
Abstract
Hearing loss (HL) can be caused by a number of different genetic factors. Non-syndromic HL refers that HL occurs as an isolated symptom in an individual, whereas syndromic HL refers that HL is associated with other symptoms or abnormalities. To date, more than 140 genes have been identified as being associated with non-syndromic HL, and approximately 400 genetic syndromes can include HL as one of the clinical symptoms. However, no gene therapeutic approaches are currently available to restore or improve hearing. Therefore, there is an urgent necessity to elucidate the possible pathogenesis of specific mutations in HL-associated genes and to investigate the promising therapeutic strategies for genetic HL. The development of the CRISPR/Cas system has revolutionized the field of genome engineering, which has become an efficacious and cost-effective tool to foster genetic HL research. Moreover, several in vivo studies have demonstrated the therapeutic efficacy of the CRISPR/Cas-mediated treatments for specific genetic HL. In this review, we briefly introduce the progress in CRISPR/Cas technique as well as the understanding of genetic HL, and then we detail the recent achievements of CRISPR/Cas technique in disease modeling and therapeutic strategies for genetic HL. Furthermore, we discuss the challenges for the application of CRISPR/Cas technique in future clinical treatments.
Similar content being viewed by others

Introduction
Hearing loss (HL) is one of the most prevalent sensory-deficit forms in humans, currently affecting over 5% of the global population (466 million people) (http://www.who.int/mediacentre/factsheets/fs300/en/). Congenital HL impacts about 1 in 500 newborns, and it is estimated that over half of the cases can be attributable to genetic factors (genetic HL), with the remaining caused by environmental factors (non-genetic/acquired HL) [1,2,3]. To date, hearing devices (e.g., hearing aids and cochlear implants) are the most available option for HL patients [4]. However, these devices cannot restore or improve hearing to normal levels and no pharmacological therapy is currently available for genetic HL.
The promise of genome editing was demonstrated when the precise modifications of DNA were achieved by the introduction of nucleases, including zinc finger nucleases (ZFNs) and transcription-activator-like effector nucleases (TALENs) [5,6,7]. However, both ZFNs- and TALENs-mediated genome editing techniques are costly, labor-intensive, and time-consuming [8,9,10]. Fortunately, the recently emerged genome-editing platform, the clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein (Cas) system, has been used to edit specific genomic sites in different species [11]. The discovery of the CRISPR/Cas system has driven a biotechnological revolution as its simplicity and efficiency allow affordable genome editing [12]. In recent years, CRISPR/Cas-mediated genome editing has gained mounting attention as a prospective approach for modeling and treating genetic HL [13,14,15,16].
In this review, we provide an overview of the CRISPR/Cas technique and review progress in the current understanding of genetic HL. Furthermore, we summarize the current achievements of CRISPR/Cas-mediated genome editing applied to the research of genetic HL, highlighting its important role in disease modeling and therapeutic strategies. Moreover, we discuss the challenges for the application in future clinical treatments.
The principles and applications of CRISPR/Cas technique
The clustered palindromic sequence with short spacers was first observed in Escherichia coli in 1987 [17], and such a sequence family was officially named CRISPR by Jansen et al. in 2002. Since 2011, the mechanism of CRISPR/Cas system in bacteria and archaea against invasive plasmids and viral particles was basically elucidated, and the systems have subsequently been utilized as a powerful gene-editing tool [11, 18,19,20]. The system is categorized into two classes (Class 1 and 2) that are composed of one or more arrays of alternating repeat sequences and spacers, a leader sequence, and a set of CRISPR-associated (cas) genes [21, 22]. Cas genes produce CRISPR-RNAs (crRNAs) and Cas proteins (a family of endonucleases), subsequently assembling to form ‘crRNA–effectors’, which monitor the cell in search of target nucleic acids [23]. Class 1 systems (types I, III, and IV) use a multisubunit crRNA–effector complex, whereas Class 2 systems (types II, V, and VI) use a single subunit crRNA–effector protein [24]. Cas 1 and Cas 2 are universal in all systems, whereas Cas3, Cas9, Cas10, Cas12, and Cas13 are specific for Type I, II, III, V, and VI CRISPR/Cas systems, respectively [21, 22, 24,25,26,27]. Among all types of CRISPR/Cas systems, Type II, V, and VI systems have recently dominated the field of genome editing [28, 29], and natural Cas nucleases, including Cas9, Cas12, and Cas13, have been adopted for use as gene editing tools and their variants have been engineered with improved performance (Table 1) [22, 30, 31].
CRISPR/Cas9 system
Type II CRISPR/Cas system consists of three key components: the Cas9 protein, crRNA, and trans-activating crRNA (tracrRNA). Specifically, Cas9 cleaves the target DNA through interaction with crRNA and tracrRNA. To date, multiple Cas9 orthologs and engineered variants have been discovered and developed as a genome editing tool, with distinct sizes, editing efficacy, and recognition motifs. Furthermore, target recognition requires a short and conserved DNA sequence (usually 3–8 bp) adjacent to the target DNA, namely the protospacer adjacent motif (PAM) [32]. The PAM sequence varies between diverse Cas9 nucleases produced by the different bacterial strains [33, 34], and the most commonly used PAM sequence is 5′-NGG-3′ (N is any nucleotide) for Streptococcus pyogenes Cas9 (SpCas9) [35, 36].
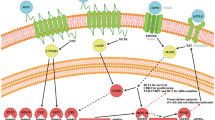
Generally, the crRNA-tracrRNA complex can be engineered as a single guide RNA (sgRNA) that joins to Cas9 and links the Cas9 to target genes. Therefore, CRISPR/Cas9-mediated genome editing can be achieved by supplying a cell with Cas9 proteins and specifically designed sgRNAs. Briefly, the sgRNA binds with and activates Cas9. Active Cas9 will search for the target site and unwind double-strand DNA, then sgRNA will anneal to one of DNA strands. If the complementary region of sgRNA and the target DNA sequence pair properly, Cas9 will cut the target DNA, causing double strand breaks (DSB) approximately 3 bp upstream of the PAM. DSBs will be commonly recovered by endogenous cellular repair pathways: non-homologous end joining (NHEJ) or homology-directed repair (HDR) (Fig. 1A) [37,38,39,40]. In the absence of any homologous sequences, the cell will undergo NHEJ. Through NHEJ, the two halves of DNA will join together, leading to insertions/deletions at the DSB site, which disrupts the target gene. If a donor homologous DNA template containing homologous arms matching the target DNA is supplied, it will be incorporated into the genome via HDR, which is desired to repair the mutated gene.

CRISPR/Cas9 mechanism. A Cas9 nuclease combines with a sgRNA to create a DSB in the targeted DNA sequence, which can be repaired by NHEJ or HDR. B Fusing an effector domain to dCas9 will regulate target gene expression. C CBEs or ABEs are engineered by fusing a nCas9 and a single-stranded DNA modifying enzyme, which are used to induce a C to T transversion or an A to G transversion. PEs encompass an engineered reverse transcriptase, a nCas9, and a pegRNA, which are the ability to generate the permanent incorporation of the desired edit into target DNA
In addition to the use of Cas9 for DNA cleavage, the catalytically inactive modification of SpCas9 (dead Cas9, dCas9) was developed for improving genome editing strategies. The dCas9 retains its ability to bind to a target DNA sequence in combination with a sgRNA but does not create DSBs [42]. By fusing to an effector domain, dCas9 can affect transcriptional machinery (e.g., transcription factors or RNA Polymerase), altering the expression level of a target gene (Fig. 1B) [88,89,90]. dCas9 that is fused with a transcriptional repressor (e.g., KRAB) can block transcription of the gene thus creating a reversible knockdown, which is a gene repression technique named CRISPR interference (CRISPRi) [91]. Alternatively, by fusing with a transcriptional activator (e.g., VP64), dCas9 can upregulate expression via CRISPR activation (CRISPRa) [92].
Base editors (BEs) and prime editors (PEs) are newly emerging genome-editing tools (Fig. 1C) [93, 94]. BEs are formed by fusing a nickase Cas9 (nCas9) to different deaminases to directly edit a single base pair of a gene without the need for DNA cleavage [95], which aim to correct point mutations in single-nucleotide variants (SNVs). There are two established classes of BEs: Cytosine BEs (CBEs) that enables a C to T transversion and Adenine BEs (ABEs) that enable an A to G transversion [95, 96]. PEs are made by fusing a Cas9 to an engineered reverse transcriptase. Compared to BEs, PEs can copy genetic information from a prime editing guide RNA (pegRNA) into a specific target genomic locus, leading to precise modification of all 12 possible classes of point mutations in SNVs, as well as small insertion/deletion mutations [97].
CRISPR/Cas12 system
Cas12 is a versatile protein that has been used as an alternative DNA endonuclease to Cas9 for gene editing. Cas12 can be guided by its crRNA to recognize the target DNA strand with PAM sequences [63]. Upon PAM recognition, Cas12 cleaves both target and non-target DNA strands via its RuvC domain and generates a staggered double-stranded break beside the PAM sequence [63]. The Cas12 protein family contains various subtypes including Cas12a (formerly known as Cpf1), Cas12b, Cas12c, Cas12d (formerly known as CasY), Cas12e (formerly known as CasX), Cas12f, Cas12g, Cas12h, Cas12i, and Cas12j (formerly known as Cas14) [63,64,65,66,67,68,69,70,71,72,73,74,75,76,77]. Distinct types of identified natural Cas12 orthologs have broader PAM recognition sites, and several Cas12a variants with weakened PAM constraints have also been developed (Table 1). CRISPR/Cas12 system is also considered as an attractive type of the CRISPR/Cas family for genome editing. Moreover, since Cas12 not only can cleave both double-strand DNA and single-strand DNA via its RuvC domain but also have trans-cleavage activity on [98], CRISPR/Cas12 system has been successfully employed for rapid and sensitive nucleic acid detection [99, 131]. Previous studies reported that Cx30 knockout mice had severe hearing loss along with a 90% reduction in Cx26 [132], while another Cx30 knockout mouse model showed normal hearing with almost half of Cx26 preserved [133]. These studies indicated that Cx30 appeared to be dispensable for cochlear functions and GJB6 might not be associated with HL. Recently, Chen et al. used CRISPR/Cas9 technology to establish a new Cx30 knockout mouse model (Cx30−/−), which retained approximately 70% of Cx26 [134]. They found that the Cx30−/− mouse models showed mild full-frequency HL in 1, 3, and 6 months. Moreover, Cx30 deficiency reduced the production of endocochlear potential and the release of ATP, which may be responsible for the induction of HL. This study suggested that Cx30 may play an important rather than redundant role in hearing development. The pathogenic variants in KCNQ4 cause DFNA2. However, the understanding of genotype–phenotype correlations between KCNQ4 and hearing is limited. KCNQ4 gene encodes a voltage-gated potassium channel (Kv7.4) that is highly expressed in the basolateral membrane of outer hair cells and mediates ionic homeostasis, in which the pathogenic mutations can lead to DFNA2 [135]. To understand the genotype–phenotype correlation between a novel KCNQ4 mutation p.G228D and hearing, Cui et al. used CRISPR/Cas9-mediated gene knock-in technique to generate the Kcnq4G229D mouse model [136]. Kcnq4G229D mice showed progressive high-frequency HL with progressive degeneration of outer hair cells in the basal turn, which could recapitulate the DFNA2 phenotype of patients and contribute a better understanding of the genotype–phenotype correlation [136].
Myosin VI (MYO6) is also vital for proper differentiation and development of stereocilia bundles. Pathogenic variants in the MYO6 gene can cause either DFNA22 or DFNB37 [137, 138]. The Myo6 c.1325G > A mutation mouse model was generated by HDR of CRISPR/Cas9 mediated DSB, which could mimic the p.C442Y variant found in human DFNA22 patients [139]. The results of immunohistochemistry experiments indicate that auditory hair cells and degeneration of stereocilia bundles on vestibular hair cells may underlie progressive HL and vestibular dysfunction of patients harboring MYO6 p.C442Y mutations [139]. Mechanoelectrical transduction plays a key role in transmitting sensory information, and the abnormality of inner ear can affect the perception of sound. Transmembrane channel-like 1 (TMC1) gene encodes a pore-forming subunit of mechanosensory transduction channels in inner hair cells, which is important for hearing function, and TMC1 mutations are associated with DFNA36 and DFNB7/11 [140, 141]. However, a lack of appropriate mouse models of recessive DFNB7/11 HL bearing a human TMC1 mutation limited the development of gene therapy for the type of genetic HL. To establish mouse models harboring recessive Tmc1 mutations, CRISPR/Cas9 technology was used to specifically introduce an A > C substitution, which resulted in a p.N193I point mutation of Tmc1 protein that is homologous to the p.N199I mutation of human TMC1 protein [142]. The results of hearing test showed that the Tmc1N193I/+ mice had normal hearing thresholds, while the Tmc1N193I/N193I mice were profoundly deaf with fewer outer hair cells at the cochlea middle and base. Moreover, viral gene therapy (AAV9-PHP.B-CB6-hTMC1 + WPRE) can restore auditory function in mice, further demonstrating the crucial role of TMC1 protein in cochlear hair cells [142]. Cadherin 23 (CDH23) gene encoding CDH23 protein that is necessary for intercellular adhesion. Different mutations in the CDH23 gene have been related to either syndromic (USH1D) or non-syndromic (DFNB12) forms of deafness in humans. Zhao et al. generated two novel mouse models with Cdh23 mutations in the CBA/CaJ mice, including Cdh23V2J2/V2J2, which consists of a single base pair deletion (c.235delG), and Cdh23erl2/erl2, which consists of a missense mutation (c.208T > C) [143]. The two mutant mice exhibit a broad frequency of hearing impairment. Structural abnormalities in the stereocilia were observed in the cochlear hair cells of the two mutant mice. The two novel mutant mouse models provide novel data for us to better understand the genotype–phenotype correlation of mutant Cdh23 alleles. MYO3A encoding myosin IIIa is expressed in cochlear hair cells and retinal cells, and MYO3A mutations are responsible for human DFNB30 [144]. To establish an animal model for studying DFNB30-type deafness and investigate its mechanism, Li et al. generated a mouse model of Myo3a mutation (c.410A > G) using the CRISPR/Cas9 tools [145]. The results show that Myo3a is essential for normal hearing by maintaining the intact structure of hair cell stereocilia, and loss of Myo3a in mice can cause stereocilium degeneration in inner ear hair cells, which leads to progressive HL [145]. Mutations in the human CIB2 (encodes calcium and integrin-binding protein 2) gene have been associated with DFNB48 and USH1J [146, 147]. To further explore the function of the CIB2 gene in hearing, Wang et al. used the CRISPR/Cas9 technique to establish Cib2 knockout mice [148]. They found that loss of Cib2 in mice abolishes mechanoelectrical transduction currents in auditory hair cells, resulting in HL [148]. In humans, TPRN (encodes the taperin protein) mutations lead to DFNB79 by an unknown mechanism [149]. To determine the role of Tprn in hearing function, Men et al. generated Tprn-null mice by CRISPR/Cas9 technology from a CBA/CaJ background, which could be ideal models of human DFNB79 [150]. Functional assays reveal that loss of Tprn in mice results in the disruption of the stereociliary rootlet, which leads to damage to stereociliary bundles and hearing impairments [150]. ELMO domain-containing 3 (ELMOD3) was identified as a new deafness gene implicated in causing HL in humans [151, 152]. Nevertheless, the specific role of ELMOD3 in auditory function remains to be elucidated. Li et al. used the CRISPR/Cas9 technology to establish an Elmod3 knockout mice line in the C57BL/6 background to investigate the role of Elmod3 in the cochlea and auditory function [153]. Their finding reveals that Elmod3 deficiencies might play roles in the actin cytoskeleton dynamics in cochlear hair cells, relating to hearing impairment [153]. Glutaredoxin domain-containing cysteine-rich protein 2 (GRXCR2) and chloride intracellular channel protein 5 (CLIC5) are both localized at the base of stereocilia and are required for normal hearing in humans and mice. However, the detailed functions of GRXCR2 or CLIC5 in hair cells remain unclear. Using the CRISPR/Cas9 system, Li et al. deleted 60 amino acids near the N-terminus of GRXCR2 that are essential for its interaction with CLIC5 [154]. More importantly, mice harboring this in-frame deletion in Grxcr2 exhibit moderate low-frequency HL and severe high-frequency HL but without significant stereocilia morphogenesis. The study reveals that the interaction between GRXCR2 and CLIC5 is crucial for normal hearing.
Given most genes are expressed in diverse parts of the body including the inner ear and have various physiological functions in addition to the auditory function, mutations in these genes will result in syndromic HL. Rac/Cdc42 guanine nucleotide exchange factor 6 (ARHGEF6) is the X-linked intellectual disability gene, and in some cases, patients carrying ARHGEF6 mutations show sensorineural HL [155]. However, the role of ARHGEF6 in inner ear development and hearing function remains unclear. Zhu et al. established Arhgef6 knockdown mice using the CRISPR/Cas9 technique [156]. The results suggest that ARHGEF6 loss leads to the inhibition of the Rho GTPases CDC42 and RAC1, which causes progressive hair cell loss and subsequent HL [156]. Song et al. characterized a family with deafness-dystonia-optic neuronopathy syndrome, in which the affected members carried a novel hemizygous variation (c.82C > T) in translocase of the inner membrane 8A (TIMM8A) gene [157]. They then generated a mouse line with the hemizygous mutation in the Timm8a1 gene using the CRISPR/Cas9 technology, which bears loss-of-function mutation in Timm8a1. The results suggest that the Timm8a1 mutation in mice leads to an abnormal mitochondrial structure in the brain, correlating with hearing and memory impairment.
Zebrafish models
Since the inner ear of Zebrafish has similar functions to that of humans, it has become an excellent model for exploring the development of the inner ear. CRISPR/Cas9 system has revolutionized the ability to generate zebrafish mutants, and previous studies have been discussed by Vona et al. in detail [158]. Mafb is a component of the Maf transcription factor family, which participates in multiple biological processes, while its role in inner-ear development remains unclear [159, 160]. To address the specific mechanism of how mafba (homologous to mammalian mafb) mutants cause inner-ear defects, Chen et al. constructed a zebrafish mafba knockout (mafba−/−) model using CRISPR/Cas9 technology [160]. Loss of mafba impairs inner-ear development of zebrafish embryos. The inner-ear deficiencies in mafba−/− embryos are related to cell apoptosis and G0/G1 cell cycle arrest caused by DNA damage. The study provides novel insights into the pathogenic mechanisms of mafba, and mafba−/− zebrafish could be an ideal model for develo** novel therapeutic approaches for inner-ear defects [160]. CATSHL (camptodactyly, tall stature, and HL) syndrome is caused by loss-of-function mutations in the fibroblast growth factor receptors 3 (FGFR3) gene [161]. However, the pathogenesis of these phenotypes remains poorly understood and there are no effective therapies. Based on CRISPR/Cas9 technology, Sun et al. generated fgfr3 knockout zebrafish exhibiting craniofacial bone malformation with microcephaly and delayed closure of cranial sutures, chondroma-like lesion, and abnormal development of auditory sensory organs, which partially resemble the clinical features of CATSHL patients [162]. Further experiments showed that loss of fgfr3 upregulates canonical Wnt/β-catenin signaling, and the phenotypes of fgfr3 mutants could be partially alleviated by pharmacologically inhibiting Wnt/β-catenin [162]. The findings provide the zebrafish model for CATSHL syndrome to deepen our understanding of pathogenetic mechanisms of the FGFR3 mutantions and explore the possible therapies.
Zebrafish is also widely used to investigate candidate genes for human genetic HL. Recently, based on CRISPR/Cas9 system, Gou et al. proposed a novel multiplex genome editing strategy that could simultaneously target five genes and rapidly generate individual homozygous zebrafish mutants for functional genetics research [163]. According to the results of the C-start assay and the AMI-43 staining, a new gene mutation (tmem183a) was identified to be associated with HL, which may affect the normal state of mechanoelectrical transduction channels in hair cells [163]. By linkage analysis and exome sequencing, Rodrigo et al. identified a rare missense variant (c.2810C > G) in the NCOA3 gene as the best candidate to be causative of bilateral, progressive, non-syndromic, and sensorineural HL in a large Brazilian family with autosomal dominant inheritance [164]. CRISPR/Cas9 system was used to generate a stable homozygous zebrafish mutant line (ncoa3−/−) that showed subtle and abnormal skeletal behavior (cartilage behavior and bone density) in the ears, suggesting that skeletal abnormalities might be responsible for the pathogenesis of NCOA3 mutations [164]. By genome-wide linkage analysis and whole exome sequencing, a heterozygous variant (c.547C > G) in THOC1 was identified as the probable cause of the late-onset, progressive, non-syndromic HL that segregates as an autosomal dominant condition in a large family [165]. The Thoc1 knockout zebrafish generated by the gRNA-Cas9 system lacks the C-startle response, indicating hearing impairment. Functional studies showed that Thoc1 deficiency promotes the expression of pro-apoptotic genes in the p53 signaling pathway that induces hair cell apoptosis in zebrafish, leading to late-onset progressive HL.
Other animal models
Compared to rodent animals, pigs are more similar to humans in the otic structure and function, thus, the pig model has become an important tool for otology and audiology research [166, 167]. Through whole-exome sequencing, oxysterol binding protein like 2 (OSBPL2) was identified as a novel DFNA-causal gene in a large affected Chinese family [177,178,179]. The zebrafish model can also undergo genetic modifications for the research of genetic HL with several advantages, such as a much faster life cycle than that of the mouse and the transparency of the inner ear, which facilitate their applications for hearing-related research [180]. Nevertheless, the significant genetic disparities between zebrafish and mammals make the zebrafish-related certain data for the purpose of understanding human HL challenging [179]. The pig is the closest species to humans in evolution except for primates, and the structure of its auditory organ is highly similar to that of humans, which makes the pig very suitable for the model of auditory studies [181]. As a non-human primate, rhesus macaque is commonly used to study sensory and perceptual processing [182]. However, these larger mammalian models (pig and rhesus macaque) bear inter-species differences to their human counterparts, which may compromise the relevance of the gathered data [177]. Despite all this, these cell and animal models generated by CRISPR/Cas-based technique provide good platforms to further study the molecular mechanism of genetic HL and play a role in the identification of possible HL-associated mutations, which might promise to revolutionize curative approaches to hearing restoration and improvement.
CRISPR/Cas in the treatment of genetic hearing loss
CRISPR/Cas9 technology, as a precise yet versatile approach, is supposed to make accurate modifications and overcome the heterogeneity in genetic HL. Therapeutic approaches targeting genetic HL are based on an increasingly detailed knowledge of the biological and molecular mechanisms underlying auditory defects. Here, this section details the recent CRISPR/Cas9-mediated treatments that were applied to genetic HL (Table 3).
NHEJ-based treatment
NHEJ as the major DSB repair mechanism tends to lead to the formation of small insertion or deletion mutations [183]. Therefore, the common use of CRISPR/Cas9 in the treatment of dominant genetic HL is the direct silencing of dominant negative pathogenic mutations via the NHEJ pathway. As mentioned above, the KCNQ4 mutations are associated with DFNA2.To explore whether in vivo gene editing is applicable to the treatment of DFNA2, a Kcnq4W276S/+ mouse model that exhibited progressive HL accompanied with outer hair cell degeneration was created and used as the mouse model of DFNA2 [184]. To disrupt the dominant-negative allele in Kcnq4, CRISPR/SpCas9-based gene therapy was applied to prevent progressive HL in the Kcnq4W276S/+ mouse models. The results suggest that in vivo gene editing targeting outer hair cells significantly improved auditory thresholds in auditory brainstem response (ABR) and distortion-product otoacoustic emission (DPOAE) [184]. Another research reported that the treatment of SaCas9-KKH-sgRNA-g3 agents targeting the Kcnq4G229D allele could significantly improve the auditory function of the Kcnq4+/G229D mouse models [185]. As mentioned previously, pathogenic variants in the MYO6 gene can cause DFNA22 [137, 138]. In a recent study, Xue et al. explored a possible treatment approach for the dominant inheritance of MYO6 gene mutations (p.C442Y) in Myo6WT/C442Y mouse models [186]. The CRISPR-SaCas9 therapeutic system was delivered into Myo6WT/C442Y mouse ears at P0–2, where it specifically knocked out the Myo6C442Y mutant allele. Consequently, specific disruption of Myo6C442Y alleles results in an overall hearing improvement in the treated Myo6WT/C442Y mice, including shorter latencies of ABR wave I, lower DPOAE thresholds, increased cell survival rates, more regular hair bundle morphology, and recovery of inward calcium levels [186].
Dominant genetic HL involves a heterozygous mutation that results in a distinct mutant allele and an unaffected wild-type allele. To achieve allele-specific CRISPR/Cas9 binding, different sgRNAs or novel PAM sites are used to distinguish the mutant allele from the wild-type allele. As mentioned previously, TMC1 mutations are also associated with DFNA36 [140]. As a model for DFNA36, Beethoven mice harbor a point mutation (c.1253T > A, namely Bth mutation) in the Tmc1 gene, which is identical to the TMC1 p.M412K mutation of human DFNA36 patients [187]. Injection of Cas9-sgRNA-lipid complexes targeting the Tmc1Bth/WT allele into the cochlea of neonatal Beethoven mice substantially reduced progressive HL, with higher hair cell survival rates and lower ABR thresholds [112]. To expand the targeting range, variants of Cas9 have also been engineered to recognize different PAM sites [43, 52]. It has been reported that the PAM sequence itself might distinguish mutant from wild-type alleles [188]. Recently, Bence et al. screened 14 Cas9/sgRNA combinations for specific and efficient disruption of a nucleotide substitution in TMC1 that causes DFNA36 [189]. A PAM variant of SaCas9 (SaCas9-KKH) was identified to selectively and efficiently disrupt the mutant allele, but not affect the wild-type Tmc1/TMC1 allele, in Beethoven mice and a DFNA36 human HAP1 cell line. Moreover, treated Beethoven mice exhibited normal or near-normal thresholds at 5–8 kHz at 24 weeks, while untreated mice were profoundly deaf. This study suggested that the PAM-selective strategy has the potential and broad application to selectively target dominant human mutations [189]. Additionally, Wu et al. used the synthetic AAV9-PHP.B dual vectors to deliver CRISPR-Cas9 systems into the inner ear of Beethoven mice, which could effectively and selectively target the Tmc1Bth/WT allele, thus rescuing hair cell survival and preserving the hearing function of Beethoven mice [190].
Recombinant protocadherin 15 (PCDH15) is one of two constituents that form the tip junction to gate the mechano-transduction channel in hair cells [191]. Homozygous Pcdh15av−3 J mice with deficient Pcdh15 are used as the mouse model of DFNB23, which show profound congenital HL and vestibular dysfunction [192]. Based on the CRISPR/Cas9-induced precise cleavage, the NHEJ-mediated frame-restoration strategy was reported to partially correct frameshift mutations in the postmitotic cells of an organ, which is helpful to improve auditory responses and restore balance function in the Pcdh15av−3J mice [192].
HDR-based treatment
CRISPR/Cas9-mediated HDR-based therapies have the potential to cure many genetic diseases because this class of therapeutics can achieve arbitrary base changes as well as the insertion or deletion of designated nucleotides [183]. The Cdh23ahl allele refers to a synonymous single nucleotide polymorphism influencing the last nucleotide of exon 7 of the Cdh23 gene, resulting in an increased frequency of exon 7 skip**, which predisposes inbred mouse strains carrying this allele to HL [193]. C57BL/6NTac mice strains are generated in a single inbred strain background (Cdh23alh/ahl) that exhibits a high-frequency HL at 3–6 months. Joffrey et al. used targeted CRISPR/Cas9-mediated HDR to successfully repair the Cdh23ahl allele repair in C57BL/6NTac zygotes [194]. For their experimental design, in-vitro transcribed offset-nicking Cas9 (D10A) nickase mRNA with two paired sgRNA and a single-stranded oligonucleotide (ssODN) as a donor template were co-injected into one-cell-stage mouse embryos. Their sequencing data suggest the approach is highly specific, with no lesions identified at any of the predicted off-target sites. Importantly, the authors demonstrated that the repair Cdh23ahl/753A>G mice exhibited normal hearing function, without either the progressive HL or sensory cell degeneration phenotypes common to the Cdh23ahl/ahl mice [194].
Solute carrier family 26, member 4 (SLC26A4) gene encoding the multifunctional anion exchanger pendrin is abundantly expressed in the inner ear, thyroid, and kidney. SLC26A4 mutations are one of the most frequent causative factors of congenital HL, including Pendred syndrome and DFNB4, and the splicing mutation (c.919-2A > G) in intron 7 of SLC26A4 is a hotspot mutation among Asian populations [195]. Candidate SaCas9-specific sgRNAs were designed to target c.919-2A within the Slc26a4 locus [13]. In vitro experiments show that the introduction of a plasmid co-expressing SaCas9 and engineered sgRNAs would suffice to induce HDR-mediated genome modification of the c.919-2A splicing site in the Slc26a4 gene. Importantly, ex vivo experiments in primary mouse embryonic fibroblasts reveal that CRISPR/Cas9 system can be used to precisely edit the causative gene of HL [13].
In a current study, based on a pig model that carries the c.740T > C mutation in the MITF gene with an inheritance pattern and clinical pathology that mimics Waardenburg syndrome 2A (WS2A), Yao et al. performed precise gene correction with CRISPR/Cas9-mediated HDR therapy [15]. Using ssODN and plasmid DNA with long homology arms as donor DNAs, precise correction of the c.740T > C point mutation was achieved, and the corrected cells were then used as the donor cell for somatic cell nuclear transfer to produce piglets. The results showed that the CRISPR/Cas9-mediated HDR therapy successfully rescued the anophthalmia and HL phenotype of WS2A in pig models [15].
HMEJ-based treatment
Recently, a homology-mediated end joining (HMEJ)-based strategy has been devised to generate animal models and for targeted gene therapies [196]. This strategy is based on CRISPR/Cas9-mediated cleavage of both transgene donor vector that contains guide RNA target sites and ∼800 bp of homology arms, and the targeted genome [196]. Kelch-like family member 18 (KLHL18) gene, encoding a 574 amino acid protein with a BTB/POZ domain, a BACK domain, and six Kelch repeats, play roles in extracellular communication, cell morphology, and actin binding [197]. Homozygous Klhl18lowf mice were used as a model of recessive genetic HL. Klhl18lowf mutant allele contains a missense point mutation of the Klhl18 gene that leads to the dysfunction of inner hair cells [197]. However, the Klhl18lowf mutant allele cannot be corrected using current base-editing strategies [14]. Using the HMEJ-based strategy, the Klhl18lowf mutation sites in inner hair cells in vivo could be accurately corrected [14]. In the treated cochleae of homozygous mutants, a part of the inner hair cells in the apical and middle turns exhibited normal or near normal stereocilia bundles, and the sustained inner hair cell exocytosis after 200 ms depolarization pulses were restored. Moreover, the HMEJ-based therapies significantly improve the auditory function of Klhl18lowf mice up to 6 months after treatment [14]. This study shows promise for further development of HMEJ-based strategies for the repair of point mutations that cause genetic HL as well as other human genetic diseases.
Base editor-based treatment
Base editors can provide therapeutic restoration of gene function by efficiently and permanently correcting pathogenic mutations without disrupting the target gene [198]. Recently, in vivo base editing by CBE (SpCas9-based AID-BE4max) has been used to genetically correct the Tmc1 c.A545G point mutation in Baringo mice [114]. The Baringo (Tmc1Y182C/Y182C) mouse is a mouse model of recessive HL that harbors a recessive loss-of-function c.A545G mutation in Tmc1 that substitutes p.Y182C and shows profound deafness by 4 weeks of age [199]. In vivo delivery of dual AID-BE3.9max AAVs resulted in ~ 51% base editing efficiency in hair cells in Baringo mice, which preserved the stereocilia morphology of inner hair cells and restored hair cell sensory transduction current [114]. However, the results of ABR tests showed that CBE-mediated gene therapy partially and transiently rescued the auditory function of Baringo mice, which might arise from incomplete base editing [114]. Therefore, further improvements in the base editor are needed to enhance editing efficiency for the permanent recovery of auditory function. In addition, Gao et al. summarized a list of HL-associated gene variants that is base-editable with a 5’-NGG/NG-3’ PAM positioned appropriately [179], which will inspire more research on base editor-based treatment for genetic HL.
CRISPR/Cas13-based treatment
Since DNA editing might induce off-target mutations in the genome, its therapeutic and clinical applications are limited. RNA editing technologies only modify the expression of target RNA without affecting the DNA, providing potential therapeutic approaches for genetic HL. As a novel RNA targeting tool, CRISPR/Cas13 system has been used to explore the potential therapeutic effects for genetic HL [201], HDR-based treatment demonstrates its therapeutic potential via precisely correcting the mutation in HL-associated genes. Moreover, newly-developed base editing tools (e.g., CBEs) and RNA targeting tools (CRISPR/Cas13 system) have also been successfully utilized for the treatment of genetic HL in animal models. In addition, other new technologies, including PEs and CRISPR/Cas12 systems, may provide more opportunities to improve the efficiency and effectiveness of gene therapies. Taken together, these findings make us believe that the use of CRISPR/Cas-mediated genome editing technologies will increase our knowledge of genetic HL processes and contribute to the development of their treatment in the near future.
Challenges and perspectives
Although CRISPR/Cas-mediated gene editing has been reported to have the potential for the treatment of genetic HL in many studies, there is still a long way to go before its clinical application.
Editing efficiency and safety of CRISPR/Cas-mediated therapy. The efficiency of CRISPR/Cas-mediated in vivo gene editing is likely to be key to sustained hearing recovery. The editing efficiency may be influenced by the type of Cas, the design of the sgRNA, the delivery method, the disease model, and other factors [34]. It is reported that the application of fully chemically modified sgRNAs with improved stability contributes to increasing the editing efficiency of CRISPR/Cas-based therapeutics [202]. Novel delivery modalities, including viral vectors, liposomes, and nanoparticles, have been applied to improve transduction efficiencies and safety and reviewed by Philipp et al. [203]. Moreover, off-target effects of the CRISPR/Cas technique remain a major concern, which might reduce the specificity of gene editing, possibly leading to unwanted mutations and potential toxicity. To reduce the off-target effects and enhance the editing specificity, the Cas9 proteins have been modified to alter PAM preferences or enhance target DNA recognition [43, 45, 48, 52]. Moreover, the immunogenicity of Cas proteins is another potential limitation to their clinical application. Theoretically, transient delivering the appropriate number of Cas proteins might help to reduce immunogenicity-induced immune responses [204, 205]. Regardless of the success rate of the CRISPR/Cas gene editing, in vivo studies of CRISPR/Cas treatment are reporting improvements in auditory function [14, 113, 114, 184, 189, 192, 194, 200], suggesting its positive impacts on a patient’s quality of life.
Specific delivery towards the inner ear Since most HL-related genes are uniquely expressed in specific inner-ear cell types and play roles in specific inner-ear environments, the specific delivery towards the inner ear is of importance. The inner ear-specific delivery facilitates CRISPR/Cas genome-editing agents to reach target inner ear hair cells. Several approaches of inner ear-specific delivery have been established, including perilymph delivery via a cochleostomy, canalostomy, or trans-round window membrane; as well as endolymph delivery via cochleostomy of the scala media space [206]. Although the cochleostomy-based approach promotes the distribution of therapeutic agents, the inevitable cochlear damage makes it clinically unfeasible [206]. The round window membrane injection carries a risk of perilymphatic leakage [1]. The canalostomy-based approach is relatively safe, and it is reported to result in robust transduction of hair cells throughout the cochlea [207]. Moreover, the cochlear aqueduct makes possible the leakage of the therapeutic agents from the perilymph into the cerebrospinal fluid and the vasculature [208], which may cause off-target editing of the brain or whole body, leading to unintended outcomes. Therefore, further investigations are needed to evaluate the security of inner ear-specific delivery.
The regeneration of auditory hair cells CRISPR/Cas-mediated therapy can correct mutation genes to prevent cell death and rescue dying cells. However, the loss of auditory hair cells still limits the recovery of the earing threshold. The iPSCs derived from patients provide potential cell sources for replenishing the hair cells that are lost before therapeutic intervention. The results of the combination of iPSC technology and CRISPR/Cas technology currently underway show promising therapeutic prospects for genetic HL [122,123,124]. However, regenerated hair cells are needed to establish appropriate mechanical coupling with the surrounding support cells (e.g., fibrocytes, epithelial cells, mesenchyme cells) and innervating neurons to reproduce cochlear tonotopy [209]. The approaches and technologies of tissue engineering, including biomedical materials and bioreactors, may help to accelerate the development of inner ear organoids.
The appropriate personalized CRISPR/Cas-mediated therapy is needed Since the heterogeneity of HL-related genes with diverse protein functions and different spatiotemporal expressions, the appropriate personalized CRISPR/Cas-mediated therapy for each type of HL-related gene still needs more discussion [210]. It is necessary to further investigate more details of each HL-related gene variant, including the age at onset, the natural course, the genotype, the pathophysiological mechanism, and the target cell populations. Such knowledge raises hopes for the possibility of future personalized CRISPR/Cas-mediated therapeutic Intervention with appropriate operations, specific therapeutic agents, and the optimal temporal window.
Despite these limitations, CRISPR/Cas-mediated therapies remain a tempting strategy in genetic HL research because they are a promising option to restore or improve hearing. More and more researchers from multidisciplinary fields put their efforts together to accelerate the development of CRISPR/Cas-mediated gene therapy. It is worth expecting that the ultimate goal in the clinical application of the CRISPR/Cas9 technique for the treatment of genetic HL may not be far away.
Conclusions
CRISPR/Cas is promoting a broad range of innovative applications from basic biology to biotechnology and medical interventions. Its favorable characteristics (e.g., easy use and high efficiency) distinguish it from other existing genome editing technologies, and its great advances in hearing research are foreseeable. Different types of genetic hearing diseases are likely to be one of the ideal targets of CRISPR/Cas-mediated therapy. With CRISPR/Cas genome editing tool, various disease models of genetic HL have been established to further study the mechanism of these diseases and explore the way to restore impaired hearing. Besides, increasing in vivo studies demonstrate that CRISPR/Cas-mediated therapy could be a promising approach to tackling these debilitating diseases. However, there are still many challenges before its clinical application, such as editing efficiency, off-target effect, immunogenicity, and so on. Given the unremitting efforts of the researchers and the rapid progress in the field, we fully anticipate that these challenges will be overcome in the future, thus potentiating novel therapeutic strategies for genetic HL.
Availability of data and materials
Not applicable.
References
Omichi R, Shibata SB, Morton CC, Smith RJH. Gene therapy for hearing loss. Hum Mol Genet. 2019;28(R1):R65-r79.
Yang T, Guo L, Wang L, Yu X. Diagnosis, intervention, and prevention of genetic hearing loss. Adv Exp Med Biol. 2019;1130:73–92.
Lieu JEC, Kenna M, Anne S, Davidson L. Hearing loss in children: a review. JAMA. 2020;324(21):2195–205.
D'Haese P, Rompaey VV, Bodt M, Heyning P. The need to increase awareness and access to cochlear implantation: advances in aural rehabilitation; 2019.
Bibikova M, Golic M, Golic KG, Carroll D. Targeted chromosomal cleavage and mutagenesis in Drosophila using zinc-finger nucleases. Genetics. 2002;161(3):1169–75.
Christian M, Cermak T, Doyle EL, Schmidt C, Zhang F, Hummel A, et al. Targeting DNA double-strand breaks with TAL effector nucleases. Genetics. 2010;186(2):757–61.
Wood AJ, Lo TW, Zeitler B, Pickle CS, Ralston EJ, Lee AH, et al. Targeted genome editing across species using ZFNs and TALENs. Sci. 2011;333(6040):307.
Hockemeyer D, Jaenisch R. Induced pluripotent stem cells meet genome editing. Cell Stem Cell. 2016;18(5):573–86.
Wiles MV, Qin W, Cheng AW, Wang H. CRISPR-Cas9-mediated genome editing and guide RNA design. Mamm Genome. 2015;26(9–10):501–10.
Stojkovic M, Han D, Jeong M, Stojkovic P, Stankovic KM. Human induced pluripotent stem cells and CRISPR/Cas-mediated targeted genome editing: platforms to tackle sensorineural hearing loss. Stem Cells. 2021;39(6):673–96.
**ek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Sci. 2012;337(6096):816–21.
Kim H, Kim JS. A guide to genome engineering with programmable nucleases. Nat Rev Genet. 2014;15(5):321–34.
Ryu N, Kim MA, Choi DG, Kim YR, Sonn JK, Lee KY, et al. CRISPR/Cas9-mediated genome editing of splicing mutation causing congenital hearing loss. Gene. 2019;703:83–90.
Gu X, Hu X, Wang D, Xu Z, Wang F, Li D, et al. Treatment of autosomal recessive hearing loss via in vivo CRISPR/Cas9-mediated optimized homology-directed repair in mice. Cell Res. 2022;32(7):699–702.
Yao J, Wang Y, Cao C, Song R, Bi D, Zhang H, et al. CRISPR/Cas9-mediated correction of MITF homozygous point mutation in a Waardenburg syndrome 2A pig model. Mol Ther Nucleic Acids. 2021;24:986–99.
Zou B, Mittal R, Grati M, Lu Z, Shu Y, Tao Y, et al. The application of genome editing in studying hearing loss. Hear Res. 2015;327:102–8.
Ishino Y, Shinagawa H, Makino K, Amemura M, Nakata A. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J Bacteriol. 1987;169(12):5429–33.
Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, et al. Multiplex genome engineering using CRISPR/Cas systems. Sci. 2013;339(6121):819–23.
Mali P, Aach J, Stranges PB, Esvelt KM, Moosburner M, Kosuri S, et al. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat Biotechnol. 2013;31(9):833–8.
Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, et al. RNA-guided human genome engineering via Cas9. Sci. 2013;339(6121):823–6.
Makarova KS, Haft DH, Barrangou R, Brouns SJ, Charpentier E, Horvath P, et al. Evolution and classification of the CRISPR-Cas systems. Nat Rev Microbiol. 2011;9(6):467–77.
Koonin EV, Makarova KS. Origins and evolution of CRISPR-Cas systems. Philos Trans R Soc Lond B Biol Sci. 2019;374(1772):20180087.
Brouns SJ, Jore MM, Lundgren M, Westra ER, Slijkhuis RJ, Snijders AP, et al. Small CRISPR RNAs guide antiviral defense in prokaryotes. Sci. 2008;321(5891):960–4.
Makarova KS, Wolf YI, Alkhnbashi OS, Costa F, Shah SA, Saunders SJ, et al. An updated evolutionary classification of CRISPR-Cas systems. Nat Rev Microbiol. 2015;13(11):722–36.
Beloglazova NV, Goryacheva IY, Mikhirev DA, de Saeger S, Niessner R, Knopp D. New immunochemically-based field test for monitoring benzo[a]pyrene in aqueous samples. Anal Sci. 2008;24(12):1613–7.
Marraffini LA, Sontheimer EJ. Invasive DNA, chopped and in the CRISPR. Structure. 2009;17(6):786–8.
Sinkunas T, Gasiunas G, Fremaux C, Barrangou R, Horvath P, Siksnys V. Cas3 is a single-stranded DNA nuclease and ATP-dependent helicase in the CRISPR/Cas immune system. EMBO J. 2011;30(7):1335–42.
Sapranauskas R, Gasiunas G, Fremaux C, Barrangou R, Horvath P, Siksnys V. The Streptococcus thermophilus CRISPR/Cas system provides immunity in Escherichia coli. Nucleic Acids Res. 2011;39(21):9275–82.
Liu H, Wang L, Luo Y. Blossom of CRISPR technologies and applications in disease treatment. Synthet Syst Biotechnol. 2018;3(4):217–28.
O’Connell MR. Molecular mechanisms of RNA targeting by Cas13-containing type VI CRISPR-Cas systems. J Mol Biol. 2019;431(1):66–87.
Pickar-Oliver A, Gersbach CA. The next generation of CRISPR-Cas technologies and applications. Nat Rev Mol Cell Biol. 2019;20(8):490–507.
Mojica FJM, Díez-Villaseñor C, García-Martínez J, Almendros C. Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology (Reading). 2009;155(Pt 3):733–40.
Khadempar S, Familghadakchi S, Motlagh RA, Farahani N, Dashtiahangar M, Rezaei H, et al. CRISPR-Cas9 in genome editing: its function and medical applications. J Cell Physiol. 2019;234(5):5751–61.
Peddle CF, MacLaren RE. The application of CRISPR/Cas9 for the treatment of retinal diseases. Yale J Biol Med. 2017;90(4):533–41.
Deveau H, Garneau JE, Moineau S. CRISPR/Cas system and its role in phage-bacteria interactions. Annu Rev Microbiol. 2010;64:475–93.
Makarova KS, Wolf YI, Iranzo J, Shmakov SA, Alkhnbashi OS, Brouns SJJ, et al. Evolutionary classification of CRISPR-Cas systems: a burst of class 2 and derived variants. Nat Rev Microbiol. 2020;18(2):67–83.
Maruyama T, Dougan SK, Truttmann MC, Bilate AM, Ingram JR, Ploegh HL. Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat Biotechnol. 2015;33(5):538–42.
Haapaniemi E, Botla S, Persson J, Schmierer B, Taipale J. CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response. Nat Med. 2018;24(7):927–30.
Zhang F, Gong Z. Regulation of DNA double-strand break repair pathway choice: a new focus on 53BP1. J Zhejiang Univ Sci B. 2021;22(1):38–46.
Mali P, Esvelt KM, Church GM. Cas9 as a versatile tool for engineering biology. Nat Methods. 2013;10(10):957–63.
Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 2013;31(9):827–32.
Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 2013;152(5):1173–83.
Kleinstiver BP, Prew MS, Tsai SQ, Topkar VV, Nguyen NT, Zheng Z, et al. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature. 2015;523(7561):481–5.
Anders C, Bargsten K, **ek M. Structural plasticity of PAM recognition by engineered variants of the RNA-guided endonuclease Cas9. Mol Cell. 2016;61(6):895–902.
Kleinstiver BP, Pattanayak V, Prew MS, Tsai SQ, Nguyen NT, Zheng Z, et al. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature. 2016;529(7587):490–5.
Nishimasu H, Shi X, Ishiguro S, Gao L, Hirano S, Okazaki S, et al. Engineered CRISPR-Cas9 nuclease with expanded targeting space. Sci. 2018;361(6408):1259–62.
Endo M, Mikami M, Endo A, Kaya H, Itoh T, Nishimasu H, et al. Genome editing in plants by engineered CRISPR-Cas9 recognizing NG PAM. Nat Plants. 2019;5(1):14–7.
Slaymaker IM, Gao L, Zetsche B, Scott DA, Yan WX, Zhang F. Rationally engineered Cas9 nucleases with improved specificity. Sci. 2016;351(6268):84–8.
Chen JS, Dagdas YS, Kleinstiver BP, Welch MM, Sousa AA, Harrington LB, et al. Enhanced proofreading governs CRISPR-Cas9 targeting accuracy. Nature. 2017;550(7676):407–10.
Hu JH, Miller SM, Geurts MH, Tang W, Chen L, Sun N, et al. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature. 2018;556(7699):57–63.
Ran FA, Cong L, Yan WX, Scott DA, Gootenberg JS, Kriz AJ, et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520(7546):186–91.
Kleinstiver BP, Prew MS, Tsai SQ, Nguyen NT, Topkar VV, Zheng Z, et al. Broadening the targeting range of Staphylococcus aureus CRISPR-Cas9 by modifying PAM recognition. Nat Biotechnol. 2015;33(12):1293–8.
Steinert J, Schiml S, Fauser F, Puchta H. Highly efficient heritable plant genome engineering using Cas9 orthologues from Streptococcus thermophilus and Staphylococcus aureus. Plant J. 2015;84(6):1295–305.
Chatterjee P, Jakimo N, Jacobson JM. Minimal PAM specificity of a highly similar SpCas9 ortholog. Sci Adv. 2018;4(10): eaau0766.
Richards VP, Palmer SR, Pavinski Bitar PD, Qin X, Weinstock GM, Highlander SK, et al. Phylogenomics and the dynamic genome evolution of the genus Streptococcus. Genome Biol Evol. 2014;6(4):741–53.
Chatterjee P, Lee J, Nip L, Koseki SRT, Tysinger E, Sontheimer EJ, et al. A Cas9 with PAM recognition for adenine dinucleotides. Nat Commun. 2020;11(1):2474.
Kim E, Koo T, Park SW, Kim D, Kim K, Cho HY, et al. In vivo genome editing with a small Cas9 orthologue derived from Campylobacter jejuni. Nat Commun. 2017;8:14500.
Zhang X, Lv S, Luo Z, Hu Y, Peng X, Lv J, et al. MiniCAFE, a CRISPR/Cas9-based compact and potent transcriptional activator, elicits gene expression in vivo. Nucleic Acids Res. 2021;49(7):4171–85.
Esvelt KM, Mali P, Braff JL, Moosburner M, Yaung SJ, Church GM. Orthogonal Cas9 proteins for RNA-guided gene regulation and editing. Nat Methods. 2013;10(11):1116–21.
Edraki A, Mir A, Ibraheim R, Gainetdinov I, Yoon Y, Song CQ, et al. A compact, high-accuracy Cas9 with a dinucleotide PAM for in vivo genome editing. Mol Cell. 2019;73(4):714-26.e4.
Hirano H, Gootenberg JS, Horii T, Abudayyeh OO, Kimura M, Hsu PD, et al. Structure and engineering of Francisella novicida Cas9. Cell. 2016;164(5):950–61.
Gao N, Zhang C, Hu Z, Li M, Wei J, Wang Y, et al. Characterization of Brevibacillus laterosporus Cas9 (BlatCas9) for mammalian genome editing. Front Cell Dev Biol. 2020;8: 583164.
Zetsche B, Gootenberg JS, Abudayyeh OO, Slaymaker IM, Makarova KS, Essletzbichler P, et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell. 2015;163(3):759–71.
Kleinstiver BP, Sousa AA, Walton RT, Tak YE, Hsu JY, Clement K, et al. Engineered CRISPR-Cas12a variants with increased activities and improved targeting ranges for gene, epigenetic and base editing. Nat Biotechnol. 2019;37(3):276–82.
Nishimasu H, Yamano T, Gao L, Zhang F, Ishitani R, Nureki O. Structural basis for the altered PAM recognition by engineered CRISPR-Cpf1. Mol Cell. 2017;67(1):139-47.e2.
Schunder E, Rydzewski K, Grunow R, Heuner K. First indication for a functional CRISPR/Cas system in Francisella tularensis. Int J Med Microbiol. 2013;303(2):51–60.
Zhong Z, Zhang Y, You Q, Tang X, Ren Q, Liu S, et al. Plant genome editing using FnCpf1 and LbCpf1 nucleases at redefined and altered PAM sites. Mol Plant. 2018;11(7):999–1002.
Li S, Zhang X, Wang W, Guo X, Wu Z, Du W, et al. Expanding the scope of CRISPR/Cpf1-mediated genome editing in rice. Mol Plant. 2018;11(7):995–8.
Teng F, Cui T, Feng G, Guo L, Xu K, Gao Q, et al. Repurposing CRISPR-Cas12b for mammalian genome engineering. Cell discovery. 2018;4:63.
Ming M, Ren Q, Pan C, He Y, Zhang Y, Liu S, et al. CRISPR-Cas12b enables efficient plant genome engineering. Nature plants. 2020;6(3):202–8.
Strecker J, Jones S, Koopal B, Schmid-Burgk J, Zetsche B, Gao L, et al. Engineering of CRISPR-Cas12b for human genome editing. Nat Commun. 2019;10(1):212.
Yan WX, Hunnewell P, Alfonse LE, Carte JM, Keston-Smith E, Sothiselvam S, et al. Functionally diverse type V CRISPR-Cas systems. Sci. 2019;363(6422):88–91.
Chen LX, Al-Shayeb B, Méheust R, Li WJ, Doudna JA, Banfield JF. Candidate phyla radiation roizmanbacteria from hot springs have novel and unexpectedly abundant CRISPR-Cas systems. Front Microbiol. 2019;10:928.
Liu JJ, Orlova N, Oakes BL, Ma E, Spinner HB, Baney KLM, et al. CasX enzymes comprise a distinct family of RNA-guided genome editors. Nature. 2019;566(7743):218–23.
Kim DY, Lee JM, Moon SB, Chin HJ, Park S, Lim Y, et al. Efficient CRISPR editing with a hypercompact Cas12f1 and engineered guide RNAs delivered by adeno-associated virus. Nat Biotechnol. 2022;40(1):94–102.
Karvelis T, Bigelyte G, Young JK, Hou Z, Zedaveinyte R, Budre K, et al. PAM recognition by miniature CRISPR-Cas12f nucleases triggers programmable double-stranded DNA target cleavage. Nucleic Acids Res. 2020;48(9):5016–23.
Carabias A, Fuglsang A, Temperini P, Pape T, Sofos N, Stella S, et al. Structure of the mini-RNA-guided endonuclease CRISPR-Cas12j3. Nat Commun. 2021;12(1):4476.
Liu L, Li X, Ma J, Li Z, You L, Wang J, et al. The molecular architecture for RNA-Guided RNA cleavage by Cas13a. Cell. 2017;170(4):714-26.e10.
Abudayyeh OO, Gootenberg JS, Essletzbichler P, Han S, Joung J, Belanto JJ, et al. RNA targeting with CRISPR-Cas13. Nature. 2017;550(7675):280–4.
Liu L, Li X, Wang J, Wang M, Chen P, Yin M, et al. Two distant catalytic sites are responsible for C2c2 RNase activities. Cell. 2017;168(1–2):121-34.e12.
Knott GJ, East-Seletsky A, Cofsky JC, Holton JM, Charles E, O’Connell MR, et al. Guide-bound structures of an RNA-targeting A-cleaving CRISPR-Cas13a enzyme. Nat Struct Mol Biol. 2017;24(10):825–33.
Smargon AA, Cox DBT, Pyzocha NK, Zheng K, Slaymaker IM, Gootenberg JS, et al. Cas13b is a type VI-B CRISPR-associated RNA-guided RNase differentially regulated by accessory proteins Csx27 and Csx28. Mol Cell. 2017;65(4):618-30.e7.
Abudayyeh OO, Gootenberg JS, Franklin B, Koob J, Kellner MJ, Ladha A, et al. A cytosine deaminase for programmable single-base RNA editing. Sci. 2019;365(6451):382–6.
Cox DBT, Gootenberg JS, Abudayyeh OO, Franklin B, Kellner MJ, Joung J, et al. RNA editing with CRISPR-Cas13. Sci. 2017;358(6366):1019–27.
Yan WX, Chong S, Zhang H, Makarova KS, Koonin EV, Cheng DR, et al. Cas13d is a compact RNA-targeting type VI CRISPR effector positively modulated by a WYL-domain-containing accessory protein. Mol Cell. 2018;70(2):327–39.
Konermann S, Lotfy P, Brideau NJ, Oki J, Shokhirev MN, Hsu PD. Transcriptome engineering with RNA-targeting type VI-D CRISPR effectors. Cell. 2018;173(3):665-76.e14.
Xu C, Zhou Y, **ao Q, He B, Geng G, Wang Z, et al. Programmable RNA editing with compact CRISPR-Cas13 systems from uncultivated microbes. Nat Methods. 2021;18(5):499–506.
Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH, et al. Genome-scale CRISPR-mediated control of gene repression and activation. Cell. 2014;159(3):647–61.
Cheng AW, Wang H, Yang H, Shi L, Katz Y, Theunissen TW, et al. Multiplexed activation of endogenous genes by CRISPR-on, an RNA-guided transcriptional activator system. Cell Res. 2013;23(10):1163–71.
Kampmann M. CRISPRi and CRISPRa screens in mammalian cells for precision biology and medicine. ACS Chem Biol. 2018;13(2):406–16.
Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE, et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 2013;154(2):442–51.
Bikard D, Jiang W, Samai P, Hochschild A, Zhang F, Marraffini LA. Programmable repression and activation of bacterial gene expression using an engineered CRISPR-Cas system. Nucleic Acids Res. 2013;41(15):7429–37.
Anzalone AV, Koblan LW, Liu DR. Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat Biotechnol. 2020;38(7):824–44.
Newby GA, Liu DR. In vivo somatic cell base editing and prime editing. Mol Ther. 2021;29(11):3107–24.
Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533(7603):420–4.
Gaudelli NM, Komor AC, Rees HA, Packer MS, Badran AH, Bryson DI, et al. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature. 2017;551(7681):464–71.
Anzalone AV, Randolph PB, Davis JR, Sousa AA, Koblan LW, Levy JM, et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 2019;576(7785):149–57.
Li SY, Cheng QX, Liu JK, Nie XQ, Zhao GP, Wang J. CRISPR-Cas12a has both cis- and trans-cleavage activities on single-stranded DNA. Cell Res. 2018;28(4):491–3.
Qiu M, Zhou XM, Liu L. Improved strategies for CRISPR-Cas12-based nucleic acids detection. J Anal Testing. 2022;6(1):44–52.
Leung RK, Cheng QX, Wu ZL, Khan G, Liu Y, **a HY, et al. CRISPR-Cas12-based nucleic acids detection systems. Methods. 2022;203:276–81.
East-Seletsky A, O’Connell MR, Knight SC, Burstein D, Cate JH, Tjian R, et al. Two distinct RNase activities of CRISPR-C2c2 enable guide-RNA processing and RNA detection. Nature. 2016;538(7624):270–3.
Lin P, Qin S, Pu Q, Wang Z, Wu Q, Gao P, et al. CRISPR-Cas13 inhibitors block RNA editing in bacteria and mammalian cells. Mol Cell. 2020;78(5):850-61.e5.
Ackerman CM, Myhrvold C, Thakku SG, Freije CA, Metsky HC, Yang DK, et al. Massively multiplexed nucleic acid detection with Cas13. Nature. 2020;582(7811):277–82.
Farooq R, Hussain K, Tariq M, Farooq A, Mustafa M. CRISPR/Cas9: targeted genome editing for the treatment of hereditary hearing loss. J Appl Genet. 2020;61(1):51–65.
Van Camp G SR. Hereditary Hearing Loss Homepage. https://hereditaryhearingloss.org.
DiStefano MT, Hemphill SE, Oza AM, Siegert RK, Grant AR, Hughes MY, et al. ClinGen expert clinical validity curation of 164 hearing loss gene-disease pairs. Genet Med. 2019;21(10):2239–47.
Korver AM, Smith RJ, Van Camp G, Schleiss MR, Bitner-Glindzicz MA, Lustig LR, et al. Congenital hearing loss. Nat Rev Dis Primers. 2017;3:16094.
Long C, McAnally JR, Shelton JM, Mireault AA, Bassel-Duby R, Olson EN. Prevention of muscular dystrophy in mice by CRISPR/Cas9-mediated editing of germline DNA. Sci. 2014;345(6201):1184–8.
Yin H, Xue W, Chen S, Bogorad RL, Benedetti E, Grompe M, et al. Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nat Biotechnol. 2014;32(6):551–3.
Zuris JA, Thompson DB, Shu Y, Guilinger JP, Bessen JL, Hu JH, et al. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat Biotechnol. 2015;33(1):73–80.
Rees HA, Komor AC, Yeh WH, Caetano-Lopes J, Warman M, Edge ASB, et al. Improving the DNA specificity and applicability of base editing through protein engineering and protein delivery. Nat Commun. 2017;8:15790.
Gao X, Tao Y, Lamas V, Huang M, Yeh WH, Pan B, et al. Treatment of autosomal dominant hearing loss by in vivo delivery of genome editing agents. Nature. 2018;553(7687):217–21.
**ao Q, Xu Z, Xue Y, Xu C, Han L, Liu Y, et al. Rescue of autosomal dominant hearing loss by in vivo delivery of mini dCas13X-derived RNA base editor. Sci Transl Med. 2022;14(654): eabn0449.
Yeh WH, Shubina-Oleinik O, Levy JM, Pan B, Newby GA, Wornow M, et al. In vivo base editing restores sensory transduction and transiently improves auditory function in a mouse model of recessive deafness. Sci Transl Med. 2020;12(546): eaay9101.
Zhao HB. Hypothesis of K(+)-recycling defect is not a primary deafness mechanism for Cx26 (GJB2) deficiency. Front Mol Neurosci. 2017;10:162.
Jagger DJ, Forge A. Connexins and gap junctions in the inner ear–it’s not just about K+ recycling. Cell Tissue Res. 2015;360(3):633–44.
Maslova EA, Orishchenko KE, Posukh OL. Functional evaluation of a rare variant c.516G>C (p.Trp172Cys) in the GJB2 (Connexin 26) gene associated with nonsyndromic hearing loss. Biomolecules. 2021;11(1):61.
**ng G, Yao J, Liu C, Wei Q, Qian X, Wu L, et al. GPRASP2, a novel causative gene mutated in an X-linked recessive syndromic hearing loss. J Med Genet. 2017;54(6):426–30.
Lu Y, Zhang M, Wei Q, Chen Z, **ng G, Yao J, et al. Disruption of Gprasp2 down-regulates Hedgehog signaling and leads to apoptosis in auditory cells. Biochem Biophys Res Commun. 2021;574:1–7.
Broly M, Polevoda BV, Awayda KM, Tong N, Lentini J, Besnard T, et al. THUMPD1 bi-allelic variants cause loss of tRNA acetylation and a syndromic neurodevelopmental disorder. Am J Hum Genet. 2022;109(4):587–600.
Yap ZY, Efthymiou S, Seiffert S, Vargas Parra K, Lee S, Nasca A, et al. Bi-allelic variants in OGDHL cause a neurodevelopmental spectrum disease featuring epilepsy, hearing loss, visual impairment, and ataxia. Am J Hum Genet. 2021;108(12):2368–84.
Tang ZH, Chen JR, Zheng J, Shi HS, Ding J, Qian XD, et al. Genetic correction of induced pluripotent stem cells from a deaf patient with MYO7A mutation results in morphologic and functional recovery of the derived hair cell-like cells. Stem Cells Transl Med. 2016;5(5):561–71.
Chen JR, Tang ZH, Zheng J, Shi HS, Ding J, Qian XD, et al. Effects of genetic correction on the differentiation of hair cell-like cells from iPSCs with MYO15A mutation. Cell Death Differ. 2016;23(8):1347–57.
Chen C, Guan MX. Genetic correction of TRMU allele restored the mitochondrial dysfunction-induced deficiencies in iPSCs-derived hair cells of hearing-impaired patients. Hum Mol Genet. 2022;31(18):3068–82.
Roccio M, Perny M, Ealy M, Widmer HR, Heller S, Senn P. Molecular characterization and prospective isolation of human fetal cochlear hair cell progenitors. Nat Commun. 2018;9(1):4027.
Liu XZ, Walsh J, Tamagawa Y, Kitamura K, Nishizawa M, Steel KP, et al. Autosomal dominant non-syndromic deafness caused by a mutation in the myosin VIIA gene. Nat Genet. 1997;17(3):268–9.
Weil D, Küssel P, Blanchard S, Lévy G, Levi-Acobas F, Drira M, et al. The autosomal recessive isolated deafness, DFNB2, and the Usher 1B syndrome are allelic defects of the myosin-VIIA gene. Nat Genet. 1997;16(2):191–3.
Miyagawa M, Nishio SY, Hattori M, Moteki H, Kobayashi Y, Sato H, et al. Mutations in the MYO15A gene are a significant cause of nonsyndromic hearing loss: massively parallel DNA sequencing-based analysis. Ann Otol Rhinol Laryngol. 2015;124(Suppl 1):158s-s168.
Woo HM, Park HJ, Baek JI, Park MH, Kim UK, Sagong B, et al. Whole-exome sequencing identifies MYO15A mutations as a cause of autosomal recessive nonsyndromic hearing loss in Korean families. BMC Med Genet. 2013;14:72.
Meng F, Cang X, Peng Y, Li R, Zhang Z, Li F, et al. Biochemical Evidence for a Nuclear Modifier Allele (A10S) in TRMU (Methylaminomethyl-2-thiouridylate-methyltransferase) related to mitochondrial tRNA Modification in the phenotypic manifestation of deafness-associated 12S rRNA mutation. J Biol Chem. 2017;292(7):2881–92.
Adadey SM, Wonkam-Tingang E, Twumasi Aboagye E, Nayo-Gyan DW, Boatemaa Ansong M, Quaye O, et al. Connexin genes variants associated with non-syndromic hearing impairment: a systematic review of the global burden. Life. 2020;10(11):258.
Teubner B, Michel V, Pesch J, Lautermann J, Cohen-Salmon M, Söhl G, et al. Connexin30 (Gjb6)-deficiency causes severe hearing impairment and lack of endocochlear potential. Hum Mol Genet. 2003;12(1):13–21.
Boulay AC, del Castillo FJ, Giraudet F, Hamard G, Giaume C, Petit C, et al. Hearing is normal without connexin30. J Neurosci. 2013;33(2):430–4.
Chen J, Chen P, He B, Gong T, Li Y, Zhang J, et al. Connexin30-deficiency causes mild hearing loss with the reduction of endocochlear potential and ATP release. Front Cell Neurosci. 2021;15: 819194.
Nie L. KCNQ4 mutations associated with nonsyndromic progressive sensorineural hearing loss. Curr Opin Otolaryngol Head Neck Surg. 2008;16(5):441–4.
Cui C, Zhang L, Qian F, Chen Y, Huang B, Wang F, et al. A humanized murine model, demonstrating dominant progressive hearing loss caused by a novel KCNQ4 mutation (p.G228D) from a large Chinese family. Clin Genet. 2022;102(2):149–54.
Ahmed ZM, Morell RJ, Riazuddin S, Gropman A, Shaukat S, Ahmad MM, et al. Mutations of MYO6 are associated with recessive deafness, DFNB37. Am J Hum Genet. 2003;72(5):1315–22.
Melchionda S, Ahituv N, Bisceglia L, Sobe T, Glaser F, Rabionet R, et al. MYO6, the human homologue of the gene responsible for deafness in Snell’s Waltzer mice, is mutated in autosomal dominant nonsyndromic hearing loss. Am J Hum Genet. 2001;69(3):635–40.
Wang J, Shen J, Guo L, Cheng C, Chai R, Shu Y, et al. A humanized mouse model, demonstrating progressive hearing loss caused by MYO6 p.C442Y, is inherited in a semi-dominant pattern. Hear Res. 2019;379:79–88.
Kurima K, Peters LM, Yang Y, Riazuddin S, Ahmed ZM, Naz S, et al. Dominant and recessive deafness caused by mutations of a novel gene, TMC1, required for cochlear hair-cell function. Nat Genet. 2002;30(3):277–84.
Pan B, Akyuz N, Liu XP, Asai Y, Nist-Lund C, Kurima K, et al. TMC1 forms the pore of mechanosensory transduction channels in vertebrate inner ear hair cells. Neuron. 2018;99(4):736–53.
Marcovich I, Baer NK, Shubina-Oleinik O, Eclov R, Beard CW, Holt JR. Optimized AAV vectors for TMC1 gene therapy in a humanized mouse model of DFNB7/11. Biomolecules. 2022;12(7):914.
Zhao T, Ma P, Zhao F, Zheng T, Yan B, Zhang Q, et al. Phenotypic differences in the inner ears of CBA/CaJ and C57BL/6J mice carrying missense and single base pair deletion mutations in the Cdh23 gene. J Neurosci Res. 2021;99(10):2743–58.
Walsh T, Walsh V, Vreugde S, Hertzano R, Shahin H, Haika S, et al. From flies’ eyes to our ears: mutations in a human class III myosin cause progressive nonsyndromic hearing loss DFNB30. Proc Natl Acad Sci U S A. 2002;99(11):7518–23.
Li P, Wen Z, Zhang G, Zhang A, Fu X, Gao J. Knock-in mice with Myo3a Y137C mutation displayed progressive hearing loss and hair cell degeneration in the inner ear. Neural Plast. 2018;2018:4372913.
Riazuddin S, Belyantseva IA, Giese AP, Lee K, Indzhykulian AA, Nandamuri SP, et al. Alterations of the CIB2 calcium- and integrin-binding protein cause Usher syndrome type 1J and nonsyndromic deafness DFNB48. Nat Genet. 2012;44(11):1265–71.
Patel K, Giese AP, Grossheim JM, Hegde RS, Delio M, Samanich J, et al. A Novel C-Terminal CIB2 (calcium and integrin binding protein 2) mutation associated with non-syndromic hearing loss in a hispanic family. PLoS ONE. 2015;10(10): e0133082.
Wang Y, Li J, Yao X, Li W, Du H, Tang M, et al. Loss of CIB2 causes profound hearing loss and abolishes mechanoelectrical transduction in mice. Front Mol Neurosci. 2017;10:401.
Li Y, Pohl E, Boulouiz R, Schraders M, Nürnberg G, Charif M, et al. Mutations in TPRN cause a progressive form of autosomal-recessive nonsyndromic hearing loss. Am J Hum Genet. 2010;86(3):479–84.
Men Y, Li X, Tu H, Zhang A, Fu X, Wang Z, et al. Tprn is essential for the integrity of stereociliary rootlet in cochlear hair cells in mice. Front Med. 2019;13(6):690–704.
Jaworek TJ, Richard EM, Ivanova AA, Giese AP, Choo DI, Khan SN, et al. An alteration in ELMOD3, an Arl2 GTPase-activating protein, is associated with hearing impairment in humans. PLoS Genet. 2013;9(9): e1003774.
Li W, Sun J, Ling J, Li J, He C, Liu Y, et al. ELMOD3, a novel causative gene, associated with human autosomal dominant nonsyndromic and progressive hearing loss. Hum Genet. 2018;137(4):329–42.
Li W, Feng Y, Chen A, Li T, Huang S, Liu J, et al. Elmod3 knockout leads to progressive hearing loss and abnormalities in cochlear hair cell stereocilia. Hum Mol Genet. 2019;28(24):4103–12.
Li J, Liu C, Zhao B. N-Terminus of GRXCR2 interacts with CLIC5 and is essential for auditory perception. Front Cell Dev Biol. 2021;9: 671364.
Kutsche K, Yntema H, Brandt A, Jantke I, Nothwang HG, Orth U, et al. Mutations in ARHGEF6, encoding a guanine nucleotide exchange factor for Rho GTPases, in patients with X-linked mental retardation. Nat Genet. 2000;26(2):247–50.
Zhu C, Cheng C, Wang Y, Muhammad W, Liu S, Zhu W, et al. Loss of ARHGEF6 causes hair cell stereocilia deficits and hearing loss in mice. Front Mol Neurosci. 2018;11:362.
Song P, Guan Y, Chen X, Wu C, Qiao A, Jiang H, et al. Frameshift mutation of Timm8a1 gene in mouse leads to an abnormal mitochondrial structure in the brain, correlating with hearing and memory impairment. J Med Genet. 2021;58(9):619–27.
Vona B, Doll J, Hofrichter MAH, Haaf T, Varshney GK. Small fish, big prospects: using zebrafish to unravel the mechanisms of hereditary hearing loss. Hear Res. 2020;397: 107906.
Moriguchi T, Hamada M, Morito N, Terunuma T, Hasegawa K, Zhang C, et al. MafB is essential for renal development and F4/80 expression in macrophages. Mol Cell Biol. 2006;26(15):5715–27.
Chen X, Huang Y, Gao P, Lv Y, Jia D, Sun K, et al. Knockout of mafba causes inner-ear developmental defects in zebrafish via the impairment of proliferation and differentiation of ionocyte progenitor cells. Biomedicines. 2021;9(11):1699.
Toydemir RM, Brassington AE, Bayrak-Toydemir P, Krakowiak PA, Jorde LB, Whitby FG, et al. A novel mutation in FGFR3 causes camptodactyly, tall stature, and hearing loss (CATSHL) syndrome. Am J Hum Genet. 2006;79(5):935–41.
Sun X, Zhang R, Chen H, Du X, Chen S, Huang J, et al. Fgfr3 mutation disrupts chondrogenesis and bone ossification in zebrafish model mimicking CATSHL syndrome partially via enhanced Wnt/β-catenin signaling. Theranostics. 2020;10(16):7111–30.
Guo S, Gao G, Zhang C, Peng G. Multiplexed genome editing for efficient phenotypic screening in Zebrafish. Vet Sci. 2022;9(2):92.
Salazar-Silva R, Dantas VLG, Alves LU, Batissoco AC, Oiticica J, Lawrence EA, et al. NCOA3 identified as a new candidate to explain autosomal dominant progressive hearing loss. Hum Mol Genet. 2021;29(22):3691–705.
Zhang L, Gao Y, Zhang R, Sun F, Cheng C, Qian F, et al. THOC1 deficiency leads to late-onset nonsyndromic hearing loss through p53-mediated hair cell apoptosis. PLoS Genet. 2020;16(8): e1008953.
Lovell JM, Harper GM. The morphology of the inner ear from the domestic pig (Sus scrofa). J Microsc. 2007;228(Pt 3):345–57.
Guo W, Yi H, Ren L, Chen L, Zhao L, Sun W, et al. The morphology and electrophysiology of the cochlea of the miniature pig. Anat Rec. 2015;298(3):494–500.
**ng G, Yao J, Wu B, Liu T, Wei Q, Liu C, et al. Identification of OSBPL2 as a novel candidate gene for progressive nonsyndromic hearing loss by whole-exome sequencing. Genet Med. 2015;17(3):210–8.
Yao J, Zeng H, Zhang M, Wei Q, Wang Y, Yang H, et al. OSBPL2-disrupted pigs recapitulate dual features of human hearing loss and hypercholesterolaemia. J Genet Genomics. 2019;46(8):379–87.
Lekmine F, Chang CK, Sethakorn N, Das Gupta TK, Salti GI. Role of microphthalmia transcription factor (Mitf) in melanoma differentiation. Biochem Biophys Res Commun. 2007;354(3):830–5.
Song J, Feng Y, Acke FR, Coucke P, Vleminckx K, Dhooge IJ. Hearing loss in Waardenburg syndrome: a systematic review. Clin Genet. 2016;89(4):416–25.
Hai T, Guo W, Yao J, Cao C, Luo A, Qi M, et al. Creation of miniature pig model of human Waardenburg syndrome type 2A by ENU mutagenesis. Hum Genet. 2017;136(11–12):1463–75.
Simmons HA. Age-associated pathology in rhesus macaques (Macaca mulatta). Vet Pathol. 2016;53(2):399–416.
Kremer H, van Wijk E, Märker T, Wolfrum U, Roepman R. Usher syndrome: molecular links of pathogenesis, proteins and pathways. Hum Mol Genet. 2006;15:R262-70.
Ryu J, Statz JP, Chan W, Burch FC, Brigande JV, Kempton B, et al. CRISPR/Cas9 editing of the MYO7A gene in rhesus macaque embryos to generate a primate model of Usher syndrome type 1B. Sci Rep. 2022;12(1):10036.
Connolly K, Gonzalez-Cordero A. Modelling inner ear development and disease using pluripotent stem cells—a pathway to new therapeutic strategies. Dis Model Mech. 2022;15(11): dmm049593.
Durán-Alonso MB, Petković H. Induced pluripotent stem cells, a step** stone to in vitro human models of hearing loss. Cells. 2022;11(20):3331.
Plum A, Winterhager E, Pesch J, Lautermann J, Hallas G, Rosentreter B, et al. Connexin31-deficiency in mice causes transient placental dysmorphogenesis but does not impair hearing and skin differentiation. Dev Biol. 2001;231(2):334–47.
Ding N, Lee S, Lieber-Kotz M, Yang J, Gao X. Advances in genome editing for genetic hearing loss. Adv Drug Deliv Rev. 2021;168:118–33.
Nicolson T. The genetics of hair-cell function in zebrafish. J Neurogenet. 2017;31(3):102–12.
Guo W, Yang SM. Advantages of a miniature pig model in research on human hereditary hearing loss. J Otol. 2015;10(3):105–7.
Rogenmoser L, Kuśmierek P, Archakov D, Rauschecker JP. The blinking eye as a window into tinnitus: a new animal model of tinnitus in the macaque. Hear Res. 2022;420: 108517.
Cox DBT, Platt RJ, Zhang F. Therapeutic genome editing: prospects and challenges. Nat Med. 2015;21(2):121–31.
Noh B, Rim JH, Gopalappa R, Lin H, Kim KM, Kang MJ, et al. In vivo outer hair cell gene editing ameliorates progressive hearing loss in dominant-negative Kcnq4 murine model. Theranostics. 2022;12(5):2465–82.
Cui C, Wang D, Huang B, Wang F, Chen Y, Lv J, et al. Precise detection of CRISPR-Cas9 editing in hair cells in the treatment of autosomal dominant hearing loss. Mol Ther Nucleic Acids. 2022;29:400–12.
Xue Y, Hu X, Wang D, Li D, Li Y, Wang F, et al. Gene editing in a Myo6 semi-dominant mouse model rescues auditory function. Mol Ther. 2022;30(1):105–18.
Zhao Y, Wang D, Zong L, Zhao F, Guan L, Zhang P, et al. A novel DFNA36 mutation in TMC1 orthologous to the Beethoven (Bth) mouse associated with autosomal dominant hearing loss in a Chinese family. PLoS ONE. 2014;9(5): e97064.
Christie KA, Courtney DG, DeDionisio LA, Shern CC, De Majumdar S, Mairs LC, et al. Towards personalised allele-specific CRISPR gene editing to treat autosomal dominant disorders. Sci Rep. 2017;7(1):16174.
György B, Nist-Lund C, Pan B, Asai Y, Karavitaki KD, Kleinstiver BP, et al. Allele-specific gene editing prevents deafness in a model of dominant progressive hearing loss. Nat Med. 2019;25(7):1123–30.
Wu J, Solanes P, Nist-Lund C, Spataro S, Shubina-Oleinik O, Marcovich I, et al. Single and dual vector gene therapy with AAV9-PHPB rescues hearing in Tmc1 mutant mice. Mol Ther. 2021;29(3):973–88.
Ahmed ZM, Riazuddin S, Ahmad J, Bernstein SL, Guo Y, Sabar MF, et al. PCDH15 is expressed in the neurosensory epithelium of the eye and ear and mutant alleles are responsible for both USH1F and DFNB23. Hum Mol Genet. 2003;12(24):3215–23.
Liu L, Zou L, Li K, Hou H, Hu Q, Liu S, et al. Template-independent genome editing in the Pcdh15(av-3j) mouse, a model of human DFNB23 nonsyndromic deafness. Cell Rep. 2022;40(2): 111061.
Johnson KR, Zheng QY, Noben-Trauth K. Strain background effects and genetic modifiers of hearing in mice. Brain Res. 2006;1091(1):79–88.
Mianné J, Chessum L, Kumar S, Aguilar C, Codner G, Hutchison M, et al. Correction of the auditory phenotype in C57BL/6N mice via CRISPR/Cas9-mediated homology directed repair. Genome Med. 2016;8(1):16.
Park HJ, Shaukat S, Liu XZ, Hahn SH, Naz S, Ghosh M, et al. Origins and frequencies of SLC26A4 (PDS) mutations in east and south Asians: global implications for the epidemiology of deafness. J Med Genet. 2003;40(4):242–8.
Yao X, Wang X, Hu X, Liu Z, Liu J, Zhou H, et al. Homology-mediated end joining-based targeted integration using CRISPR/Cas9. Cell Res. 2017;27(6):801–14.
Ingham NJ, Banafshe N, Panganiban C, Crunden JL, Chen J, Lewis MA, et al. Inner hair cell dysfunction in Klhl18 mutant mice leads to low frequency progressive hearing loss. PLoS ONE. 2021;16(10): e0258158.
Rees HA, Liu DR. Base editing: precision chemistry on the genome and transcriptome of living cells. Nat Rev Genet. 2018;19(12):770–88.
Manji SS, Miller KA, Williams LH, Dahl HH. Identification of three novel hearing loss mouse strains with mutations in the Tmc1 gene. Am J Pathol. 2012;180(4):1560–9.
Zheng Z, Li G, Cui C, Wang F, Wang X, Xu Z, et al. Preventing autosomal-dominant hearing loss in Bth mice with CRISPR/CasRx-based RNA editing. Signal Transduct Target Ther. 2022;7(1):79.
Di Stazio M, Foschi N, Athanasakis E, Gasparini P, d’Adamo AP. Systematic analysis of factors that improve homologous direct repair (HDR) efficiency in CRISPR/Cas9 technique. PLoS ONE. 2021;16(3): e0247603.
Mir A, Alterman JF, Hassler MR, Debacker AJ, Hudgens E, Echeverria D, et al. Heavily and fully modified RNAs guide efficient SpyCas9-mediated genome editing. Nat Commun. 2018;9(1):2641.
Niggemann P, György B, Chen ZY. Genome and base editing for genetic hearing loss. Hear Res. 2020;394: 107958.
Chen M, Mao A, Xu M, Weng Q, Mao J, Ji J. CRISPR-Cas9 for cancer therapy: opportunities and challenges. Cancer Lett. 2019;447:48–55.
Maeder ML, Stefanidakis M, Wilson CJ, Baral R, Barrera LA, Bounoutas GS, et al. Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nat Med. 2019;25(2):229–33.
Shibata SB, Yoshimura H, Ranum PT, Goodwin AT, Smith RJH. Intravenous rAAV2/9 injection for murine cochlear gene delivery. Sci Rep. 2017;7(1):9609.
Zhao Y, Zhang L, Wang D, Chen B, Shu Y. Approaches and vectors for efficient cochlear gene transfer in adult mouse models. Biomolecules. 2022;13(1):38.
Salt AN, Hirose K. Communication pathways to and from the inner ear and their contributions to drug delivery. Hear Res. 2018;362:25–37.
Delmaghani S, El-Amraoui A. Inner ear gene therapies take off: current promises and future challenges. J Clin Med. 2020;9(7):2309.
Taiber S, Avraham KB. Genetic therapies for hearing loss: accomplishments and remaining challenges. Neurosci Lett. 2019;713: 134527.
Acknowledgements
We are grateful to some materials in the figures that are produced by BioRender (https://biorender.com).
Funding
This work was supported by West China Hospital, Sichuan University (Nos. 2019HXFH003, ZYJC21027, 2019HXBH079), Chengdu Science and Technology Bureau (No. 2019-YF05-00461-SN), Sichuan University (Nos. GSALK2020021, 2020SCU12049), The Science and Technology Department of Sichuan Province (Nos. 2020YFH0090, 2020YFS0111, 2022YFS0066), The Health Department of Sichuan Province (No. 20PJ030), China Postdoctoral Science Foundation (No. 2020M673250), and National Natural Youth Science Foundation of China (No. 82002868).
Author information
Authors and Affiliations
Contributions
YZ and ZM conceived the manuscript. JW, YT and DD contributed towards conception, design and revision of the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wu, J., Tao, Y., Deng, D. et al. The applications of CRISPR/Cas-mediated genome editing in genetic hearing loss. Cell Biosci 13, 93 (2023). https://doi.org/10.1186/s13578-023-01021-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13578-023-01021-7