
Abstract
Crotonylation of proteins is a newly found type of post-translational modifications (PTMs) which occurs leadingly on the lysine residue, namely, lysine crotonylation (Kcr). Kcr is conserved and is regulated by a series of enzymes and co-enzymes including lysine crotonyltransferase (writer), lysine decrotonylase (eraser), certain YEATS proteins (reader), and crotonyl-coenzyme A (donor). Histone Kcr has been substantially studied since 2011, but the Kcr of non-histone proteins is just an emerging field since its finding in 2017. Recent advances in the identification and quantification of non-histone protein Kcr by mass spectrometry have increased our understanding of Kcr. In this review, we summarized the main proteomic characteristics of non-histone protein Kcr and discussed its biological functions, including gene transcription, DNA damage response, enzymes regulation, metabolic pathways, cell cycle, and localization of heterochromatin in cells. We further proposed the performance of non-histone protein Kcr in diseases and the prospect of Kcr manipulators as potential therapeutic candidates in the diseases.
Similar content being viewed by others

Introduction
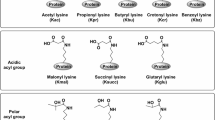
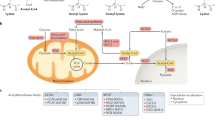
Post-translational modifications (PTMs) of proteins make up most of the proteome and to a large extent, and establish the impressive level of functional diversity in higher multi-cellular organisms. Mounting evidence suggests that PTMs provide an elegant mechanism to govern protein function in diverse biological processes including cell differentiation and organismal development, and aberrant protein modification may contribute to diseases such as cancer. As the only amino acid with a side chain amine [1], lysine can be covalently modified by glycosyl [2], propionyl [3], butyryl [4], acetyl [5], hydroxyl [6], crotonyl [7], ubiquitinyl and ubiquitinyl-like [8], formyl [9], malonyl [Uncovering lysine crotonylation It is increasingly appreciated that combinations of PTMs can generate distinct protein isoforms with varying functions, which vastly expand the functional diversity of mammalian proteomes. The widespread occurrence of PTMs only started to become clear in the first years of the twenty-first century, when advances in high-resolution mass spectrometry enabled detection of thousands of low-abundance PTM sites. In this context, protein Kcr on histones was first described in 2011 by Zhao and colleagues [7], who designed a comprehensive method to systematically analyze histone PTM, using PTMap, an algorithm that can recognize all possible PTMs of proteins [22]. In this method, mass spectrometry (MS) analysis of histone hydrolyzed peptides maximizes sequence coverage and sensitivity, resulting in recognition of many new PTM sites, including Kcr identified as a new type of histone modification. Histone crotonylation is an evolutionarily conserved histone post-translational modification appearing in eukaryotic cells from a wide range of species. Using a pan antibody against Kcr, Kcr signals in the core histones of sapiens (HeLa) cells, mouse, cerevisiae, elegans, melanogaster, as well as plant, have been detected [7, 23,24,Quantitative mass spectrometry for crotonylomics analysis Various MS instruments and methodological approaches can be used to perform crotonylomics analysis for proteins and peptides. Nearly all large-scale crotonylation studies use the “shotgun” or bottom‑up proteomics approach, which involves enzymatic digestion of all proteins (the proteome) followed by liquid chromatography coupled to tandem MS (LC–MS/MS) (Fig. 2). source before entering the mass spectrometer. MS and MS/MS spectra are then computationally processed to deduce peptide sequences, including the presence and location of crotonylation, and to quantify the abundance of crotonylated peptides and proteins Schematic diagram of the experimental procedures for mass spectrometry-based global analysis. Proteins are extracted from cells or tissues and digested into peptides with a protease such as trypsin. The tryptic peptides are then separated and fractionated by high pH reverse-phase high performance liquid chromatography (HPLC). Proteolysis of whole-cell protein extracts generates numerous peptides, but only a small fraction is crotonylated. To enrich lysine crotonylated peptides, pan-Kcr antibodies are applied to identify the crotonylated peptides in complex peptide mixtures using immunoaffinity purification. The resulting peptides are ionized in the electrospray Crotonylation of some protein sites cannot be detected in wild types cells. In order to find out which lysine sites may be crotonylated, some studies use drugs that promote crotonylation or knock down the expression of “modulators” of crotonylation during cell culture, such as sodium crotonate (NACR) which promotes non-histone crotonylation by conversion to crotonyl-coenzyme A [49], and HDAC inhibitor SAHA [21] or crotonyl-CoA hydrolase CDYL [52] which negatively regulate Kcr. But more researchers collect biological samples under interested conditions for modification (Table 3). In the next workflow, proteins are extracted from biological samples and digested into large numbers of peptides by enzymatic hydrolysis (usually trypsin), only a small part of the peptides is crotonylated. In order to identify the modified sites under specific conditions, quantitative analysis should be used. The most commonly used methods are to identify modified sites by comparing the intensity of crotonylated peptides among different samples, including metabolic labeling, chemical labeling, and label-free quantification. Each method has its advantages and disadvantages. SILAC (stable isotope labelling by amino acids in cell culture), which belongs to metabolic labeling, is one of the most commonly used methods in quantitative proteomics [53]. Natural isotopes (light) or stable isotopes (medium/heavy) are used to replace the corresponding amino acids during cell culture. Its advantage is the high efficiency for protein labeling, while its disadvantage is time consuming. Metabolic labeling has been successfully used to reveal the relative changes of crotonylation of non-histone proteins in homologous recombination-mediated DNA repair [52]. Chemical labeling can be used in samples that cannot be metabolically labeled, and several samples can be quantified in parallel, for example, by tandem mass tags (TMTs) [54] or by isobaric tags for relative and absolute quantification (iTRAQ) [55, 56]. However, due to the influence of labeled groups, the identification flux was lower than the label-free approach. Label-free quantification allows direct identification and quantification of proteins in a large scale, usually requiring analysis of a sample in triplicate to ensure that the measured differences are statistically significant [17, 20]. However, the accuracy of label-free quantification is slightly worse than that of the labeled quantification, because the former may be affected by the stability of mass spectrometry and other factors. In recent years, mass spectrometry techniques with increased stability and repeatability, as well as greatly improved computational algorithms for quantification of MS data, have made label-free quantification an attractive option [57,58,59]. To reduce the complexity of samples, after digestion, the trypsin polypeptides can be divided into several components by high-pH reversed-phase fractionation (RPF), known as sample fractionation. To increase the depth of the crotonylation analysis, immunoaffinity purification is usually used, in which the pan-Kcr antibody is immobilized to a resin bead and selectively bound to the crotonylated tryptic peptides and is then eluted [17, 52, 60, 61]. Enrichment of crotonylated peptides is usually combined with sample fractionation to improve the efficiency of the next mass spectrometry (MS). Finally, the enriched peptides are usually separated by liquid chromatography and ionized in the electrospray source, and entered into the mass spectrometer for analysis. High-resolution and high-quality precision analyzers can detect hundreds or thousands of different molecular features in a single LC–MS experiment, but only a small fraction of which can be identified and quantified [62]. The abundance of these eluted peptides, which range over many orders of magnitude, is a formidable analytical challenge that has been driving the progress of faster and more sensitive instruments and detection modes over the past decades [63,64,65]. For instance, a scan mode termed parallel accumulation-serial fragmentation (PASEF) has recently been demonstrated to increase sequencing speed exponentially without loss of sensitivity [66, 67]. All these technological advances are high performance additions to the technology toolbox in crotonylomics. Other experiments can also be used to verify the results of crotonylomics, such as western blotting and immunofluorescent staining. Immunofluorescent staining shows that crotonylated proteins are widely located in the cytoplasm and nuclei of H1299 and HeLa cells [17]. In addition, crotonylated proteins are widely found in a variety of tissues of mice, including lung, kidney, liver, colon, uterus, and ovary. Although these methods are not as efficient as MS, they can be used to verify the conclusions of large-scale experiments, such as NPM1, FHL1, ACTN1, integrin β1, ERK2, and CDK1, which are considered to be crotonylated as assayed by MS and can also be detected by western blotting [17]. Due to the progress of technology, the field of lysine crotonylation has developed rapidly in the past few years. Crotonylation was initially found to occur mainly in histones. However, a significant conclusion from recent proteomic studies is that most crotonylation events occur on non-histone proteins [17, 52, 56, 61, 68]. MS-based proteomics methods are now used in a variety of organisms not limited to humans, resulting in the identification of thousands of new crotonylation sites (Table 3). These studies show that crotonylation sites are often conserved in different organisms [18, 60, 69,70,71], thus it is obvious that crotonylation can be regarded as a protein modification that exists prevalently in all fields of life. Crotonylated non-histone proteins are widely distributed in subcellular compartments and participate in a variety of important cellular functions, signal pathways, and variant biological activities (Table 3). Motifs refer to some specific amino acids sequences which localize near the lysine acylation site and are generally highly conserved. The identification of the sequence of modification sites and the study of the corresponding model peptides provides clues for predicting the potential modification sites of new proteins. For instance, motif analysis is often used to predict potential kinase phosphorylation sites in bioinformatics [68, 72]. The amino acid sequences of motifs have been extracted from the upstream and downstream of the crotonylated lysine residue sites, which can describe the sequence commonness around the crotonylation sites. The studies given in Table 3 describe the amino acid sequence background of the crotonylation sites in eukaryotes. The highly conserved amino acids in these motifs, that is, E and D [17, Not applicable. Acyl-CoA synthetase short chain family member 2 ALL1 fused gene from chromosome 9 Chromobox homolog 5 Chromodomain Y-like Coenzyme A Camptothecin Cutaneous T-cell lymphoma DNA damage response DEAD-box helicase 5 Double plant homeodomain finger Double-strand break General control nonrepressed-protein 5 GCN5-related N-acetyltransferase Histone deacetylase Human males absent on the first Heterochromatin protein 1α Homologous recombination Heat shock protein Immunoglobulin A nephropathy Inositol phosphates Ionizing radiation Lysine acetylation Lysine crotonylation Lysine crotonyltransferase Lysine decrotonylase Liquid chromatography coupled to tandem MS Long-terminal repeat Minichromosome maintenance Monocytic leukemia zinc-finger protein Medial prefrontal cortex Mass spectrometry Moz Ybf2 Sas2 and Tip60 Sodium crotonate Nuclear magnetic resonance Nucleophosmin-1 Non-small cell lung cancer Patulin Peripheral blood mononuclear cells P300/CREB-binding protein P300/CBP-associated factor Plant homeodomain finger Post-translational modification Replicative protein A Suberoylanilide hydroxamic acid Short-chain fatty acid Sirtuin family deacetylase Single-stranded DNA TATA binding protein-associated factor-1 TATA binding protein-associated factor-14 Toxoplasma gondii parasites Tumor lymph node metastasis Trichostatin A Wang ZA, Kurra Y, Wang X, Zeng Y, Lee YJ, Sharma V, et al. A versatile approach for site-specific lysine acylation in proteins. Angew Chem Int Ed Engl. 2017;56(6):1643–7. Yan SB, Wold F. Neoglycoproteins: in vitro introduction of glycosyl units at glutamines in beta-casein using transglutaminase. Biochemistry. 1984;23(16):3759–65. Garrity J, Gardner JG, Hawse W, Wolberger C, Escalante-Semerena JC. N-lysine propionylation controls the activity of propionyl-CoA synthetase. J Biol Chem. 2007;282(41):30239–45. van Slyke DD, Sinex FM. The course of hydroxylation of lysine to form hydroxylysine in collagen. J Biol Chem. 1958;232(2):797–806. Sutendra G, Kinnaird A, Dromparis P, Paulin R, Stenson TH, Haromy A, et al. A nuclear pyruvate dehydrogenase complex is important for the generation of acetyl-CoA and histone acetylation. Cell. 2014;158(1):84–97. Das D, Bandyopadhyay D, Banerjee RK. Oxidative inactivation of gastric peroxidase by site-specific generation of hydroxyl radical and its role in stress-induced gastric ulceration. Free Radic Biol Med. 1998;24(3):460–9. Tan M, Luo H, Lee S, ** F, Yang JS, Montellier E, et al. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell. 2011;146(6):1016–28. Hochstrasser M. Origin and function of ubiquitin-like proteins. Nature. 2009;458(7237):422–9. Wisniewski JR, Zougman A, Mann M. Nepsilon-formylation of lysine is a widespread post-translational modification of nuclear proteins occurring at residues involved in regulation of chromatin function. Nucleic Acids Res. 2008;36(2):570–7. Peng C, Lu Z, **e Z, Cheng Z, Chen Y, Tan M, et al. The first identification of lysine malonylation substrates and its regulatory enzyme. Mol Cell Proteomics. 2011. https://doi.org/10.1074/mcp.M111.012658. Zhang Z, Tan M, **e Z, Dai L, Chen Y, Zhao Y. Identification of lysine succinylation as a new post-translational modification. Nat Chem Biol. 2011;7(1):58–63. Lan F, Shi Y. Epigenetic regulation: methylation of histone and non-histone proteins. Sci China C Life Sci. 2009;52(4):311–22. Ruiz-Andres O, Sanchez-Niño MD, Cannata-Ortiz P, Ruiz-Ortega M, Egido J, Ortiz A, et al. Histone lysine crotonylation during acute kidney injury in mice. Dis Model Mech. 2016;9(6):633–45. Liu Y, Li M, Fan M, Song Y, Yu H, Zhi X, et al. Chromodomain y-like protein-mediated histone crotonylation regulates stress-induced depressive behaviors. Biol Psychiatry. 2019;85(8):635–49. Jiang G, Nguyen D, Archin NM, Yukl SA, Méndez-Lagares G, Tang Y, et al. HIV latency is reversed by ACSS2-driven histone crotonylation. J Clin Invest. 2018;128(3):1190–8. Berger K, Moeller MJ. Mechanisms of epithelial repair and regeneration after acute kidney injury. Semin Nephrol. 2014;34(4):394–403. Xu W, Wan J, Zhan J, Li X, He H, Shi Z, et al. Global profiling of crotonylation on non-histone proteins. Cell Res. 2017;27(7):946–9. Kwon OK, Kim SJ, Lee S. First profiling of lysine crotonylation of myofilament proteins and ribosomal proteins in zebrafish embryos. Sci Rep. 2018;8(1):3652. Wan J, Liu H, Chu J, Zhang H. Functions and mechanisms of lysine crotonylation. J Cell Mol Med. 2019;23(11):7163–9. Liu JF, Wu SF, Liu S, Sun X, Wang XM, Xu P, et al. Global lysine crotonylation profiling of mouse liver. Proteomics. 2020;20(19–20):e2000049. Wu Q, Li W, Wang C, Fan P, Cao L, Wu Z, et al. Ultradeep Lysine Crotonylome Reveals the Crotonylation Enhancement on Both Histones and Nonhistone Proteins by SAHA Treatment. J Proteome Res. 2017;16(10):3664–71. Chen Y, Chen W, Cobb MH, Zhao Y. PTMap–a sequence alignment software for unrestricted, accurate, and full-spectrum identification of post-translational modification sites. Proc Natl Acad Sci U S A. 2009;106(3):761–6. Kotliński M, Rutowicz K, Kniżewski Ł, Palusiński A, Olędzki J, Fogtman A, et al. Histone H1 variants in arabidopsis are subject to numerous post-translational modifications, both conserved and previously unknown in histones, suggesting complex functions of H1 in plants. PLoS ONE. 2016;11(1):e0147908. Montellier E, Boussouar F, Rousseaux S, Zhang K, Buchou T, Fenaille F, et al. Chromatin-to-nucleoprotamine transition is controlled by the histone H2B variant TH2B. Genes Dev. 2013;27(15):1680–92. Sun H, Liu X, Li F, Li W, Zhang J, **ao Z, et al. First comprehensive proteome analysis of lysine crotonylation in seedling leaves of Nicotiana tabacum. Sci Rep. 2017;7(1):3013. Tweedie-Cullen RY, Brunner AM, Grossmann J, Mohanna S, Sichau D, Nanni P, et al. Identification of combinatorial patterns of post-translational modifications on individual histones in the mouse brain. PLoS ONE. 2012;7(5):e36980. Tanner KG, Landry J, Sternglanz R, Denu JM. Silent information regulator 2 family of NAD- dependent histone/protein deacetylases generates a unique product, 1-O-acetyl-ADP-ribose. Proc Natl Acad Sci U S A. 2000;97(26):14178–82. Tanny JC, Moazed D. Coupling of histone deacetylation to NAD breakdown by the yeast silencing protein Sir2: Evidence for acetyl transfer from substrate to an NAD breakdown product. Proc Natl Acad Sci U S A. 2001;98(2):415–20. Li Y, Sabari BR, Panchenko T, Wen H, Zhao D, Guan H, et al. Molecular coupling of histone crotonylation and active transcription by AF9 YEATS domain. Mol Cell. 2016;62(2):181–93. Zhao S, Zhang X, Li H. Beyond histone acetylation-writing and erasing histone acylations. Curr Opin Struct Biol. 2018;53:169–77. Sabari BR, Tang Z, Huang H, Yong-Gonzalez V, Molina H, Kong HE, et al. Intracellular Crotonyl-CoA stimulates transcription through p300-catalyzed histone crotonylation. Mol Cell. 2018;69(3):533. Liu X, Wei W, Liu Y, Yang X, Wu J, Zhang Y, et al. MOF as an evolutionarily conserved histone crotonyltransferase and transcriptional activation by histone acetyltransferase-deficient and crotonyltransferase-competent CBP/p300. Cell Discov. 2017;3:17016. Kollenstart L, de Groot A, Janssen G, Cheng X, Vreeken K, Martino F, et al. Gcn5 and Esa1 function as histone crotonyltransferases to regulate crotonylation-dependent transcription. J Biol Chem. 2019;294(52):20122–34. Zhao D, Guan H, Zhao S, Mi W, Wen H, Li Y, et al. YEATS2 is a selective histone crotonylation reader. Cell Res. 2016;26(5):629–32. Andrews FH, Shinsky SA, Shanle EK, Bridgers JB, Gest A, Tsun IK, et al. The Taf14 YEATS domain is a reader of histone crotonylation. Nat Chem Biol. 2016;12(6):396–8. Wei W, Liu X, Chen J, Gao S, Lu L, Zhang H, et al. Class I histone deacetylases are major histone decrotonylases: evidence for critical and broad function of histone crotonylation in transcription. Cell Res. 2017;27(7):898–915. Feldman JL, Baeza J, Denu JM. Activation of the protein deacetylase SIRT6 by long-chain fatty acids and widespread deacylation by mammalian sirtuins. J Biol Chem. 2013;288(43):31350–6. Bao X, Wang Y, Li X, Li XM, Liu Z, Yang T, et al. Identification of “erasers” for lysine crotonylated histone marks using a chemical proteomics approach. Elife. 2014. https://doi.org/10.7554/eLife.02999. Martinez-Moreno JM, Fontecha-Barriuso M, Martín-Sánchez D, Sánchez-Niño MD, Ruiz-Ortega M, Sanz AB, et al. The contribution of histone crotonylation to tissue health and disease: focus on kidney health. Front Pharmacol. 2020;11:393. Sabari BR, Tang Z, Huang H, Yong-Gonzalez V, Molina H, Kong HE, et al. Intracellular crotonyl-CoA stimulates transcription through p300-catalyzed histone crotonylation. Mol Cell. 2015;58(2):203–15. Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. 2009;10(1):32–42. Houtkooper RH, Pirinen E, Auwerx J. Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol Cell Biol. 2012;13(4):225–38. Sauve AA, Wolberger C, Schramm VL, Boeke JD. The biochemistry of sirtuins. Annu Rev Biochem. 2006;75:435–65. Flynn EM, Huang OW, Poy F, Oppikofer M, Bellon SF, Tang Y, et al. A subset of human bromodomains recognizes butyryllysine and crotonyllysine histone peptide modifications. Structure. 2015;23(10):1801–14. **ong X, Panchenko T, Yang S, Zhao S, Yan P, Zhang W, et al. Selective recognition of histone crotonylation by double PHD fingers of MOZ and DPF2. Nat Chem Biol. 2016;12(12):1111–8. Zhang Q, Zeng L, Zhao C, Ju Y, Konuma T, Zhou MM. Structural insights into histone crotonyl-lysine recognition by the AF9 YEATS domain. Structure. 2016;24(9):1606–12. Stilling RM, van de Wouw M, Clarke G, Stanton C, Dinan TG, Cryan JF. The neuropharmacology of butyrate: the bread and butter of the microbiota-gut-brain axis. Neurochem Int. 2016;99:110–32. Lin H, Su X, He B. Protein lysine acylation and cysteine succination by intermediates of energy metabolism. ACS Chem Biol. 2012;7(6):947–60. Wei W, Mao A, Tang B, Zeng Q, Gao S, Liu X, et al. Large-scale identification of protein crotonylation reveals its role in multiple cellular functions. J Proteome Res. 2017;16(4):1743–52. Caron C, Pivot-Pajot C, van Grunsven LA, Col E, Lestrat C, Rousseaux S, et al. Cdyl: a new transcriptional co-repressor. EMBO Rep. 2003;4(9):877–82. Liu S, Yu H, Liu Y, Liu X, Zhang Y, Bu C, et al. Chromodomain protein CDYL acts as a Crotonyl-CoA hydratase to regulate histone crotonylation and spermatogenesis. Mol Cell. 2017;67(5):853-66.e5. Yu H, Bu C, Liu Y, Gong T, Liu X, Liu S, et al. Global crotonylome reveals CDYL-regulated RPA1 crotonylation in homologous recombination-mediated DNA repair. Sci Adv. 2020. https://doi.org/10.1126/sciadv.aay4697. Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, et al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 2002;1(5):376–86. Yang Q, Li Y, Apaliya MT, Zheng X, Serwah B, Zhang X, et al. The Response of Rhodotorula mucilaginosa to patulin based on lysine crotonylation. Front Microbiol. 2018;9:2025. Evans C, Noirel J, Ow SY, Salim M, Pereira-Medrano AG, Couto N, et al. An insight into iTRAQ: where do we stand now. Anal Bioanal Chem. 2012;404(4):1011–27. Chen W, Tang D, Xu Y, Zou Y, Sui W, Dai Y, et al. Comprehensive analysis of lysine crotonylation in proteome of maintenance hemodialysis patients. Medicine. 2018;97(37):e12035. Cox J, Hein MY, Luber CA, Paron I, Nagaraj N, Mann M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol Cell Proteomics. 2014;13(9):2513–26. Zhao L, Cong X, Zhai L, Hu H, Xu JY, Zhao W, et al. Comparative evaluation of label-free quantification strategies. J Proteomics. 2020;215:103669. Lv H, Dao FY, Guan ZX, Yang H, Li YW, Lin H. Deep-Kcr: accurate detection of lysine crotonylation sites using deep learning method. Brief Bioinform. 2020. https://doi.org/10.1093/bib/bbaa255. Sun J, Qiu C, Qian W, Wang Y, Sun L, Li Y, et al. Ammonium triggered the response mechanism of lysine crotonylome in tea plants. BMC Genomics. 2019;20(1):340. Huang H, Wang DL, Zhao Y. Quantitative crotonylome analysis expands the roles of p300 in the regulation of lysine crotonylation pathway. Proteomics. 2018;18(15):e1700230. Michalski A, Cox J, Mann M. More than 100,000 detectable peptide species elute in single shotgun proteomics runs but the majority is inaccessible to data-dependent LC-MS/MS. J Proteome Res. 2011;10(4):1785–93. Altelaar AF, Munoz J, Heck AJ. Next-generation proteomics: towards an integrative view of proteome dynamics. Nat Rev Genet. 2013;14(1):35–48. Aebersold R, Mann M. Mass-spectrometric exploration of proteome structure and function. Nature. 2016;537(7620):347–55. Eliuk S, Makarov A. Evolution of orbitrap mass spectrometry instrumentation. Annu Rev Anal Chem (Palo Alto Calif). 2015;8:61–80. Meier F, Beck S, Grassl N, Lubeck M, Park MA, Raether O, et al. Parallel accumulation-serial fragmentation (PASEF): multiplying sequencing speed and sensitivity by synchronized scans in a trapped ion mobility device. J Proteome Res. 2015;14(12):5378–87. Meier F, Brunner AD, Koch S, Koch H, Lubeck M, Krause M, et al. Online parallel accumulation-serial fragmentation (PASEF) with a novel trapped ion mobility mass spectrometer. Mol Cell Proteomics. 2018;17(12):2534–45. Lin H, Tang D, Xu Y, Zhang R, Ou M, Zheng F, et al. Quantitative analysis of protein crotonylation identifies its association with immunoglobulin A nephropathy. Mol Med Rep. 2020;21(3):1242–50. Liu K, Yuan C, Li H, Chen K, Lu L, Shen C, et al. A qualitative proteome-wide lysine crotonylation profiling of papaya (Carica papaya L.). Sci Rep. 2018;8(1):8230. Liu S, Xue C, Fang Y, Chen G, Peng X, Zhou Y, et al. Global involvement of lysine crotonylation in protein modification and transcription regulation in rice. Mol Cell Proteomics. 2018;17(10):1922–36. Yin D, Jiang N, Zhang Y, Wang D, Sang X, Feng Y, et al. Global lysine crotonylation and 2-hydroxyisobutyrylation in phenotypically different Toxoplasma gondii parasites. Mol Cell Proteomics. 2019;18(11):2207–24. Kemp BE, Pearson RB. Protein kinase recognition sequence motifs. Trends Biochem Sci. 1990;15(9):342–6. Montellier E, Rousseaux S, Zhao Y, Khochbin S. Histone crotonylation specifically marks the haploid male germ cell gene expression program: post-meiotic male-specific gene expression. BioEssays. 2012;34(3):187–93. Kolthur-Seetharam U, Martianov I, Davidson I. Specialization of the general transcriptional machinery in male germ cells. Cell Cycle. 2008;7(22):3493–8. Sassone-Corsi P. Unique chromatin remodeling and transcriptional regulation in spermatogenesis. Science. 2002;296(5576):2176–8. Vogelstein B, Kinzler KW. p53 function and dysfunction. Cell. 1992;70(4):523–6. Dai C, Gu W. p53 post-translational modification: deregulated in tumorigenesis. Trends Mol Med. 2010;16(11):528–36. Brooks CL, Gu W. The impact of acetylation and deacetylation on the p53 pathway. Protein Cell. 2011;2(6):456–62. Reed SM, Hagen J, Tompkins VS, Thies K, Quelle FW, Quelle DE. Nuclear interactor of ARF and Mdm2 regulates multiple pathways to activate p53. Cell Cycle. 2014;13(8):1288–98. Ito A, Kawaguchi Y, Lai CH, Kovacs JJ, Higashimoto Y, Appella E, et al. MDM2-HDAC1-mediated deacetylation of p53 is required for its degradation. EMBO J. 2002;21(22):6236–45. Chae YK, Anker JF, Carneiro BA, Chandra S, Kaplan J, Kalyan A, et al. Genomic landscape of DNA repair genes in cancer. Oncotarget. 2016;7(17):23312–21. Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40(2):179–204. Fanning E, Klimovich V, Nager AR. A dynamic model for replication protein A (RPA) function in DNA processing pathways. Nucleic Acids Res. 2006;34(15):4126–37. Toledo LI, Altmeyer M, Rask MB, Lukas C, Larsen DH, Povlsen LK, et al. ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell. 2013;155(5):1088–103. Prakash R, Zhang Y, Feng W, Jasin M. Homologous recombination and human health: the roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harb Perspect Biol. 2015;7(4):a016600. Syed A, Tainer JA. The MRE11-RAD50-NBS1 complex conducts the orchestration of damage signaling and outcomes to stress in DNA replication and repair. Annu Rev Biochem. 2018;87:263–94. Tammaro M, Liao S, McCane J, Yan H. The N-terminus of RPA large subunit and its spatial position are important for the 5’->3’ resection of DNA double-strand breaks. Nucleic Acids Res. 2015;43(18):8790–800. Dou H, Huang C, Singh M, Carpenter PB, Yeh ET. Regulation of DNA repair through deSUMOylation and SUMOylation of replication protein A complex. Mol Cell. 2010;39(3):333–45. Elia AE, Wang DC, Willis NA, Boardman AP, Hajdu I, Adeyemi RO, et al. RFWD3-dependent ubiquitination of rpa regulates repair at stalled replication forks. Mol Cell. 2015;60(2):280–93. He H, Wang J, Liu T. UV-induced RPA1 acetylation promotes nucleotide excision repair. Cell Rep. 2017;20(9):2010–25. Liu Y, Liu S, Yuan S, Yu H, Zhang Y, Yang X, et al. Chromodomain protein CDYL is required for transmission/restoration of repressive histone marks. J Mol Cell Biol. 2017;9(3):178–94. Abu-Zhayia ER, Awwad SW, Ben-Oz BM, Khoury-Haddad H, Ayoub N. CDYL1 fosters double-strand break-induced transcription silencing and promotes homology-directed repair. J Mol Cell Biol. 2018;10(4):341–57. Lei M. The MCM complex: its role in DNA replication and implications for cancer therapy. Curr Cancer Drug Targets. 2005;5(5):365–80. Verschure PJ, van der Kraan I, de Leeuw W, van der Vlag J, Carpenter AE, Belmont AS, et al. In vivo HP1 targeting causes large-scale chromatin condensation and enhanced histone lysine methylation. Mol Cell Biol. 2005;25(11):4552–64. Qiu Y, Zhao Y, Becker M, John S, Parekh BS, Huang S, et al. HDAC1 acetylation is linked to progressive modulation of steroid receptor-induced gene transcription. Mol Cell. 2006;22(5):669–79. Wan J, Liu H, Ming L. Lysine crotonylation is involved in hepatocellular carcinoma progression. Biomed Pharmacother. 2019;111:976–82. Pant K, Peixoto E, Richard S, Gradilone SA. Role of histone deacetylases in carcinogenesis: potential role in cholangiocarcinoma. Cells. 2020;9(3):780. Olsen EA, Kim YH, Kuzel TM, Pacheco TR, Foss FM, Parker S, et al. Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. J Clin Oncol. 2007;25(21):3109–15. Chen MY, Liao WS, Lu Z, Bornmann WG, Hennessey V, Washington MN, et al. Decitabine and suberoylanilide hydroxamic acid (SAHA) inhibit growth of ovarian cancer cell lines and xenografts while inducing expression of imprinted tumor suppressor genes, apoptosis, G2/M arrest, and autophagy. Cancer. 2011;117(19):4424–38. Konstantinopoulos PA, Wilson AJ, Saskowski J, Wass E, Khabele D. Suberoylanilide hydroxamic acid (SAHA) enhances olaparib activity by targeting homologous recombination DNA repair in ovarian cancer. Gynecol Oncol. 2014;133(3):599–606. Munster PN, Troso-Sandoval T, Rosen N, Rifkind R, Marks PA, Richon VM. The histone deacetylase inhibitor suberoylanilide hydroxamic acid induces differentiation of human breast cancer cells. Cancer Res. 2001;61(23):8492–7. Li X, Li XM, Jiang Y, Liu Z, Cui Y, Fung KY, et al. Structure-guided development of YEATS domain inhibitors by targeting π-π-π stacking. Nat Chem Biol. 2018;14(12):1140–9. Wang M, Chang Q, Yang H, Liu Y, Wang C, Hu F, et al. Elevated lysine crotonylation and succinylation in the brains of BTBR mice. Int J Dev Neurosci. 2019;76:61–4. Fellows R, Denizot J, Stellato C, Cuomo A, Jain P, Stoyanova E, et al. Microbiota derived short chain fatty acids promote histone crotonylation in the colon through histone deacetylases. Nat Commun. 2018;9(1):105. Koh A, De Vadder F, Kovatcheva-Datchary P, Bäckhed F. From dietary fiber to host physiology: short-chain fatty acids as key bacterial metabolites. Cell. 2016;165(6):1332–45. Nath A, Chan C. Genetic alterations in fatty acid transport and metabolism genes are associated with metastatic progression and poor prognosis of human cancers. Sci Rep. 2016;6:18669. Donohoe DR, Garge N, Zhang X, Sun W, O’Connell TM, Bunger MK, et al. The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell Metab. 2011;13(5):517–26. Cai L, Sutter BM, Li B, Tu BP. Acetyl-CoA induces cell growth and proliferation by promoting the acetylation of histones at growth genes. Mol Cell. 2011;42(4):426–37. Gowans GJ, Bridgers JB, Zhang J, Dronamraju R, Burnetti A, King DA, et al. Recognition of Histone Crotonylation by Taf14 Links Metabolic State to Gene Expression. Mol Cell. 2019;76(6):909-21.e3. Ju Z, He JJ. Prediction of lysine crotonylation sites by incorporating the composition of k-spaced amino acid pairs into Chou’s general PseAAC. J Mol Graph Model. 2017;77:200–4. Qiu WR, Sun BQ, Tang H, Huang J, Lin H. Identify and analysis crotonylation sites in histone by using support vector machines. Artif Intell Med. 2017;83:75–81. Qiu WR, Sun BQ, **ao X, Xu ZC, Chou KC. iPTM-mLys: identifying multiple lysine PTM sites and their different types. Bioinformatics. 2016;32(20):3116–23. Theillet FX, Smet-Nocca C, Liokatis S, Thongwichian R, Kosten J, Yoon MK, et al. Cell signaling, post-translational protein modifications and NMR spectroscopy. J Biomol NMR. 2012;54(3):217–36. **e X, Li XM, Qin F, Lin J, Zhang G, Zhao J, et al. Genetically encoded photoaffinity histone marks. J Am Chem Soc. 2017;139(19):6522–5. The authors thank members in Dr. JM Cao’s laboratory for their helps in the studies related to this review. This work and related studies are supported by Shanxi “1331 Project” Key Subjects Construction (1331KSC), Applied Basic Research Program of Shanxi Province (201801D221269), Scientific and Technological Innovation Programs of Higher Education Institutions in Shanxi (STIP) (2019L0437), and partially by a fund from the National Natural Science Foundation of China (81670313). JH: conception and writing of manuscript draft; JL: references preparation; LZ, DW: reading and revising manuscript; JC: revising and supervising submission. All authors read and approved the final manuscript. Not applicable. All authors have read and consent to the publication of the article. The authors declare that they have no competing interests in this work. Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data. Hou, JY., Zhou, L., Li, JL. et al. Emerging roles of non-histone protein crotonylation in biomedicine.
Cell Biosci 11, 101 (2021). https://doi.org/10.1186/s13578-021-00616-2 Received: Accepted: Published: DOI: https://doi.org/10.1186/s13578-021-00616-2
Proteomic characteristics of crotonylation
Availability of data and materials
Abbreviations
References
Acknowledgements
Funding
Author information
Authors and Affiliations
Contributions
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Consent for publication
Competing interests
Additional information
Publisher's Note
Rights and permissions
About this article
Cite this article
Keywords
