA, Smith LE, Fagerlie S, Gurley K, Loukinov D, et al. Loss of maternal CTCF is associated with peri-implantation lethality of Ctcf null embryos. PLoS ONE. 2012;7(4):e34915. https://doi.org/10.1371/journal.pone.0034915.
Nora EP, Lajoie BR, Schulz EG, Giorgetti L, Okamoto I, Servant N, et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature. 2012;485(7398):381–5. https://doi.org/10.1038/nature11049.
Ong CT, Corces VG. CTCF: an architectural protein bridging genome topology and function. Nat Rev Genet. 2014;15(4):234–46. https://doi.org/10.1038/nrg3663.
Ong CT, Van Bortle K, Ramos E, Corces VG. Poly(ADP-ribosyl)ation regulates insulator function and intrachromosomal interactions in Drosophila. Cell. 2013;155(1):148–59. https://doi.org/10.1016/j.cell.2013.08.052.
Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods. 2017;14(4):417–9. https://doi.org/10.1038/nmeth.4197.
Phillips JE, Corces VG. CTCF: master weaver of the genome. Cell. 2009;137(7):1194–211. https://doi.org/10.1016/j.cell.2009.06.001.
Pugacheva EM, Kubo N, Loukinov D, Tajmul M, Kang S, Kovalchuk AL, et al. CTCF mediates chromatin loo** via N-terminal domain-dependent cohesin retention. Proc Natl Acad Sci U S A. 2020;117(4):2020–31. https://doi.org/10.1073/pnas.1911708117.
Ramirez F, Ryan DP, Gruning B, Bhardwaj V, Kilpert F, Richter AS, et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016;44(W1):W160–5. https://doi.org/10.1093/nar/gkw257.
Ritter N, Ali T, Kopitchinski N, Schuster P, Beisaw A, Hendrix DA, et al. The lncRNA locus handsdown regulates cardiac gene programs and is essential for early mouse development. Dev Cell. 2019;50(5):644-57.e8. https://doi.org/10.1016/j.devcel.2019.07.013.
Del Rosario BC, Kriz AJ, Del Rosario AM, Anselmo A, Fry CJ, White FM, et al. Exploration of CTCF post-translation modifications uncovers Serine-224 phosphorylation by PLK1 at pericentric regions during the G2/M transition. Elife. 2019;8:e42341. https://doi.org/10.7554/eLife.42341.
Ross-Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, Dunning MJ, et al. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature. 2012;481(7381):389–93. https://doi.org/10.1038/nature10730.
Sekiya T, Murano K, Kato K, Kawaguchi A, Nagata K. Mitotic phosphorylation of CCCTC-binding factor (CTCF) reduces its DNA binding activity. FEBS Open Bio. 2017;7(3):397–404. https://doi.org/10.1002/2211-5463.12189.
Servant N, Varoquaux N, Lajoie BR, Viara E, Chen CJ, Vert JP, et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 2015;16:259. https://doi.org/10.1186/s13059-015-0831-x.
Soneson C, Love MI, Robinson MD. Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res. 2015;4:1521. https://doi.org/10.12688/f1000research.7563.2.
Song Y, Liang Z, Zhang J, Hu G, Wang J, Li Y, et al. CTCF functions as an insulator for somatic genes and a chromatin remodeler for pluripotency genes during reprogramming. Cell Rep. 2022;39(1):110626. https://doi.org/10.1016/j.celrep.2022.110626.
Soshnikova N, Montavon T, Leleu M, Galjart N, Duboule D. Functional analysis of CTCF during mammalian limb development. Dev Cell. 2010;19(6):819–30. https://doi.org/10.1016/j.devcel.2010.11.009.
Stansfield JC, Cresswell KG, Vladimirov VI, Dozmorov MG. HiCcompare: an R-package for joint normalization and comparison of HI-C datasets. BMC Bioinformatics. 2018;19(1):279. https://doi.org/10.1186/s12859-018-2288-x.
Wolff J, Rabbani L, Gilsbach R, Richard G, Manke T, Backofen R, et al. Galaxy HiCExplorer 3: a web server for reproducible Hi-C, capture Hi-C and single-cell Hi-C data analysis, quality control and visualization. Nucleic Acids Res. 2020;48(W1):W177–84. https://doi.org/10.1093/nar/gkaa220.
**ang JF, Corces VG. Regulation of 3D chromatin organization by CTCF. Curr Opin Genet Dev. 2021;67:33–40. https://doi.org/10.1016/j.gde.2020.10.005.
Yang T, Zhang F, Yardimci GG, Song F, Hardison RC, Noble WS, et al. HiCRep: assessing the reproducibility of Hi-C data using a stratum-adjusted correlation coefficient. Genome Res. 2017;27(11):1939–49. https://doi.org/10.1101/gr.220640.117.
Yoshida M, Kijima M, Akita M, Beppu T. Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J Biol Chem. 1990;265(28):17174–9.
Yu W, Ginjala V, Pant V, Chernukhin I, Whitehead J, Docquier F, et al. Poly(ADP-ribosyl)ation regulates CTCF-dependent chromatin insulation. Nat Genet. 2004;36(10):1105–10. https://doi.org/10.1038/ng1426.
Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–7. https://doi.org/10.1089/omi.2011.0118.
Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008;9(9):R137. https://doi.org/10.1186/gb-2008-9-9-r137.
Zheng H, **e W. The role of 3D genome organization in development and cell differentiation. Nat Rev Mol Cell Biol. 2019;20(9):535–50. https://doi.org/10.1038/s41580-019-0132-4.
Acknowledgements
The authors gratefully acknowledge the support from the Guangzhou Branch of the Supercomputing Center of the Chinese Academy of Sciences.
Funding
This work was supported in part by grants from the National Key R&D Program of China (2021YFA1100300), Strategic Priority Research Program of the Chinese Academy of Sciences (XDA16010502), National Natural Science Foundation of China (31925009, U21A20195, 32000424, 32100462, 32100463, and 81902885), Science and Technology Planning Project of Guangdong Province, China (2019B020234004, 2019A050510004 and 2020B1212060052), and Macau Science and Technology Development Fund (FDCT0107/2019/A2).
Author information
Authors and Affiliations
Contributions
H.Y. initiated the study and designed the experiments. S.G. conducted most of the experiments. G.H. performed the bioinformatics analysis. R.G., J.Z., Y.Y., B.J. and G.L. contributed to the work. H.Y., S.G., and G.H. wrote the manuscript. H.Y. conceived, supervised and funded the study. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors have no competing interests to declare.
Supplementary Information
Additional file 1.
Supplementary tables
Additional file 2. Supplementary movie 1
Additional file 3. Supplementary movie 2
Additional file 4.
Supplementary figures
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Gong, S., Hu, G., Guo, R. et al. CTCF acetylation at lysine 20 is required for the early cardiac mesoderm differentiation of embryonic stem cells. Cell Regen 11, 34 (2022). https://doi.org/10.1186/s13619-022-00131-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13619-022-00131-w