
Abstract
Autism spectrum disorder (ASD) comprises a group of neurodevelopmental disorders characterized by impaired social interactions as well as the presentation of restrictive and repetitive behaviors. ASD is highly heritable but genetically heterogenous with both common and rare genetic variants collaborating to predispose individuals to the disorder. In this review, we synthesize recent efforts to develop human induced pluripotent stem cell (iPSC)-derived models of ASD-related phenotypes. We firstly address concerns regarding the relevance and validity of available neuronal iPSC-derived models. We then critically evaluate the robustness of various differentiation and cell culture protocols used for producing cell types of relevance to ASD. By exploring iPSC models of ASD reported thus far, we examine to what extent cellular and neuronal phenotypes with potential relevance to ASD can be linked to genetic variants found to underlie it. Lastly, we outline promising strategies by which iPSC technology can both enhance the power of genetic studies to identify ASD risk factors and nominate pathways that are disrupted across groups of ASD patients that might serve as common points for therapeutic intervention.
Similar content being viewed by others


Introduction
The parable of the blind men and the elephant has often been used as a metaphor for a reductionist cognitive process. Similar to the blind men trying to reconstruct the complexity of the elephant through fragmented experience, scientists aim at modeling complex human disease through functional studies of simpler, more approachable systems. In this perspective, availability of relevant experimental models is critical to further our understanding of any human disease, as well as to facilitate drug discovery. Advantages as well as potential limitations of employing human induced pluripotent stem cell (iPSC)-derived systems as an advanced technological tool to apply a reductionist approach to the study of autism spectrum disorder (ASD), have been extensively reviewed elsewhere [1,2,3,4]. Here, we focus on recent milestones in this space that are slowly enabling a more holistic outlook, and review efforts that apply iPSC-derived models to reconstruct, quantitate, and predict the complexity of the human brain. Specifically, we extensively discuss which cell types may be most vulnerable to the genetic variation underlying ASD and how their altered function might underlie behavioral changes in people with ASD. In this perspective, we critically evaluate neuronal iPSC-derived models used thus far to obtain such cell types in culture, and summarize reported cellular phenotypes that can represent partial readouts for ASD-like features. Then, we consider the few studies where iPSC technology has been coupled with human genetics to advance our understanding of the molecular makeup of neurodevelopmental complex genetic disease, including ASD. Finally, we contemplate ways that iPSC models can enhance the power of genetic studies to identify ASD risk factors and fuel discoveries of key pathways that are altered across many individuals with ASD and might serve as shared points for therapeutic intervention.
Background: the advantages of modeling disease with induced Pluripotent Stem Cells (iPSCs)
Animal models, especially mice, have historically been a key tool in basic research and therapeutics. However, because of existing species-specific differences in pathways implicated in health and disease [5], human cell-based models have always been regarded as appealing complementary systems [6]. Human primary cells are however generally unavailable for study due to their limited availability from patients [7, 8].
In the attempt to overcome this impasse, a landmark in the field of cellular disease modelling was the employment of in vitro grown embryonic stem cells (ESC) derived from human blastocysts, that have the ability to indefinitely self-renew and can give rise to any type of somatic cells [9, 10]. However, despite providing many human cell types for research and therapeutics, their employment immediately raised societal concerns over their early embryonic origin [11], as well as practical limitations due to lack of information about the donors, and their familial and medical history (including their propensity to develop ASD) [12].
Takahashi and Yamanaka’s pioneering studies in the 2000s [13] led to rapid expansion of induced pluripotent stem cell (iPSC) technologies, and opened unprecedented opportunities for disease modeling. The last decade is testimony of how iPSC-based studies can enhance biomedical research and personalized regenerative medicine [12,13,14]: somatic cells from easily accessible tissues of patients can now be routinely reprogrammed into an embryonic stem cell-like state, and subsequently differentiated into cell types that are relevant for the same patient’s disease. iPSC technology also yields virtually unlimited amounts of human tissue carrying a genetic variant of interest, that becomes easily available for manipulation and therapeutic endeavors. Furthermore, genetic variants can be examined on a genetic background sensitive to the disorder and that may account for unpredicted secondary effects [15, 16].
The considerations above are all especially relevant in the context of neuropsychiatric and neurodevelopmental diseases—including ASD—which are for the most part quintessentially human and often polygenic [17, 18]. Primary cultures of patient-derived cells are largely unavailable, because brain biopsies for establishing an in vitro neuronal cell line, are considered unethical [19], while human post mortem samples, despite being desirable alternatives, typically do not represent the developmental stage when the disease is firstly manifested, and can be confounded by other factors, including treatment for the disease of study or for some of its symptoms [20]. Additionally, neither genetically engineered animal models, nor post mortem samples, have thus far had the capacity to predict patient-specific clinical outcomes to candidate ASD therapeutics [21].
iPSCs meet all requirements to address these issues, as effectively unlimited quantities of patient-derived cells can be used to model cellular components of the human brain, to identify therapeutic targets, and to investigate said targets and design candidate therapies [2, 19,20,21] (Fig. 1). iPSCs are therefore an optimal resource to study various aspects of ASD in vitro, under the assumption that specific cell types are vulnerable to ASD, and that such cell types can be reliably derived from iPSCs using currently available protocols.

Overview of all available model systems currently employed to model disease. iPSC-based models represent a source of unlimited patient-specific material, able to recapitulate neuronal development without ethical concerns linked to use of embryonic material or patient biopsies
ASD is a complex, polygenic, and heritable disorder
Under the broad diagnosis of ASD is a variety of neurodevelopmental disorders marked by impaired social skills and restrictive-repetitive behavior [3]. Individuals diagnosed with ASD exhibit a variety of phenotypes depending on a complex interplay between genetic and environmental factors and often manifest other comorbidities, both neurological and non-neurological.
The phenotypic complexity of ASD reflects its underlying genetic architecture, made of contributions from rare variants of large effect, either CNV (e.g., 16p11.2 or 22q11.2 duplication and deletion) or point mutations (e.g., CHD8, SCN2A), and common variants each conveying small effect but collectively sha** most of its risk [4, 22,23,24,25,26].
Recently, an unprecedented expansion of genome-wide association studies (GWAS) have led to the identification of common variants associated with ASD [22, 23, 27], while large-scale exome sequencing studies of ASD have now identified over 100 high-confidence autism risk genes [24, 25, 28]. However, how disruption of such genes results in altered neurodevelopment and neurophysiology in individuals with ASD, is still largely unclear.
Nevertheless, granular understanding of ASD genetic architecture has provided a tool in determining the dynamics of ASD onset during development at the cellular level, using analysis of concerted expression of ASD risk genes [29], and has been pivotal in defining the identity of cell types most relevant to ASD physiopathology. Identifying cell types that are vulnerable to ASD can subsequently guide efforts in perfecting protocols to derive such cell types from iPSC models [30], providing a promising avenue to translate genetic information into cell modeling.
Cell types of both develo** and adult brain are vulnerable to ASD and can be modelled in vitro
The phenotypic complexity of ASD suggests that there might be multiple cell types vulnerable to ASD both during development and adulthood (Table 1).
According to co-expression studies of ASD-relevant genes, a critical window for the onset of ASD, coincides with early fetal development, as specific molecular and cellular programs depending on coordinated expression of ASD risk genes, do not persist in the mature cells of the adult brain [29, 31, 32]. This argument nominates early time-points in neuronal maturation, such as Neural Progenitor Cells (NPCs), as attractive cell models. On the other hand, mature neurons have been strongly implicated in ASD physiopathology by a number of bulk gene expression studies showing changes in the neocortex of ASD patients, and indicating functional convergence of risk-gene expression in the adult brain [33].
Similarly, Satterstrom and colleagues, who applied co-expression network analysis to BrainSpan datasets, showed that ASD genes are indeed expressed at high levels not just in the develo** brain, but also in the adult cortex [28]. The authors were able to pinpoint specific cell types, based on expression modules of 4,261 cells from the prenatal human forebrain [34]. In accordance with previous evidence that identified excitatory glutamatergic neurons [29, 31, 32], they found neuronal cell types as being prevalently recapitulating the transcriptional signature of ASD, with most genes being expressed in excitatory and, to a lesser-extent, inhibitory lineage cells [28]. Other neuronal types found to be enriched for ASD signal were striatal interneurons. Interestingly, Cogill et al. demonstrated that also lncRNAs may play a role in ASD due to their convergence on shared pathways with ASD-associated coding-genes. It will be important to follow up on this finding and include lncRNAs in future re-analyses based on co-expression modules [35]. Despite the general lack of statistical power in molecular studies involving scarcely available post-mortem tissue, Velmeshev and colleagues recently conducted a single-nucleus RNA sequencing study on cortical tissue from patients, and found that expression of synaptic and neurodevelopmental genes is especially affected in cortical neurons [28].
In conclusion, although some non-neuronal cell types were found to be vulnerable to ASD (including microglia and OPCs), most of the transcriptional modules seem to converge on neuronal lineages, and notably on maturating neurons and excitatory neurons of the adult neocortex [36].
It should also be noted that abnormal pain sensitivity is commonly reported in ASD patients [37] and developmental disorders linked to monogenic forms of ASD are also associated with defects in somato-sensation [38]. Many studies conducted in rodent models of ASD with highly penetrant monogenic mutations, indicate that abnormalities in sensory reactivity correlate with ASD-related phenotypes, in line with the hypothesis that impaired sensory perception may impact brain development and function, and results in disparate symptoms associated with ASD [39]. This hypothesis provides a potential mechanistic link between otherwise heterogeneous ASD-related phenotypes, and implicates peripheral sensory neurons, in addition to the other neuronal types discussed above, in ASD etiology [40].
State-of-the art iPSC-derived differentiation protocols that model cell types vulnerable to ASD
iPSC-based differentiation protocols offer a valuable resource to generate cell types relevant to virtually any disease of interest, with the caveat that such cell types can be reliably derived from iPSCs using currently available protocols.
A selected overview of the otherwise large number of published protocols for neural differentiation of cell types relevant to ASD, is reported in Table 2. They are all based on the premise that it is possible to mimic embryonic differentiation in a dish, with the distinction that some recapitulate intermediate NPC states, while others achieve direct differentiation to the terminal neural cell type of choice. Either way, many protocols start with dual SMAD inhibition [41]. Subsequent differences in concentration of patterning factors or in the timing of their addition can yield a variety of neural cell types and impact largely the homogeneity and nature of neurons. In order to overcome culturing heterogeneity, many protocols are now based on inducible transcription factor expression, via virus transduction or integration into a safe harbor locus [42,43,44,45,46]. These protocols generally produce highly differentiated and homogeneous cells in a shorter time frame and on a larger scale when compared to others. However, it is still essential to share detailed experimental guidelines to guarantee reproducibility of each new protocol.
High degree of reproducibility was reported by Nehme and colleagues, who combined small-molecule with transcriptional patterning, to generate cortical excitatory glutamatergic neurons [47]. After three weeks of maturation, transcriptomic analysis confirmed homogenous maturation of cultured iPSCs into upper layer cortical projection neurons. Microelectrode array and patch clamp electrophysiology also showed AMPA and NMDA-mediated synaptic transmission, which are hallmarks of postnatal cortical neurons. Of particular interest for NPC in vitro modelling, Wells and colleagues adapted the protocol developed by Nehme [47], allowing rapid (48 h) generation and maintenance of human stem cell-derived progenitor cells (SNaPs) [48].
Finally, the association between ASD and altered somato-sensation, suggests that generating iPSC models of sensory neurons from ASD patients will be a valuable system for testing the ability to reverse some ASD-related cellular phenotypes, as previously done in mouse models [49], and spinal cord [50]. Although few protocols have been previously reported (Table 2), none has been utilized in ASD research thus far.
Complex cell culturing systems to study ASD
The translational potential of iPSC-derived models can be further enhanced by complementing cell culture with the inclusion of additional components of the in vivo niche of the cell type of interest, or mimicking cell–cell and cell–matrix interactions that occur within organs and tissues [51] (Fig. 2). Complex culturing systems, including co-cultures and three-dimensional (3D) cultures, may also account for non-cell-autonomous effects on differentiation, and help modulate neuronal activity and drug response, while also promoting neuronal maturity [52].

Summary of various iPSC-based culturing systems. Bi-dimensional cultures can be adapted for co-culturing of more than one cell type at a time. Three-dimensional cultures can be supported by microfluidic devices
One of the most ASD-relevant examples of co-cultures consists of iPSC-derived neurons and glial cells, an abundant cell population in the human brain that have critical supporting roles for neurons in both health and disease [53, 54]. Co-culturing neurons with microglia has been key in studying chronic inflammation correlated with ASD and neurodegeneration [55]. Other examples of cell types that have been co-cultured with neurons in ASD modeling are oligodendrocytes (impacting neuronal myelination [56]) and astrocytes (impacting viability, synaptic function, and neurite outgrowth [57]).
3D culturing conditions are also an important development for improving physiologically-relevant in vitro disease models. For instance, it is now possible to model specific regions of the brain, allowing a more holistic comparison between ASD-derived and control cultures [58]. Although 3D cultures can be achieved through microfluidics and bioprinting, one of the most promising technologies is that of iPSC-derived brain organoids. Brain organoids consist of multicellular aggregates that differentiate and self-organize, mimicking its in vivo development [59], and offer new models for assessing the pathogenesis of ASD, especially in the context of monogenic syndromes [60,61,62,63]. Additionally, implementations of organoid protocols allow production of specific regions of the brain, including hippocampus and cerebellum, as well as cortical folding, enabling a holistic study of the human brain in development and disease [58]. Attempts have also been made to use this technology to recreate early stages of corticogenesis, particularly relevant to the study of prenatal brain organization and function [64].
Co-culture and three-dimensional (3D) cultures are therefore a promising development for iPSC modeling of complex tissues (reviewed in [65, 66]), although currently still challenging to establish and reproduce, mostly due to batch-to-batch or organoid-to-organoid heterogeneity and long differentiation periods (60–120 days to reach differentiation levels similar to mid-gestation, compared to 14–30 days of NGN2-based patterning protocols).
iPSC-based models manifest ASD-related, measurable phenotypes
NPCs and neurons derived from syndromic and idiopathic individuals diagnosed with ASD, display a wide range of phenotypes [30, 67,68,69,70]. The phenotypic diversity observed in many individuals with ASD is representative of the underlying heterogeneity of their genetic background and is also reflected on the diversity of reported cellular phenotypes observed across iPSC models. Comprehensive catalogues of ASD-relevant cellular phenotypes as well as detailed description of current Biobanks of deposited patient-derived cell material, have been compiled in a number of excellent reviews [71, 72]. However, it is often challenging to compare qualitative observations, and the field would certainly benefit form adapting standardized quantitative measures to evaluate the impact of genetic background on cellular phenotypes.
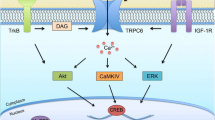
Modelling ASD with iPSC technology and classifying each model based on a well-defined subset of quantitative qualifiers or “phenotypic classes”, has proven crucial in revealing novel cellular and molecular mechanisms underlying it. Thus far, the most robust quantitative measures utilized to stratify cellular phenotypes have focused on cell proliferation and brain growth; RNA-processing; synapse density and dendritic arborization; electrophysiology; and calcium signaling (Fig. 3). Here we provide a summary of the most reproducible, robust, and representative cellular phenotypes according to these metrics (Table 3), and focus on few representative examples to discuss how they relate to ASD symptoms, and whether they can be used for translational endeavors (Fig. 4).

Overview of measurable phenotypes observed in cell types either derived from ASD patients or obtained via gene-editing. Neuronal phenotypes can be cell autonomous or mediated by interaction with co-cultured non-neuronal cells

iPSC-based models as fundamental tools to bridge human genetics and functional studies in ASD, through the employment of large-scale assays, including PPI networks, CRISPR-screens and “villages”
Cell proliferation
Several clinical studies of ASD have reported accelerated brain growth in the first three years of life of patients [73]. This translates in macrocephaly, that is in fact a characteristic phenotype of some genetic subtypes of ASD. Conversely, microcephaly, the inverse phenotype, is also associated with autism. For example, deletions and duplications at two loci, 1q21.1 and 16p11.2, have opposing brain growth phenotypes [74], as well as individual gene mutations: CHD8′s [124], and in animal models. Notably, ** and colleagues used in vivo Perturb-seq to introduce frame shift mutations in 35 genes strongly associated with ASD in humans, and studied their effect on mouse postnatal brain [125]. Given the recent advances in adapting CRISPR-screen to human neurons [126], it is easy to imagine how a similar experimental design could be soon translated to iPSC-derived human models.
Additionally, a number of studies have recently focused on employing genetic heterogeneity within populations, to exploit the multiplexing potential of single cell RNA-seq [127]. Specifically, genetic information offers a natural identifier or barcode to demultiplex pooled samples, allowing complex combinatorial experimental designs of single cell RNA-seq experiments. This approach has been successfully combined to CRISPR-screens, and iPSC-derived neurons represent an ideal cell model to analyze effects of ASD-risk genes on cellular phenotypes [128]. Another interesting application of recent advances in pooled RNA-seq technology is represented by systematic sequencing of villages of neurons obtained from patient-derived iPSCs [129, 130]. For instance, Cederquist and colleagues, pooled in a single dish 30 isogenic iPSC lines harboring de novo ASD mutations to disentangle ASD genetic heterogeneity [131]. The same approach can be used to explore polygenic risk or to assess the impact of genetic background on ASD-relevant phenotypes and eQTLs (expression quantitative trait loci). Lines carrying monogenic ASD mutations of patients that have been extensively genotyped certainly represents an attractive proof-of-principle for further developments of this approach.
Future perspectives: iPSC models to drive therapeutic intervention
In this review, we summarized how iPSCs have been utilized to model certain aspects of ASD, and to quantitatively assess ASD-associated phenotypes. Looking into the future, it will be important to enhance the translational potential of current technologies. Given the heritability of ASD, gene therapy offers a complementary alternative to small molecule-based approaches, especially in the monogenic syndrome space. This approach has been already largely explored for treatment of several diseases and notably for some neurological monogenic disorders [132, 133]. Genetic correction could be tested on cell systems to optimize both optimal carriers and efficiency in rescuing specific phenotypes. AAVs have emerged as the principal delivery candidates, and have proven effective in mice carrying a null allele for Mecp2 [134, 135]. However cell toxicity, optimal time-window for transduction, and off-target effects have not yet been determined, and need further investigation [136]. iPSC models represent an immediate venue for these types of evaluations. An appealing alternative to genetic correction is modulation of gene expression by knockdown of mRNA transcripts through antisense oligonucleotides (ASOs) or short interfering RNAs (siRNAs). Both technologies are based on Watson–Crick base pairing to particular mRNA transcripts aimed at preventing their translation (detailed mechanisms of action, are reviewed elsewhere [137]). ASOs targeting Ube3a-ATS have been used to correct cognitive deficits in a mouse engineered to model features of Angelman syndrome [138]. In the same vein, ASOs have been used to normalize MeCP2 levels and rescue the neurological defects observed in mice carrying an extra copy of MECP2 [139]. This avenue appears to be broadly attractive for treatment of many syndromes caused by haploinsufficiency, where effective therapeutics should aim at restoring normal range of gene expression rather than editing the genome.
Although humanized mouse models, circumventing the issue of potential interspecies differences across vertebrates, may be utilized for testing these studies, patient-derived iPSC models represent a more expedited and scalable tool to test transability of these results in humans and vulnerable cell types, as in the case of other neurological and neurodegenerative conditions, including ALS/FTD and AD [140, 141].
Conclusions
ASD comprises a group of highly inheritable neurodevelopmental disorders characterized by impaired social interactions as well as the presentation of restrictive and repetitive behaviors [3]. The phenotypic complexity of ASD reflects its underlying genetic architecture, made of contributions from highly penetrant rare variants, and common variants each conveying small effects but collectively sha** most of its risk [4, 22,23,24,25,26].
In this review, we discussed how iPSC technology has become central to modelling various aspects of complex human disease, and can potentially allow researchers to advance our understanding of the pathophysiology of ASD, and to test personalized drug candidates. However, since the goal of iPSC-based ASD models is to reproduce and somehow functionally break down the complexity of the human brain, they need to be highly elaborate, yet reproducible. As a consequence, there are many challenges to face, including improving reliability and robustness of iPSC culturing and differentiation protocols. In this perspective, a good balance should be found in the current attempts on advancing the complexity of cellular models [142], and the vast batch effects, that might account for the most part of measured phenotypes [143].
Development of more robust protocols, employment of isogenic lines, and use of gene editing to compare variants within the same genetic background, will certainly contribute to overcome most technical roadblocks encountered in the past [118,119,120]. The rapid development of protocols to derive three-dimensional cultures and organoids, as well as to efficiently maintain co-cultures of mixed cell-types, coupled with technological advances in engineering culturing devices, all seem to be promising paths towards obtaining more complex and accurate disease models [60,61,62, 144]. Additionally, iPSC biobanks, providing access to a plethora of established, well-characterized and well-annotated iPSC, have significantly improved our understanding of the biological basis of natural genetic variation [117,118,119].
Generating faithful models is of utmost importance, but efforts in obtaining more reliable cell models, must be matched with advances in standardizing measurable cellular phenotypes related to certain aspects of ASD. In fact, the community would immensely benefit from a standardized assessment of in vitro phenotypes that reflect disease-relate mechanisms rather than generic experimental and/or genetic variance. Individual studies, mostly performed on iPSC-derived models of monogenic syndromes, have focused on assaying cell-proliferation, altered RNA-processing, electrophysiological properties, synaptic structure and calcium signaling [68, 69, 104, 106]. Assessing these phenotypes in larger-scale studies, comparing vast numbers of iPSC-derived cell lines, and linking them to dysfunction of discrete molecular pathways, is more than ever necessary. In parallel, continuous efforts in clinical sequencing of stratified patients and broadening biobank databases, will be key in advancing our understanding of complex genotype–phenotype correlations at the individual and cellular levels [48, 129].
In conclusion, recent advances in the field of human genetics, with tens of genes being identified as concentrating ASD risk [27], and hundreds of rare-variants with different degrees of penetrance [28], emphasized the need for a better understanding of the complexity of ASD. iPSC-based cell systems, while offering an unprecedented opportunity for modeling measurable ASD-related phenotypes, also provide a unique platform to rapidly validate and enhance genetic findings by nominating pathways that are disrupted across groups of ASD patients [48, 129]. These might represent as hot-spots for ASD vulnerability and desirable targets for therapeutic intervention.
Availability of data and materials
Not applicable.
Abbreviations
- AD:
-
Alzheimer disease
- ALS:
-
Atrophy lateral sclerosis
- ASD:
-
Autism spectrum disorder
- CNV:
-
Copy number variant
- CRISPR:
-
Clusters of regularly interspaced short palindromic repeats
- eQTL:
-
Expression quantitative trait locus
- ESC:
-
Embryonic stem cell
- FXS:
-
Fragile X syndrome
- FTD:
-
Frontotemporal dementia
- GWAS:
-
Genome-Wide Association Study
- iPSC:
-
Induced pluripotent stem cell
- MPRA:
-
Massive parallel reporter assay
- NPC:
-
Neural progenitor cell
- PPI:
-
Protein–protein interaction
- OPC:
-
Oligodendrocyte progenitor cell
- scRNA-seq:
-
Single-cell RNA-sequencing
References
Soldner F, Jaenisch R. Stem cells, genome editing, and the path to translational medicine. Cell (Internet). 2018;175(3):615–32. https://doi.org/10.1016/j.cell.2018.09.010.
Griffin TA, Wolfe JH. 43. Xenotransplantation of iPSC derived neural stem cells from a patient with lysosomal storage disease. Mol Ther (Internet) 2012;20:S18. https://doi.org/10.1016/S1525-0016(16)35847-6
Quesnel-Vallières M, Weatheritt RJ, Cordes SP, Blencowe BJ. Autism spectrum disorder: insights into convergent mechanisms from transcriptomics. Nat Rev Genet (Internet). 2019;20(1):51–63. https://doi.org/10.1038/s41576-018-0066-2.
Gaugler T, Klei L, Sanders SJ, Bodea CA, Goldberg AP, Lee AB, et al. Most genetic risk for autism resides with common variation. Nat Genet (Internet). 2014;46(8):881–5.
Perlman RL. Mouse models of human disease: an evolutionary perspective. Evol Med Public Heal (Internet). 2016;2016(1):170–6. https://doi.org/10.1093/emph/eow014.
Zhao X, Bhattacharyya A. Human models are needed for studying human neurodevelopmental disorders. Am J Hum Genet (Internet). 2018;103(6):829–57. https://doi.org/10.1016/j.ajhg.2018.10.009.
Awatade NT, Wong SL, Hewson CK, Fawcett LK, Kicic A, Jaffe A, et al. Human primary epithelial cell models: promising tools in the era of cystic fibrosis personalized medicine. Front Pharmacol (Internet). 2018;9:1429.
Pamies D, Bal-Price A, Chesné C, Coecke S, Dinnyes A, Eskes C, et al. Advanced good cell culture practice for human primary, stem cell-derived and organoid models as well as microphysiological systems. ALTEX Altern Anim Exp (Internet). 2018;2018:35.
Martin GR. Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proc Natl Acad Sci (Internet) 1981;78(12):7634 LP–7638.
Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, et al. Embryonic stem cell lines derived from human blastocysts. Science (80–) (Internet) 1998;282(5391):1145 LP–1147.
Aach J, Lunshof J, Iyer E, Church GM. Addressing the ethical issues raised by synthetic human entities with embryo-like features. Watt FM, editor. Elife (Internet) 2017;6:e20674. https://doi.org/10.7554/eLife.20674
de Almeida PE, Ransohoff JD, Nahid A, Wu JC. Immunogenicity of pluripotent stem cells and their derivatives. Circ Res (Internet). 2013;112(3):549–61. https://doi.org/10.1161/CIRCRESAHA.111.249243.
Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell (Internet). 2006;126(4):663–76. https://doi.org/10.1016/j.cell.2006.07.024.
Shi Y, Inoue H, Wu JC, Yamanaka S. Induced pluripotent stem cell technology: a decade of progress. Nat Rev Drug Discov (Internet). 2017;16(2):115–30. https://doi.org/10.1038/nrd.2016.245.
Avior Y, Sagi I, Benvenisty N. Pluripotent stem cells in disease modelling and drug discovery. Nat Rev Mol Cell Biol (Internet). 2016;17(3):170–82. https://doi.org/10.1038/nrm.2015.27.
Engle SJ, Blaha L, Kleiman RJ. Best practices for translational disease modeling using human iPSC-derived neurons. Neuron (Internet). 2018;100(4):783–97. https://doi.org/10.1016/j.neuron.2018.10.033.
Kaiser T, Zhou Y, Feng G. Animal models for neuropsychiatric disorders: prospects for circuit intervention. Curr Opin Neurobiol (Internet). 2017;45:59–65.
Ebert AD, Liang P, Wu JC. Induced pluripotent stem cells as a disease modeling and drug screening platform. J Cardiovasc Pharmacol (Internet). 2012;60(4):408–16.
Prilutsky D, Palmer NP, Smedemark-margulies N, Schlaeger TM, Margulies DM, Kohane IS. iPSC-derived neurons as a higher-throughput readout for autism : promises and pitfalls. Trends Mol Med (Internet). 2014;20(2):91–104. https://doi.org/10.1016/j.molmed.2013.11.004.
Deep-Soboslay A, Benes FM, Haroutunian V, Ellis JK, Kleinman JE, Hyde TM. Psychiatric brain banking: three perspectives on current trends and future directions. Biol Psychiatry (Internet). 2011;69(2):104–12. https://doi.org/10.1016/j.biopsych.2010.05.025.
Liu C, Oikonomopoulos A, Sayed N, Wu JC. Modeling human diseases with induced pluripotent stem cells: from 2D to 3D and beyond. Development (Internet) 2018;145(5):dev156166.
De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature (Internet). 2014;515(7526):209–15.
Iossifov I, O’Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature (Internet). 2014;515(7526):216–21.
Sanders SJ, He X, Willsey AJ, Ercan-Sencicek AG, Samocha KE, Cicek AE, et al. Insights into autism spectrum disorder genomic architecture and biology from 71 Risk Loci. Neuron. 2015;
Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, et al. Strong association of de novo copy number mutations with autism. Science (80–) (Internet) 2007;316(5823):445–9.
Pinto D, Delaby E, Merico D, Barbosa M, Merikangas A, Klei L, et al. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am J Hum Genet (Internet). 2014;94(5):677–94.
Grove J, Ripke S, Als TD, Mattheisen M, Walters RK, Won H, et al. Identification of common genetic risk variants for autism spectrum disorder. Nat Genet (Internet) 2019/02/25. 2019 Mar;51(3):431–44.
Satterstrom FK, Kosmicki JA, Wang J, Breen MS, De Rubeis S, An J-Y, et al. Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell (Internet). 2020;180(3):568-584.e23. https://doi.org/10.1016/j.cell.2019.12.036.
Parikshak NN, Luo R, Zhang A, Won H, Lowe JK, Chandran V, et al. Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell (Internet). 2013;155(5):1008–21.
Russo FB, Brito A, De FAM, Castanha A, De FBC, Cristina P, et al. Neurobiology of disease the use of iPSC technology for modeling autism spectrum disorders. Neurobiol Dis (Internet). 2019;130(May):104483. https://doi.org/10.1016/j.nbd.2019.104483.
Xu X, Wells AB, O'Brien DR, Nehorai A, Dougherty JD. Cell type-specific expression analysis to identify putative cellular mechanisms for neurogenetic disorders. J Neurosci (Internet) 2014;34(4):1420 LP–1431.
Chang J, Gilman SR, Chiang AH, Sanders SJ, Vitkup D. Genotype to phenotype relationships in autism spectrum disorders. Nat Neurosci (Internet). 2015;18(2):191–8. https://doi.org/10.1038/nn.3907.
Kwan KY. Transcriptional dysregulation of neocortical circuit assembly in ASD. Int Rev Neurobiol (Internet). 2013;113:167–205.
Nowakowski TJ, Bhaduri A, Pollen AA, Alvarado B, Mostajo-Radji MA, Di Lullo E, et al. Spatiotemporal gene expression trajectories reveal developmental hierarchies of the human cortex. Science (80–) (Internet) 2017;358(6368):1318 LP–1323.
Cogill SB, Srivastava AK, Yang MQ, Wang L. Co-expression of long non-coding RNAs and autism risk genes in the develo** human brain. BMC Syst Biol (Internet). 2018;12(7):91. https://doi.org/10.1186/s12918-018-0639-x.
Velmeshev D, Schirmer L, Jung D, Haeussler M, Perez Y, Mayer S, et al. Single-cell genomics identifies cell type—specific molecular changes in autism. 2019;689(May):685–9.
Campbell RE, Tour O, Palmer AE, Steinbach PA, Baird GS, Zacharias DA, et al. A monomeric red fluorescent protein. Proc Natl Acad Sci USA (Internet). 2002;99(12):7877–82.
Tomchek SD, Dunn W. Sensory processing in children with and without autism: a comparative study using the short sensory profile. Am J Occup Ther (Internet). 2007;61(2):190–200. https://doi.org/10.5014/ajot.61.2.190.
Orefice LL. Peripheral somatosensory neuron dysfunction: emerging roles in autism spectrum disorders. Neuroscience (Internet) 2020;
Orefice LL. Outside-in: Rethinking the etiology of autism spectrum disorders. Science (80–) (Internet) 2019;366(6461):45 LP–46.
Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M, Studer L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat Biotechnol. 2009;27(3):275–80.
Busskamp V, Lewis NE, Guye P, Ng AHM, Shipman SL, Byrne SM, et al. Rapid neurogenesis through transcriptional activation in human stem cells. Mol Syst Biol (Internet) 2014;10(11):760. https://doi.org/10.15252/msb.20145508
Cheng J, Kapranov P, Drenkow J, Dike S, Brubaker S, Patel S, et al. Transcriptional maps of 10 human chromosomes at 5-nucleotide resolution. Science (Internet). 2005;308(5725):1149–54.
Ho Y-S, Tsai W-H, Lin F-C, Huang W-P, Lin L-C, Wu SM, et al. Cardioprotective actions of TGF$β$RI inhibition through stimulating autocrine/paracrine of survivin and inhibiting wnt in cardiac progenitors. Stem Cells (Internet). 2016;34(2):445–55. https://doi.org/10.1002/stem.2216.
Gifford C a, Ziller MJ, Gu H, Trapnell C, Donaghey J, Tsankov A, et al. Transcriptional and epigenetic dynamics during specification of human embryonic stem cells. Cell (Internet) 2013;153(5):1149–63.
Zhao Z, Xu M, Wu M, Tian X, Zhang C, Fu X. Transdifferentiation of fibroblasts by defined factors. Vol. 17, Cellular Reprogramming. Mary Ann Liebert Inc.; 2015. p. 151–9.
Nehme R, Zuccaro E, Ghosh SD, Li C, Sherwood JL, Pietilainen O, et al. Combining NGN2 programming with developmental patterning generates human excitatory neurons with NMDAR-mediated synaptic transmission. Cell Rep (Internet). 2018;23(8):2509–23.
Wells MF, Salick MR, Piccioni F, Hill EJ, Mitchell JM, Worringer KA, et al. Genome-wide screens in accelerated human stem cell-derived neural progenitor cells identify Zika virus host factors and drivers of proliferation. bioRxiv (Internet) 2018 Jan 1;476440. Available from: http://biorxiv.org/content/early/2018/11/22/476440.abstract
Orefice LL, Mosko JR, Morency DT, Wells MF, Tasnim A, Mozeika SM, et al. Targeting peripheral somatosensory neurons to improve tactile-related phenotypes in ASD models. Cell (Internet). 2019;178(4):867-886.e24. https://doi.org/10.1016/j.cell.2019.07.024.
Han Q, Kim YH, Wang X, Liu D, Zhang Z-J, Bey AL, et al. SHANK3 Deficiency impairs heat hyperalgesia and TRPV1 signaling in primary sensory neurons. Neuron (Internet). 2016;92(6):1279–93.
Skardal A, Shupe T, Atala A. Organoid-on-a-chip and body-on-a-chip systems for drug screening and disease modeling. Vol. 21, Drug Discovery Today. Elsevier Ltd; 2016. p. 1399–411.
Choi SH, Kim YH, Quinti L, Tanzi RE, Kim DY. 3D culture models of Alzheimer’s disease: a road map to a “cure-in-a-dish”. Mol Neurodegeneration. 2016;11:75. https://doi.org/10.1186/s13024-016-0139-7.
Barres BA. The mystery and magic of glia: a perspective on their roles in health and disease. Vol. 60, Neuron. Cell Press; 2008. p. 430–40.
Fields RD, Stevens-Graham B. New insights into neuron-glia communication. Science (Internet). 2002;298(5593):556–62.
Colonna M, Butovsky O. Microglia function in the central nervous system during health and neurodegeneration. Annu Rev Immunol (Internet). 2017;35(1):441–68.
García-León JA, Kumar M, Boon R, Chau D, One J, Wolfs E, et al. SOX10 single transcription factor-based fast and efficient generation of oligodendrocytes from human pluripotent stem cells. Stem Cell Rep. 2018;10(2):655–72.
Zhang J, Liu Q. Cholesterol metabolism and homeostasis in the brain. Protein Cell (Internet). 2015;6(4):254–64.
Quadrato G, Brown J, Arlotta P. The promises and challenges of human brain organoids as models of neuropsychiatric disease. Nat Med (Internet). 2016;22(11):1220–8. https://doi.org/10.1038/nm.4214.
Dutta D, Heo I, Clevers H. Disease Modeling in Stem Cell-Derived 3D Organoid Systems. Trends Mol Med. 2017;23(5):393–410. https://doi.org/10.1016/j.molmed.2017.02.007.
Lancaster MA, Renner M, Martin C-A, Wenzel D, Bicknell LS, Hurles ME, et al. Cerebral organoids model human brain development and microcephaly. Nature (Internet). 2013;501(7467):373–9. https://doi.org/10.1038/nature12517.
Lancaster MA, Knoblich JA. Generation of cerebral organoids from human pluripotent stem cells. Nat Protoc. 2014;9(10):2329–40. https://doi.org/10.1038/nprot.2014.158.
Sasai Y. Next-generation regenerative medicine: organogenesis from stem cells in 3D culture. Cell Stem Cell (Internet). 2013;12(5):520–30. https://doi.org/10.1016/j.stem.2013.04.009.
Yin X, Mead BE, Safaee H, Langer R, Karp JM, Levy O. Engineering Stem Cell Organoids. Cell Stem Cell. 2016;18(1)25–38. https://doi.org/10.1016/j.stem.2015.12.005.
Birey F, Andersen J, Makinson CD, Islam S, Wei W, Huber N, et al. Assembly of functionally integrated human forebrain spheroids. Nature. 2017;545(7652):54–9.
Velasco S, Paulsen B, Arlotta P. 3D Brain organoids: studying brain development and disease outside the Embryo (Internet) Vol. 43, Annual Review of Neuroscience. Annual Reviews Inc.; 2020 [cited 2020 Nov 29]. p. 375–89.
Rossi G, Manfrin A, Lutolf MP. Progress and potential in organoid research (Internet) Vol. 19, Nature Reviews Genetics. Nature Publishing Group; 2018 [cited 2020 Nov 29]. p. 671–87. Available from: https://pubmed.ncbi.nlm.nih.gov/30228295/
Griesi-Oliveira K, Acab A, Gupta AR, Sunaga DY, Chailangkarn T, Nicol X, et al. Modeling non-syndromic autism and the impact of TRPC6 disruption in human neurons. Mol Psychiatry. 2015;20(11):1350–65.
Marchetto MCN, Carromeu C, Acab A, Yu D, Yeo GW, Mu Y, et al. A Model for Neural Development and Treatment of Rett Syndrome Using Human Induced Pluripotent Stem Cells. Cell (Internet). 2010;143(4):527–39. https://doi.org/10.1016/j.cell.2010.10.016.
Marchetto MC, Belinson H, Tian Y, Freitas BC, Fu C, Vadodaria K, et al. Altered proliferation and networks in neural cells derived from idiopathic autistic individuals. Mol Psychiatry (Internet). 2017;22(6):820–35.
Shcheglovitov A, Shcheglovitova O, Yazawa M, Portmann T, Shu R, Sebastiano V, et al. SHANK3 and IGF1 restore synaptic deficits in neurons from 22q13 deletion syndrome patients. Nature. 2013;503(7475):267–71.
Nehme R, Barrett LE. Using human pluripotent stem cell models to study autism in the era of big data. Mol Autism (Internet). 2020;11(1):21. https://doi.org/10.1186/s13229-020-00322-9.
Gordon A, Geschwind DH. Human in vitro models for understanding mechanisms of autism spectrum disorder (Internet) Vol. 11, Molecular Autism. BioMed Central Ltd.; 2020 [cited 2020 Nov 29]. Available from: https://pubmed.ncbi.nlm.nih.gov/32299488/
Courchesne E, PR M, ME C, al et. Neuron number and size in prefrontal cortex of children with autism. JAMA (Internet) 2011;306(18):2001–10.
Malhotra D, Sebat J. CNVs: Harbingers of a rare variant revolution in psychiatric genetics. Vol. 148, Cell. NIH Public Access; 2012. p. 1223–41.
Bernier R, Golzio C, **ong B, Stessman HA, Coe BP, Penn O, et al. Disruptive CHD8 mutations define a subtype of autism early in development. Cell. 2014;158(2):263–76.
Butler MG, Dazouki MJ, Zhou XP, Talebizadeh Z, Brown M, Takahashi TN, et al. Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J Med Genet. 2005;42(4):318–21.
Courcet JB, Faivre L, Malzac P, Masurel-Paulet A, Lopez E, Callier P, et al. The DYRK1A gene is a cause of syndromic intellectual disability with severe microcephaly and epilepsy. J Med Genet. 2012;49(12):731–6.
Archer HL, Evans J, Edwards S, Colley J, Newbury-Ecob R, O’Callaghan F, et al. CDKL5 mutations cause infantile spasms, early onset seizures, and severe mental retardation in female patients. J Med Genet. 2006;43(9):729–34.
Deshpande A, Yadav S, Dao DQ, Wu ZY, Hokanson KC, Cahill MK, et al. Cellular Phenotypes in human iPSC-Derived Neurons from a Genetic Model of Autism Spectrum Disorder. Cell Rep. 2017;21(10):2678–87.
Wang M, Wei PC, Lim CK, Gallina IS, Marshall S, Marchetto MC, et al. Increased neural progenitor proliferation in a hiPSC model of autism induces replication stress-associated genome instability. Cell Stem Cell (Internet). 2020;26(2):221-233.e6.
Abrahams BS, Geschwind DH. Advances in autism genetics: on the threshold of a new neurobiology. Nat Rev Genet (Internet). 2008;9(5):341–55.
Tran SS, Jun H-I, Bahn JH, Azghadi A, Ramaswami G, Van Nostrand EL, et al. Widespread RNA editing dysregulation in brains from autistic individuals. Nat Neurosci (Internet). 2019;22(1):25–36. https://doi.org/10.1038/s41593-018-0287-x.
Griesi-Oliveira K, Fogo MS, Pinto BGG, Alves AY, Suzuki AM, Morales AG, et al. Transcriptome of iPSC-derived neuronal cells reveals a module of co-expressed genes consistently associated with autism spectrum disorder. Mol Psychiatry (Internet). 2020. https://doi.org/10.1038/s41380-020-0669-9.
Stamova BS, Tian Y, Nordahl CW, Shen MD, Rogers S, Amaral DG, et al. Evidence for differential alternative splicing in blood of young boys with autism spectrum disorders. Mol Autism (Internet). 2013;4(1):30. https://doi.org/10.1186/2040-2392-4-30.
Hicks SD, Rajan AT, Wagner KE, Barns S, Carpenter RL, Middleton FA. Validation of a Salivary RNA Test for Childhood Autism Spectrum Disorder (Internet) Vol. 9, Frontiers in Genetics . 2018. p. 534. https://doi.org/10.3389/fgene.2018.00534
Parikshak NN, Gandal MJ, Geschwind DH. Systems biology and gene networks in neurodevelopmental and neurodegenerative disorders. Nat Rev Genet (Internet). 2015;16:441. https://doi.org/10.1038/nrg3934.
Fernandez BA, Scherer SW. Syndromic autism spectrum disorders: moving from a clinically defined to a molecularly defined approach. Dialogues Clin Neurosci (Internet). 2017;19(4):353–71.
C Yuen RK, Merico D, Bookman M, L Howe J, Thiruvahindrapuram B, Patel R V, et al. Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nat Neurosci (Internet) 2017;20(4):602–11
Schmidt D, Wilson MD, Ballester B, Schwalie PC, Brown GD, Marshall A, et al. Five-vertebrate ChIP-seq reveals the evolutionary dynamics of transcription factor binding. Science (Internet). 2010;328(5981):1036–40.
Sunamura N, Iwashita S, Enomoto K, Kadoshima T, Isono F. Loss of the fragile X mental retardation protein causes aberrant differentiation in human neural progenitor cells. Sci Rep (Internet). 2018;8(1):11585. https://doi.org/10.1038/s41598-018-30025-4.
Telias M, Mayshar Y, Amit A, Ben-Yosef D. Molecular mechanisms regulating impaired neurogenesis of Fragile X syndrome human embryonic stem cells. Stem Cells Dev (Internet) 2015;24(20):2353–65. Available from: https://pubmed.ncbi.nlm.nih.gov/26393806/
Lu P, Chen X, Feng Y, Zeng Q, Jiang C, Zhu X, et al. Integrated transcriptome analysis of human iPS cells derived from a fragile X syndrome patient during neuronal differentiation. Sci China Life Sci (Internet). 2016;59(11):1093–105. https://doi.org/10.1007/s11427-016-0194-6.
Frye RE, Vassall S, Kaur G, Lewis C, Karim M, Rossignol D. Emerging biomarkers in autism spectrum disorder: a systematic review. Ann Transl Med (Internet). 2019;7(23):792.
Lugtenberg D, Kleefstra T, Oudakker AR, Nillesen WM, Yntema HG, Tzschach A, et al. Structural variation in Xq28: MECP2 duplications in 1% of patients with unexplained XLMR and in 2% of male patients with severe encephalopathy. Eur J Hum Genet. 2009;17(4):444–53.
Nageshappa S, Carromeu C, Trujillo CA, Mesci P, Espuny-Camacho I, Pasciuto E, et al. Altered neuronal network and rescue in a human MECP2 duplication model. Mol Psychiatry. 2016;21(2):178–88.
Yi F, Danko T, Botelho SC, Patzke C, Pak C, Wernig M, et al. Autism-associated SHANK3 haploinsufficiency causes Ih channelopathy in human neurons. Science (80–). 2016;352(6286):aaf2669.
Doers ME, Musser MT, Nichol R, Berndt ER, Baker M, Gomez TM, et al. IPSC-derived forebrain neurons from FXS individuals show defects in initial Neurite outgrowth. Stem Cells Dev. 2014;23(15):1777–87.
Krey JF, Paşca SP, Shcheglovitov A, Yazawa M, Schwemberger R, Rasmusson R, et al. Timothy syndrome is associated with activity-dependent dendritic retraction in rodent and human neurons. Nat Neurosci. 2013;16(2):201–9.
Chailangkarn T, Trujillo CA, Freitas BC, Hrvoj-Mihic B, Herai RH, Yu DX, et al. A human neurodevelopmental model for Williams syndrome. Nature. 2016;536(7616):338–43.
Zaslavsky K, Zhang WB, McCready FP, Rodrigues DC, Deneault E, Loo C, et al. SHANK2 mutations associated with autism spectrum disorder cause hyperconnectivity of human neurons. Nat Neurosci. 2019;22(4):556–64.
Deneault E, White SH, Rodrigues DC, Ross PJ, Faheem M, Zaslavsky K, et al. Complete disruption of autism-susceptibility genes by gene editing predominantly reduces functional connectivity of isogenic human neurons. Stem Cell Reports. 2018;11(5):1211–25.
Pak C, Danko T, Zhang Y, Aoto J, Anderson G, Maxeiner S, et al. Human neuropsychiatric disease modeling using conditional deletion reveals synaptic transmission defects caused by heterozygous mutations in NRXN1. Cell Stem Cell (Internet). 2015;17(3):316–28. https://doi.org/10.1016/j.stem.2015.07.017.
Ross PJ, Zhang WB, Mok RSF, Zaslavsky K, Deneault E, D’Abate L, et al. Synaptic dysfunction in human neurons with autism-associated deletions in PTCHD1-AS. Biol Psychiatry (Internet) 2020;87(2):139–49. Available from: https://pubmed.ncbi.nlm.nih.gov/31540669/
Deneault E, Faheem M, White SH, Rodrigues DC, Sun S, Wei W, et al. CNTN5-/+or EHMT2-/+human iPSC-derived neurons from individuals with autism develop hyperactive neuronal networks. Zoghbi HY, Chao M V, editors. Elife (Internet) 2019;8:e40092. https://doi.org/10.7554/eLife.40092
Südhof TC. Neuroligins and neurexins link synaptic function to cognitive disease. Nature (Internet). 2008;455(7215):903–11.
Avazzadeh S, McDonagh K, Reilly J, Wang Y, Boomkamp SD, McInerney V, et al. Increased Ca(2+) signaling in NRXN1α (+/−) neurons derived from ASD induced pluripotent stem cells. Mol Autism (Internet). 2019;10:52.
DeRosa BA, El Hokayem J, Artimovich E, Garcia-Serje C, Phillips AW, Van Booven D, et al. Convergent Pathways in idiopathic autism revealed by time course transcriptomic analysis of patient-derived neurons. Sci Rep (Internet). 2018;8(1):8423. https://doi.org/10.1038/s41598-018-26495-1.
Heo M, Maslov S, Shakhnovich E. Topology of protein interaction network shapes protein abundances and strengths of their functional and nonspecific interactions. Proc Natl Acad Sci (Internet) 2011;108(10):4258 LP–4263. Available from: http://www.pnas.org/content/108/10/4258.abstract
Safari-Alighiarloo N, Taghizadeh M, Rezaei-Tavirani M, Goliaei B, Peyvandi AA. Protein-protein interaction networks (PPI) and complex diseases. Gastroenterol Hepatol from bed to bench (Internet). 2014;7(1):17–31.
Janjić V, Pržulj N. Biological function through network topology: a survey of the human diseasome. Brief Funct Genomics (Internet). 2012;11(6):522–32. https://doi.org/10.1093/bfgp/els037.
Lage K, Karlberg EO, Størling ZM, Ólason PÍ, Pedersen AG, Rigina O, et al. A human phenome-interactome network of protein complexes implicated in genetic disorders. Nat Biotechnol (Internet). 2007;25(3):309–16. https://doi.org/10.1038/nbt1295.
Neale BM, Kou Y, Liu L, Ma’ayan A, Samocha KE, Sabo A, et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature (Internet) 2012;485:242. https://doi.org/10.1038/nature11011
Li T, Wernersson R, Hansen RB, Horn H, Mercer J, Slodkowicz G, et al. A scored human protein-protein interaction network to catalyze genomic interpretation. Nat Methods. 2016;
Lage K, Greenway SC, Rosenfeld JA, Wakimoto H, Gorham JM, Segrè A V, et al. Genetic and environmental risk factors in congenital heart disease functionally converge in protein networks driving heart development. Proc Natl Acad Sci (Internet) 2012;109(35):14035 LP–14040
Pintacuda G, Lassen FH, Hsu Y-HH, Kim A, Martín JM, Malolepsza E, et al. Genoppi: an open-source software for robust and standardized integration of proteomic and genetic data. bioRxiv (Internet) 2020 Jan 1;2020.05.04.076034. Available from: http://biorxiv.org/content/early/2020/05/05/2020.05.04.076034.abstract
Abudayyeh OO, Gootenberg JS, Essletzbichler P, Han S, Joung J, Belanto JJ, et al. RNA targeting with CRISPR-Cas13. Nature (Internet). 2017;550(7675):280–4.
Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, et al. Multiplex genome engineering using CRISPR/Cas systems. Science (80–) (Internet) 2013;339(6121):819–23
Larson MH, Gilbert LA, Wang X, Lim WA, Weissman JS, Qi LS. CRISPR interference (CRISPRi) for sequence-specific control of gene expression. Nat Protoc (Internet). 2013;8(11):2180–96.
Kampmann M. CRISPR-based functional genomics for neurological disease (Internet) Vol. 16, Nature Reviews Neurology. Nature Research; 2020 [cited 2020 Nov 29]. p. 465–80. Available from: https://pubmed.ncbi.nlm.nih.gov/32641861/
Townsley KG, Brennand KJ, Huckins LM. Massively parallel techniques for cataloguing the regulome of the human brain (Internet) Vol. 23, Nature Neuroscience. Nature Research; 2020 [cited 2020 Dec 1]. Available from: https://pubmed.ncbi.nlm.nih.gov/33199899/
Tian R, Gachechiladze MA, Ludwig CH, Laurie MT, Hong JY, Nathaniel D, et al. CRISPR Interference-Based Platform for Multimodal Genetic Screens in Human iPSC-Derived Neurons. Neuron (Internet). 2019;104(2):239–55.
Dixit A, Parnas O, Li B, Chen J, Fulco CP, Jerby-Arnon L, et al. Perturb-Seq: Dissecting Molecular Circuits with Scalable Single-Cell RNA Profiling of Pooled Genetic Screens. Cell (Internet). 2016;167(7):1853–66.
Willsey AJ, Morris MT, Wang S, Willsey HR, Sun N, Teerikorpi N, et al. The psychiatric cell map initiative: a convergent systems biological approach to illuminating key molecular pathways in neuropsychiatric disorders (internet) Vol. 174, Cell. Cell Press; 2018 [cited 2020 Nov 29]. p. 505–20. Available from: https://pubmed.ncbi.nlm.nih.gov/30053424/
Shen JP, Zhao D, Sasik R, Luebeck J, Birmingham A, Bojorquez-Gomez A, et al. Combinatorial CRISPR-Cas9 screens for de novo map** of genetic interactions. Nat Methods (Internet). 2017;14(6):573–6.
** X, Simmons SK, Guo AX, Shetty AS, Ko M, Nguyen L, et al. <em>In vivo</em> Perturb-Seq reveals neuronal and glial abnormalities associated with Autism risk genes. bioRxiv (Internet) 2019 Jan 1;791525. Available from: http://biorxiv.org/content/early/2019/10/07/791525.abstract
Tian R, Abarientos A, Hong J, Hashemi SH, Yan R, Nalls MA, et al. Genome-wide CRISPRi/a screens in human neurons link lysosomal failure to ferroptosis. bioRxiv (Internet) 2020 Jan 1;2020.06.27.175679. Available from: http://biorxiv.org/content/early/2020/07/22/2020.06.27.175679.abstract
Goldman SL, MacKay M, Afshinnekoo E, Melnick AM, Wu S, Mason CE. The Impact of Heterogeneity on Single-Cell Sequencing (Internet) Vol. 10, Frontiers in Genetics . 2019. p. 8. Available from: https://www.frontiersin.org/article/https://doi.org/10.3389/fgene.2019.00008
Datlinger P, Rendeiro AF, Schmidl C, Krausgruber T, Traxler P, Klughammer J, et al. Pooled CRISPR screening with single-cell transcriptome readout. Nat Methods (Internet). 2017;14(3):297–301. https://doi.org/10.1038/nmeth.4177.
Cuomo ASE, Seaton DD, McCarthy DJ, Martinez I, Bonder MJ, Garcia-Bernardo J, et al. Single-cell RNA-sequencing of differentiating iPS cells reveals dynamic genetic effects on gene expression. Nat Commun (Internet). 2020;11(1):810. https://doi.org/10.1038/s41467-020-14457-z.
Mitchell JM, Nemesh J, Ghosh S, Handsaker RE, Mello CJ, Meyer D, et al. Map** genetic effects on cellular phenotypes with “cell villages.” bioRxiv (Internet) 2020 Jan 1;2020.06.29.174383. Available from: http://biorxiv.org/content/early/2020/06/29/2020.06.29.174383.abstract
Cederquist GY, Tchieu J, Callahan SJ, Ramnarine K, Ryan S, Zhang C, et al. A Multiplex Human Pluripotent Stem Cell Platform Defines Molecular and Functional Subclasses of Autism-Related Genes. Cell Stem Cell (Internet). 2020;27(1):35–49.
Hoy SM. Nusinersen: first global approval. Drugs (Internet). 2017;77(4):473–9. https://doi.org/10.1007/s40265-017-0711-7.
Mendell JR, Al-Zaidy S, Shell R, Arnold WD, Rodino-Klapac LR, Prior TW, et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N Engl J Med (Internet). 2017;377(18):1713–22. https://doi.org/10.1056/NEJMoa1706198.
Gadalla KKE, Bailey MES, Spike RC, Ross PD, Woodard KT, Kalburgi SN, et al. Improved survival and reduced phenotypic severity following AAV9/MECP2 gene transfer to neonatal and juvenile male Mecp2 knockout mice. Mol Ther (Internet). 2013;21(1):18–30. https://doi.org/10.1038/mt.2012.200.
Garg SK, Lioy DT, Cheval H, McGann JC, Bissonnette JM, Murtha MJ, et al. Systemic delivery of MeCP2 rescues behavioral and cellular deficits in female mouse models of rett syndrome. J Neurosci (Internet) 2013;33(34):13612 LP–13620.
Gray SJ. Gene therapy and neurodevelopmental disorders. Neuropharmacology (Internet). 2013;68:136–42.
Kole R, Krainer AR, Altman S. RNA therapeutics: beyond RNA interference and antisense oligonucleotides. Nat Rev Drug Discov (Internet). 2012;11(2):125–40. https://doi.org/10.1038/nrd3625.
Meng L, Ward AJ, Chun S, Bennett CF, Beaudet AL, Rigo F. Towards a therapy for Angelman syndrome by targeting a long non-coding RNA. Nature (Internet). 2015;518(7539):409–12. https://doi.org/10.1038/nature13975.
Sztainberg Y, Chen H, Swann JW, Hao S, Tang B, Wu Z, et al. Reversal of phenotypes in MECP2 duplication mice using genetic rescue or antisense oligonucleotides. Nature (Internet). 2015;528(7580):123–6. https://doi.org/10.1038/nature16159.
Korecka JA, Talbot S, Osborn TM, de Leeuw SM, Levy SA, Ferrari EJ, et al. Neurite collapse and altered ER Ca2+ control in human parkinson disease patient iPSC-derived neurons with LRRK2 G2019S mutation. Stem Cell Reports (Internet). 2019;12(1):29–41. https://doi.org/10.1016/j.stemcr.2018.11.021.
Lutz C. Realizing the gains and losses in C9ORF72 ALS/FTD. Nat Neurosci (Internet). 2020;23(5):596–7. https://doi.org/10.1038/s41593-020-0622-x.
Velasco S, Kedaigle AJ, Simmons SK, Nash A, Rocha M, Quadrato G, et al. Individual brain organoids reproducibly form cell diversity of the human cerebral cortex. Nature (Internet). 2019;570(7762):523–7. https://doi.org/10.1038/s41586-019-1289-x.
Schwartzentruber J, Foskolou S, Kilpinen H, Rodrigues J, Alasoo K, Knights AJ, et al. Molecular and functional variation in iPSC-derived sensory neurons. Nat Genet (Internet). 2018;50(1):54–61. https://doi.org/10.1038/s41588-017-0005-8.
Takebe T, Zhang R-R, Koike H, Kimura M, Yoshizawa E, Enomura M, et al. Generation of a vascularized and functional human liver from an iPSC-derived organ bud transplant. Nat Protoc (Internet). 2014;9(2):396–409. https://doi.org/10.1038/nprot.2014.020.
Mariani J, Coppola G, Zhang P, Abyzov A, Provini L, Tomasini L, et al. FOXG1-dependent dysregulation of GABA/glutamate neuron differentiation in autism spectrum disorders. Cell (Internet). 2015;162(2):375–90. https://doi.org/10.1016/j.cell.2015.06.034.
Gandal MJ, Haney JR, Parikshak NN, Leppa V, Ramaswami G, Hartl C, et al. Shared molecular neuropathology across major psychiatric disorders parallels polygenic overlap. Science (80–) (Internet) 2018;359(6376):693 LP–697.
Wang P, Zhao D, Lachman HM, Zheng D. Enriched expression of genes associated with autism spectrum disorders in human inhibitory neurons. Transl Psychiatry (Internet). 2018;8(1):13. https://doi.org/10.1038/s41398-017-0058-6.
Hashemi E, Ariza J, Rogers H, Noctor SC, Martínez-Cerdeño V. The number of parvalbumin-expressing interneurons is decreased in the prefrontal cortex in Autism. Cereb Cortex (Internet). 2016;27(3):1931–43. https://doi.org/10.1093/cercor/bhw021.
Filice F, Vörckel KJ, Sungur AÖ, Wöhr M, Schwaller B. Reduction in parvalbumin expression not loss of the parvalbumin-expressing GABA interneuron subpopulation in genetic parvalbumin and shank mouse models of autism. Mol Brain (Internet). 2016;9(1):10. https://doi.org/10.1186/s13041-016-0192-8.
Phan BN, Bohlen JF, Davis BA, Ye Z, Chen H-Y, Mayfield B, et al. A myelin-related transcriptomic profile is shared by Pitt-Hopkins syndrome models and human autism spectrum disorder. Nat Neurosci (Internet). 2020;23(3):375–85. https://doi.org/10.1038/s41593-019-0578-x.
Kim H-J, Cho M-H, Shim WH, Kim JK, Jeon E-Y, Kim D-H, et al. Deficient autophagy in microglia impairs synaptic pruning and causes social behavioral defects. Mol Psychiatry (Internet). 2017;22(11):1576–84. https://doi.org/10.1038/mp.2016.103.
Russo FB, Freitas BC, Pignatari GC, Fernandes IR, Sebat J, Muotri AR, et al. Modeling the interplay between neurons and astrocytes in autism using human induced pluripotent stem cells. Biol Psychiatry. 2018;83(7):569–78.
Mead J, Ashwood P. Evidence supporting an altered immune response in ASD. Immunol Lett (Internet). 2015;163(1):49–55.
Zhang Y, Pak C, Han Y, Ahlenius H, Zhang Z, Chanda S, et al. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron (Internet). 2013;78(5):785–98.
Yang N, Chanda S, Marro S, Ng Y-H, Janas JA, Haag D, et al. Generation of pure GABAergic neurons by transcription factor programming. Nat Methods (Internet). 2017;14(6):621–8. https://doi.org/10.1038/nmeth.4291.
Mahajani S, Raina A, Fokken C, Kügler S, Bähr M. Homogenous generation of dopaminergic neurons from multiple hiPSC lines by transient expression of transcription factors. Cell Death Dis (Internet). 2019;10(12):898. https://doi.org/10.1038/s41419-019-2133-9.
Guimarães MZP, De Vecchi R, Vitória G, Sochacki JK, Paulsen BS, Lima I, et al. Generation of iPSC-derived human peripheral sensory neurons releasing substance P elicited by TRPV1 Agonists. Front Mol Neurosci (Internet). 2018;11:277.
Maroof AM, Keros S, Tyson JA, Ying S-W, Ganat YM, Merkle FT, et al. Directed differentiation and functional maturation of cortical interneurons from human embryonic stem cells. Cell Stem Cell (Internet). 2013;12(5):559–72.
Gillentine M, Yin J, Bajic A, … PZ-TAJ of, 2017 undefined. Functional consequences of CHRNA7 copy-number alterations in induced pluripotent stem cells and neural progenitor cells. Elsevier (Internet) [cited 2020 Nov 30]; Available from: https://www.sciencedirect.com/science/article/pii/S0002929717303932
Lin M, Pedrosa E, Hrabovsky A, Chen J, Puliafito BR, Gilbert SR, et al. Integrative transcriptome network analysis of iPSC-derived neurons from schizophrenia and schizoaffective disorder patients with 22q11.2 deletion. BMC Syst Biol. 2016;10, 1.
Paşca S, Portmann T, Voineagu I, medicine MY-N, 2011 undefined. Using iPSC-derived neurons to uncover cellular phenotypes associated with Timothy syndrome. nature.com (Internet) 2011 [cited 2020 Nov 30]; Available from: https://www.nature.com/articles/nm.2576.pdf?origin=ppub
Wang P, Mokhtari R, Pedrosa E, Kirschenbaum M, Bayrak C, Zheng D, et al. CRISPR/Cas9-mediated heterozygous knockout of the autism gene CHD8 and characterization of its transcriptional networks in cerebral organoids derived from iPS cells. Mol Autism. 2017 Mar 20;8(1).
Marro S, Chanda S, Yang N, Janas J, Neuron GV-, 2019 undefined. Neuroligin-4 regulates excitatory synaptic transmission in human neurons. Elsevier (Internet) [cited 2020 Nov 30]; Available from: https://www.sciencedirect.com/science/article/pii/S0896627319304969
Flaherty E, Zhu S, Barretto N, Cheng E, … PD-N, 2019 undefined. Neuronal impact of patient-specific aberrant NRXN1α splicing. nature.com (Internet) [cited 2020 Nov 30]; Available from: https://www.nature.com/articles/s41588-019-0539-z
Lam M, Moslem M, Bryois J, Pronk R, … EU-E cell, 2019 undefined. Single cell analysis of autism patient with bi-allelic NRXN1-alpha deletion reveals skewed fate choice in neural progenitors and impaired neuronal. Elsevier (Internet) [cited 2020 Nov 30]; Available from: https://www.sciencedirect.com/science/article/pii/S001448271930309X
Gouder L, Vitrac A, Goubran-Botros H, reports AD-S, 2019 undefined. Altered spinogenesis in iPSC-derived cortical neurons from patients with autism carrying de novo SHANK3 mutations. nature.com (Internet) [cited 2020 Nov 30]; Available from: https://www.nature.com/articles/s41598-018-36993-x
Huang G, Chen S, Chen X, Zheng J, Xu Z, Doostparast Torshizi A, et al. Uncovering the functional link between SHANK3 deletions and deficiency in neurodevelopment using iPSC-derived human neurons. Front Neuroanat. 2019;18:13.
Kathuria A, Nowosiad P, Jagasia R, … SA-M, 2018 undefined. Stem cell-derived neurons from autistic individuals with SHANK3 mutation show morphogenetic abnormalities during early development. nature.com (Internet) [cited 2020 Nov 30]; Available from: https://www.nature.com/articles/mp2017185
Fink J, Robinson T, Germain N, … CS-N, 2017 undefined. Disrupted neuronal maturation in Angelman syndrome-derived induced pluripotent stem cells. nature.com (Internet) [cited 2020 Nov 30]; Available from: https://www.nature.com/articles/ncomms15038
Xuyang Sun A, Yuan Q, Fukuda M, Yu W, Yan H, Gui Yin Lim G, et al. Potassium channel dysfunction in human neuronal models of Angelman syndrome (Internet) science.sciencemag.org. [cited 2020 Nov 30]. Available from: http://science.sciencemag.org/
Lewis EMA, Meganathan K, Baldridge D, Gontarz P, Zhang B, Bonni A, et al. Cellular and molecular characterization of multiplex autism in human induced pluripotent stem cell-derived neurons. Mol Autism. 2019;10:1.
Liu X, Campanac E, Cheung H-H, Ziats MN, Canterel-Thouennon L, Raygada M, et al. Idiopathic autism: cellular and molecular phenotypes in pluripotent stem cell-derived neurons. Mol Neurobiol (Internet). 2017;54(6):4507–23. https://doi.org/10.1007/s12035-016-9961-8.
Moore D, Meays B, Madduri L, FS-S cells, 2019 undefined. Downregulation of an evolutionary young miR-1290 in an iPSC-derived neural stem cell model of autism spectrum disorder. hindawi.com (Internet) [cited 2020 Nov 30]; Available from: https://www.hindawi.com/journals/sci/2019/8710180/abs/
Schafer S, Paquola A, Stern S, … DG-N, 2019 undefined. Pathological priming causes developmental gene network heterochronicity in autistic subject-derived neurons. nature.com (Internet) [cited 2020 Nov 30]; Available from: https://www.nature.com/articles/s41593-018-0295-x
Kircher M, **ong C, Martin B, Schubach M, Inoue F, Bell RJA, et al. Saturation mutagenesis of twenty disease-associated regulatory elements at single base-pair resolution. Nat Commun. 2019;10:1.
Ulirsch JC, Nandakumar SK, Wang L, Giani FC, Zhang X, Rogov P, et al. Systematic functional dissection of common genetic variation affecting red blood cell traits. Cell (Internet). 2016;165(6):1530–45.
Tian R, Abarientos A, Hong J, Hashemi SH, Yan R, Nalls MA, et al. Genome-wide CRISPRi/a screens in human neurons link lysosomal failure to ferroptosis. biorxiv.org (Internet) 2020; https://doi.org/10.1101/2020.06.27.175679
Diao Y, Fang R, Li B, Meng Z, Yu J, Qiu Y, et al. A tiling-deletion-based genetic screen for cis-regulatory element identification in mammalian cells. Nat Methods. 2017;14(6):629–35.
**e S, Duan J, Li B, Zhou P, Hon GC. Multiplexed engineering and analysis of combinatorial enhancer activity in single cells. Mol Cell. 2017;66(2):285–99.
Fulco CP, Nasser J, Jones TR, Munson G, Bergman DT, Subramanian V, et al. Activity-by-contact model of enhancer–promoter regulation from thousands of CRISPR perturbations. Vol. 51, Nature Genetics. Nature Research; 2019. p. 1664–9.
Gasperini M, Hill AJ, McFaline-Figueroa JL, Martin B, Kim S, Zhang MD, et al. A Genome-wide Framework for Map** Gene Regulation via Cellular Genetic Screens. Cell. 2019;176(1–2):377–90.
Soldner F, Jaenisch R. iPSC Disease Modeling. Science (80–) (Internet) 2012;338(6111):1155 LP–1156.
Gunaseeli I, Doss MX, Antzelevitch C, Sachinidis JH and A. Induced pluripotent stem cells as a model for accelerated patient- and disease-specific drug discovery (Internet) Vol. 17, Current Medicinal Chemistry. 2010. p. 759–66. Available from: http://www.eurekaselect.com/node/70961/article
Dimos JT, Rodolfa KT, Niakan KK, Weisenthal LM, Mitsumoto H, Chung W, et al. Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science (80–) (Internet) 2008;321(5893):1218 LP–1221.
Mandai M, Watanabe A, Kurimoto Y, Hirami Y, Morinaga C, Daimon T, et al. Autologous induced stem-cell–derived retinal cells for macular degeneration. N Engl J Med (Internet). 2017;376(11):1038–46. https://doi.org/10.1056/NEJMoa1608368.
van den Berg A, Mummery CL, Passier R, van der Meer AD. Personalised organs-on-chips: functional testing for precision medicine. Lab Chip (Internet). 2019;19(2):198–205. https://doi.org/10.1039/C8LC00827B.
Hoekstra SD, Stringer S, Heine VM, Posthuma D. Genetically-informed patient selection for iPSC studies of complex diseases may aid in reducing cellular heterogeneity (Internet) Vol. 11, Frontiers in Cellular Neuroscience . 2017. p. 164. Available from: https://doi.org/10.3389/fncel.2017.00164
Sebastiano V, Maeder ML, Angstman JF, Haddad B, Khayter C, Yeo DT, et al. In situ genetic correction of the sickle cell anemia mutation in human induced pluripotent stem cells using engineered zinc finger nucleases. Stem Cells (Internet). 2011;29(11):1717–26. https://doi.org/10.1002/stem.718.
Yusa K, Rashid ST, Strick-Marchand H, Varela I, Liu P-Q, Paschon DE, et al. Targeted gene correction of α1-antitrypsin deficiency in induced pluripotent stem cells. Nature (Internet). 2011;478(7369):391–4. https://doi.org/10.1038/nature10424.
Kilpinen H, Goncalves A, Leha A, Afzal V, Alasoo K, Ashford S, et al. Common genetic variation drives molecular heterogeneity in human iPSCs. Nature (Internet). 2017;546(7658):370–5. https://doi.org/10.1038/nature22403.
Carcamo-Orive I, Hoffman GE, Cundiff P, Beckmann ND, D’Souza SL, Knowles JW, et al. Analysis of transcriptional variability in a large human iPSC library reveals genetic and non-genetic determinants of heterogeneity. Cell Stem Cell (Internet). 2017;20(4):518-532.e9. https://doi.org/10.1016/j.stem.2016.11.005.
Acknowledgements
Not applicable.
Funding
Funding was provided by the Stanley Center for Psychiatric Research at the Broad Institute.
Author information
Authors and Affiliations
Contributions
GP, JMM, KCE reviewed existing literature and wrote the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
KCE is a co-founder of Q-State Biosciences, Quralis and Enclear Therapies, and currently employed at BioMarin Pharmaceutical.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Pintacuda, G., Martín, J.M. & Eggan, K.C. Mind the translational gap: using iPS cell models to bridge from genetic discoveries to perturbed pathways and therapeutic targets. Molecular Autism 12, 10 (2021). https://doi.org/10.1186/s13229-021-00417-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13229-021-00417-x