
Abstract
Interpretation of the multitude of variants obtained from next generation sequencing (NGS) is labor intensive and complex. Web-based interfaces such as Galaxy streamline the generation of variant lists but lack flexibility in the downstream annotation and filtering that are necessary to identify causative variants in medical genomics. To this end, we built VariantDB, a web-based interactive annotation and filtering platform that automatically annotates variants with allele frequencies, functional impact, pathogenicity predictions and pathway information. VariantDB allows filtering by all annotations, under dominant, recessive or de novo inheritance models and is freely available at http://www.biomina.be/app/variantdb/.
Similar content being viewed by others


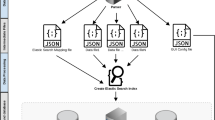
Background
Next generation sequencing (NGS) has the power to screen a whole genome for all kinds of genetic variation in a single experiment [1]. In medical genetics, NGS has proven to be a key tool to identify disease-causing mutations in individuals with Mendelian disorders. Most studies so far have concentrated on the exome or protein coding part of the genome, which comprises only 1.5% of the complete human genome. Despite the smaller target size, whole exome sequencing (WES) typically yields over 20,000 protein altering variants per sample [2],[3]. Today, several studies have proven the potential of WES to identify causal genetic defects underlying various disorders in a substantial number of patients [4]-[6]. As such, WES greatly reduces experimental costs while achieving high analytical power. Despite the proven utility of, and high diagnostic demand for, NGS-based assays, interpretation and filtering of the extensive variant lists is currently a labor-intensive and cumbersome task, and hampers the implementation of WES in routine diagnostics [3],[4].
NGS data analysis can be subdivided into two sequential subtasks. The first task comprises quality control of the raw sequencing reads, map** reads to a reference genome and generating a primary variant list [7]. The second stage comprises interpretation of the variants in relation to the patient’s phenotype. Several approaches are available to handle the read-to-variant stage. Commercial packages often offer all-in-one solutions such as SeqNext [8], CLCBio Genomic Workbench [9] or Illumina’s CASAVA [10]. Academic solutions on the other hand typically consist of the combination of sequential tools for specific steps in the analysis. These include tools for cleaning up the sequence (for example, FASTX-Toolkit [11], CutAdapt [12]), aligning reads to the genome (for example, Bowtie [13], BWA [14]) and variant calling (for example, samtools [15], Genome Analysis Toolkit (GATK) [16]). Out of this extensive collection of analysis options, the research community has converged on a BWA-GATK based pipeline as the preferred method, as it appears to have the highest sensitivity and specificity. Recently, the superiority of this consensus approach was corroborated by an in-depth performance analysis of several available methods [17]. Galaxy, a flexible and publicly available online platform, offers streamlined execution of consecutive processing steps to non-bioinformatics experts, thus providing a straightforward implementation of the first analysis stage [18]-[20].
Ideally, the second analysis stage would be able to handle identified variants of either a single sample, a family-based analysis, or a case/control study, while at the same time integrating extensive annotation with biological information and dynamic filtering. Commercial packages such as Bench Suite [21] provide turn-key solutions for variant annotation, interpretation and prioritization. However, these platforms are tailored at long-term usage in routine clinical diagnostics laboratories, and are less suitable for use in smaller laboratories or research settings that typically demand more flexible and less expensive solutions.
Currently available academic software still requires the manual inspection of variants using a combination of web tools and stand-alone packages. Many of these tools were developed for specific research questions, such as either family-based [22],[23] or case/control-based experiments [24], or provide broad annotation in text-based output without dynamic filtering options [23],[25]-[28]. Other available tools provide dynamic filtering options but can only handle a limited set of annotations [29]-[31]. Direct integration of the first and second analysis stage, bypassing manual handling of intermediate results, is a feature currently only available in the WEP platform [32]. Finally, as both genetic and phenotypic heterogeneity appear to be an emerging theme in many genetic disorders, it is clear that WES data should be evaluated in the context of large cohorts of patients and controls [33]. Hence, online collaboration between genetic centers in a protected setting, which is available only for a limited number of current tools, provides a significant advantage [29].
To overcome the limitations of currently available solutions in the complex annotation and filtering stage of NGS data analysis, we developed VariantDB. It unifies broad annotation and flexible filtering strategies in a user-friendly online interface and at the same time provides direct integration with the semi-automatic analysis capabilities of platforms such as Galaxy. Furthermore, it allows collaboration and data protection using role-based authentication.
Implementation
Interface and database
VariantDB consists of a PHP (5.3.2) based web interface, driving a CGI (5.10.1) backend. All data are stored in a MySQL (5.1.41) database on solid state drives (Figure 1). Structurally, data are ordered in sample and variant specific tables (Additional file 1). One additional table links variants to samples and holds quality information from GATK. Variant annotations are stored in separate tables based on the annotation source. This structure optionally allows VariantDB to retrieve annotation or filtering data from multiple sources in parallel, using the Perl Parallel::ForkManager library. Further improvements in performance can be achieved by enabling Memcached. The Perl Cache::Memcached::Fast library can reduce database load by caching and preloading frequently used data in memory. Queries, sources, and documentation for all filters and annotations are stored in XML files. Additional filtering rules can be specified as separate nodes in these configuration files.

Schematic representation of VariantDB implementation. Depending on the expected platform load, server elements can be hosted either on a single machine (default) or on separate physical hosts. If high performance computing (HPC) infrastructure is available, annotation processes can be distributed. HPO, Human Phenotype Ontology.
A public VariantDB instance is available for academic use. Furthermore, local installation is supported through either a downloadable virtualbox application or full installation on local infrastructure. Instructions for both approaches are available in the online documentation. To keep local installations up to date, automatic updating through the web interface is possible for the local administrator.
Data import
VCF files can be imported from an FTP server, accessible using VariantDB user credentials, or directly from a Galaxy server using the VariantDB tool (Additional file 2; for installation see [34]). Imported VCF files should comply with the VCF4.0 standards. Quality annotations generated by the GATK-based genotypers [7] are extracted and stored.
VariantDB provides the option to store the imported VCF file and associated BAM file. If available, direct links are presented to load VCF and BAM files into Integrative Genomics Viewer (IGV) for visualization of filtering results [35].
Annotation
Data annotation within VariantDB is available at sample and variant levels. With regard to sample annotation, family and experimental relations can be provided, which can later be applied to formulate inheritance patterns for variant filtering. Second, gender and phenotype information based on the Human Phenotype Ontology [36] is available. Finally, samples can be labeled as controls, which allow exclusion of common variants in filtering. Variant annotation is triggered by importing VCF files. Annotation proceeds by collecting variants missing a respective annotation, annotating the list of variants, and storing the results in the database. The annotation-specific tables in the database structure allow this process to be parallelized. If a high performance computing infrastructure is available, VariantDB can be configured to distribute these processes using the Perl Schedule::DRMAAc module (0.81). In total, 110 annotations are added to each variant (Table 1), taken from eight sources. The annotation engine utilizes ANNOVAR, snpEff, the Perl WWW::Mechanize library (for web tools) and a set of in-house parsers to retrieve the annotations [25],[28]. All annotations are presented by checkboxes in VariantDB for inclusion into the results (Figure 2). Users can also define sets of annotations that can be loaded simultaneously.

Selection of annotations. Top left: sample selection box, using either a dropdown menu, or auto-completion. Top right: when raw data files are available, hyperlinks are presented to download VCF/BAM files or load the files into IGV. Bottom left: all available annotations are listed. Users can select annotations using checkboxes for inclusion into the filtering results. Bottom right: previously saved sets of annotations can be enabled at once by selecting the checkbox and pressing `Add Annotations’.
GATK genoty** modules provide a set of quality parameters for each identified variant. VariantDB stores the values of the allelic ratio, Phred score of the polymorphism (QUAL), Phred-based genotype quality (GQ), genotype (GT), allelic depths (AD), quality by depth (QD), map** quality (MQ), strand bias (FS) and rank sums (BaseQRankSum, MQRankSum, ReadPosRankSum). If available, filter entries such as the VQSR tranches filter, are also stored.
Minor allele frequencies (MAFs) are available from the 1000 Genomes Project (v.2012apr) and the exome sequencing project (v.esp5400.2012Jul11, v.esp6500.2013Jan22), both global and population specific [42],[43]. Second, dbSNP rsIDs, MAFs and population size values are available for versions 130, 135 and 137 [44]. Starting from version 135, the clinical association label is also extracted.
Transcript information is extracted in UCSC, RefSeq and Ensembl-based format. Available information includes gene symbol or ID, transcript ID in case of multiple variants, affected position on cDNA and protein level and the effect on the protein level (intron/exon, missense/synonymous/nonsense, splicing).
Predictions with regard to pathogenicity are included from several tools. Using ANNOVAR, dbSNFP annotations for LRT, MutationTaster, PhyloP, PolyPhen2 and SIFT are included [45]. GERP++ [46] and CADD [47] scores are added from the respective tool data. Up-to-date scores of PROVEAN, SIFT, Grantham and MutationTaster are retrieved using the respective web tools [37],[38]. Finally, the SnpEff annotations also provide an estimate of the variant impact on the protein function [25].
Two sources are provided for functional annotation. First, Gene Ontology terms and the first level parental terms associated with affected genes are provided [40]. Second, a summary of the information available in ClinVar is available [41]. This summary includes hyperlinks to the ClinVar entry of variants that exactly match or overlap the variant in the queried sample, the type of variant in ClinVar (SNP/indel), the affected gene and transcript, latest update, evidence type, pathogenicity classification and associated disease. For gene, disease and alleles listed in ClinVar, hyperlinks are provided to several external databases.
Finally, users can specify additional information on inheritance, experimental validation and diagnostic classification on a per variant level.
Annotation updates
VariantDB provides two functionality layers to automatically keep annotation sources up to date. First, using scheduled execution at a frequency specified by the system administrator, third-party resources are checked for updated releases. When new data are available, all variants are re-annotated using the new release. To maintain data traceability, all discarded annotations are archived and all changes to variant annotation are logged. Finally, users are informed by email of possibly relevant novel annotations. Second, VariantDB automates the conversion between genome builds from the web interface. Upon conversion, the platform administrator needs information on the new build, including ANNOVAR, snpEff and IGV genome versions (hg19, GRC37.66 and hg19, respectively, for the current VariantDB version). Availability of the requested build is checked and, if available, all annotation tables are downloaded. Genome coordinates of currently stored variants are converted using the UCSC LiftOver tool, and failed conversions are presented to the platform administrator for manual curation [48]. Finally, all variants are re-annotated with regard to the new coordinates and users are informed. Previous genome versions remain accessible with their final annotations in read-only mode. The current genome build is always stated in the user interface. Also, when importing data from external pipelines such as galaxy, VariantDB requires the source genome build version to be passed along with the variant files, and will generate an error message on conflicting versions.
Variant filtering
VariantDB allows filtering on a combination of any of the available annotations listed in Table 1. To set filters, users select the criteria from dropdown menus (Figure 3) and optionally group them into a multi-level decision scheme (Figure 4). Successful filter settings can be saved for future usage. Next to the functional filtering criteria, parental and sibling relationships enable filtering for de novo, dominant and recessive inheritance models. Population-based variant selection can be performed on two levels. First, users can select variants that are present at least, or no more than, a specified number of times in a selection of samples. Second, genes can be selected for mutation burden by specifying the minimal or maximal number of samples containing a mutation in the same gene.

Selection of filters. Left: filtering criteria are organized in high-level categories. Filters are added by selecting the relevant filter and settings from dropdown menus. Numeric (for example, quality control values) or textual (for example, Gene Symbol) criteria can be added in text fields where appropriate. Right: previously saved filtering schemes can be enabled at once by selecting the checkbox and pressing `Apply Filter’.

Graphical representation of the selected filtering scheme. Individual filters can be grouped using logic AND/OR rules. Grou** and ordering is handled using a drag-and-drop interface.
Next to general gene and population level information, users can create in silico gene panels for targeted evaluation of candidate genes. A gene panel exists of a set of RefSeq identifiers, optionally augmented with additional comments. Gene panels are private at the user level, but can be made available as a public resource to all users.
Visualisation
By default, results are presented in a tabular overview (Figure 5) with selected annotations and IGV hyperlinks [35]. VariantDB aims at presenting all information related to a variant in a compact single screen view. Alternatively, a classic, wide table format is available, presenting all annotations on a single line per variant (Additional file 3). Results can also be exported to CSV files for downstream analysis. Finally, various charts are available to review the quality or characteristics of the resulting variant set. These charts include, among others, the Tr/Tv ratio, known versus novel ratio, MAF distribution and SNP versus indel ratio.

Results table. For each of the resulting variants, selected annotations are presented. On top, genomic position (which is also a hyperlink to the position in IGV), and other essential variant information is provided. If relevant, annotations are grouped in sub-tables on affected feature. User-specified information related to validation and classification is presented in a separate box on the left-hand side.
Results and discussion
Integration with existing NGS data processing systems
VariantDB provides a broad annotation of the detected variants, in combination with relevant filtering schemes and seamless integration with upstream data processing by means of a dedicated Galaxy tool. Communication between Galaxy and VariantDB occurs through generic HTTP-based forms. Hence, import of VCF files into VariantDB can be implemented as the endpoint of any NGS data analysis pipeline running on high performance computing infrastructure with internet access. We have chosen to support data import for VCF files only, as this format is the current community standard for NGS data. Although any generic VCF file can be loaded into VariantDB, GATK-based variant calling (Unified Genotyper, Haplotype Caller, MuTect [16],[49]) is currently regarded as the gold standard [17]. Therefore, we included specific import of various quality scores from GATK-based VCF files.
Filtering approaches
In total 110 annotations are available targeting specific aspects for selecting relevant variants. Although all annotations can be used as filtering criteria, two of the main approaches are gene-based and family/cohort-based filtering.
Gene-centric information is provided according to NCBI, Ensembl and UCSC nomenclature. To guarantee optimal sensitivity, filters to select variants that affect exonic sequence (Gene Location filter) or lead to a premature stop codon (VariantType filter) are applied in a transcript-specific manner. Using this approach, all genes where a variant introduces a stop codon in at least one transcript variant are reported. Apart from unbiased filtering, users can specify a list of candidate genes to perform in silico targeted analysis (Location Information filter). In silico gene panel analysis offers a two-step analysis for molecular diagnostics. By reducing the risk of incidental findings in initial analysis, a two-step approach lowers psychological distress for patients undergoing genetic testing [50]. If no causal variants are found in the candidate genes, whole exome or whole genome data are still available for follow-up investigation.
When family or cohort information is available, this information can be used to further refine the variant list. As an example, in a recessive disorder one would select homozygous variants (Genotype Composition filter) in a patient, which are present as heterozygous variants in both parents (Family Information filter). In the absence of such information, VariantDB can select for rare variants based on MAFs taken from dbSNP, the 1000 Genomes Project, the Exome Sequencing Project, or a private control cohort (Occurrence Information filter).
Ascertaining biological relevance
Although a selected filtering approach might already imply a certain biological relevance of the resulting variants (for example, de novo stop mutations), specific annotations are provided in VariantDB to further interpret the effect of a variant. First, known clinical associations are available in dbSNP as of version v135. More extensive information, however, is added from ClinVar (Clinvar Information filter) [41]. This database brings together genotype and phenotype data for known genetic variants, both SNP and structural variants, together with experimental data, links to external resources and relevant literature. Since its release in 2012, ClinVar rapidly became a reference resource for the interpretation of high throughput genetic data [51]. Second, information on the biological function of affected genes is presented based on Gene Ontology [40].
Finally, several prediction algorithms are available within VariantDB for the ascertainment of the variant pathogenicity (Mutation Effect Prediction filter). These predictions are typically based on evolutionary conservation [37],[39],[52], biochemical properties of the altered amino acids [53], or a combination of these [38],[54]. CADD, a novel prediction algorithm, was recently described and added to VariantDB. It integrates over 60 different annotations into a single model for variant deleteriousness, showing a significantly higher performance than previous methods [47]. With ClinVar and CADD, VariantDB thus contains two state-of-the-art annotation resources to interpret the functional impact of variants, in addition to several other widely used annotation sources.
Retrospective analysis
The development of various high-throughput screening methods resulted in an ever increasing amount of biological knowledge. Due to the continuously evolving interpretational resources, researchers are faced with the need to periodically reevaluate previous experiments for novel insights. VariantDB is, to our knowledge, the only publicly available platform that has the functionality to automatically handle such retrospective analyses. It updates all third-party resources on a preset time schedule, and notifies users when novel putatively interesting annotations are available. Here, we define putatively interesting as variants with a potential high impact on protein function (for example, frameshift or nonsense), based on both the RefSeq and the more comprehensive Ensembl gene sets, or matching variants classified as clinically relevant in ClinVar.
Performance
At the time of writing, the public VariantDB server holds over 46 million variants from almost 2,000 samples, corresponding to 2.2 million unique variants. By utilizing data caching and pre-fetching of data while users are setting filters, we achieve sufficient performance to allow interactive filtering and annotation of results (Table 2). After filtering, results are presented in batches of 100 variants to the user (Figure 5).
Data protection
VariantDB contains a user authentication module to protect stored data. Projects, defined as a collection of samples, can be shared with collaborators with rights ranging from read-only access to the ability to edit or delete whole projects. This online, role-based approach offers a major advantage over desktop solutions such as VarSifter or PriVar, and web-based but single-user approaches such as EVA [30],[31],[55]. As a centralized solution, VariantDB enables intuitive retrospective or multi-sample analysis, and collaboration between researchers from multiple laboratories. This was already successfully demonstrated in multiple published and ongoing studies [33],[56]-[58] (Proost et al., Sommen et al., unpublished results).
For an institutional setup of VariantDB, we provide private installation of the platform behind local firewalls. This can either be the deployment of a preinstalled virtual machine or full installation on private infrastructure.
Conclusions
VariantDB offers an all-in-one solution for annotation and filtering of variants obtained from NGS experiments. As summarized in Table 3, all the currently available platforms lack one or more of the essential aspects of variant interpretation present in VariantDB. It combines a broad range of annotations and filters, thereby eliminating the need for bioinformatics expertise by the user. Availability of in silico gene panel analysis reduces the risk of incidental findings, while centralized data storage enables large multi-center study designs, automated and retrospective updates of annotations and data traceability. The modularity of VariantDB offers extensibility with field-specific (for example, COSMIC for cancer research) and future (for example, ENCODE for whole genome sequencing interpretation) annotations and annotation tools in local instances. Overall, we conclude that VariantDB has a significant added value in streamlining NGS data analysis.
Availability and requirements
Project Name: VariantDB
Project homepage: http://www.biomina.be/app/variantdb
Operating system: Ubuntu Linux
Programming language: Perl, php/cgi
License: GPLv3
Restrictions for non-academics: ANNOVAR license needed
Authors’ contributions
GV designed and developed the software, and wrote the draft manuscript. LVL, TVDB, BL and RFK coordinated and participated in the design of the software and selection of included data sources, and revised the manuscript. All authors have read and approved the final manuscript.
Additional files
Abbreviations
- GATK:
-
Genome Analysis Toolkit
- IGV:
-
Integrative Genomics Viewer
- MAF:
-
minor allele frequency
- NGS:
-
next generation sequencing
- SNP:
-
single-nucleotide polymorphism
- WES:
-
whole exome sequencing
References
Vandeweyer G, Kooy RF: Detection and interpretation of genomic structural variation in health and disease. Expert Rev Mol Diagn. 2013, 13: 61-82. 10.1586/erm.12.119.
Vissers LELM, de Ligt J, Gilissen C, Janssen I, Steehouwer M, de Vries P, van Lier B, Arts P, Wieskamp N, del Rosario M, van Bon BW, Hoischen A, de Vries BB, Brunner HG, Veltman JA: A de novo paradigm for mental retardation. Nat Genet. 2010, 42: 1109-1112. 10.1038/ng.712.
Bamshad MJ, Ng SB, Bigham AW, Tabor HK, Emond MJ, Nickerson DA, Shendure J: Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet. 2011, 12: 745-755. 10.1038/nrg3031.
de Ligt J, Willemsen MH, van Bon BW, Kleefstra T, Yntema HG, Kroes T, Vulto-van Silfhout AT, Koolen DA, de Vries P, Gilissen C, del Rosario M, Hoischen A, Scheffer H, de Vries BB, Brunner HG, Veltman JA, Vissers LE: Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med. 2012, 367: 1921-1929. 10.1056/NEJMoa1206524.
O'Roak BJ, Deriziotis P, Lee C, Vives L, Schwartz JJ, Girirajan S, Karakoc E, Mackenzie AP, Ng SB, Baker C, Rieder MJ, Nickerson DA, Bernier R, Fisher SE, Shendure J, Eichler EE: Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet. 2011, 43: 585-589. 10.1038/ng.835.
Xu B, Roos JL, Dexheimer P, Boone B, Plummer B, Levy S, Gogos JA, Karayiorgou M: Exome sequencing supports a de novo mutational paradigm for schizophrenia. Nat Genet. 2011, 43: 864-868. 10.1038/ng.902.
DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ: A framework for variation discovery and genoty** using next-generation DNA sequencing data. Nat Genet. 2011, 43: 491-498. 10.1038/ng.806.
JSI Medical Systems GmbH, Kippenheim, Germany, [http://www.jsi-medisys.de]
CLC Bio, Aarhus, Denmark, [http://www.clcbio.com]
Illumina Inc., San Diego, USA, [http://www.illumina.com]
FastX-Toolkit: FastQ/A short-read pre-processing tools, [http://hannonlab.cshl.edu/fastx_toolkit/]
Martin M: Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet Jl. 2011, 17: 10-12. 10.14806/ej.17.1.200.
Langmead B, Trapnell C, Pop M, Salzberg SL: Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10: R25-10.1186/gb-2009-10-3-r25.
Li H, Durbin R: Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009, 25: 1754-1760. 10.1093/bioinformatics/btp324.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R: The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009, 25: 2078-2079. 10.1093/bioinformatics/btp352.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA: The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20: 1297-1303. 10.1101/gr.107524.110.
Yi M, Zhao Y, Jia L, He M, Kebebew E, Stephens RM: Performance comparison of SNP detection tools with illumina exome sequencing data-an assessment using both family pedigree information and sample-matched SNP array data. Nucleic Acids Res. 2014, 42 (12): e101-10.1093/nar/gku392.
Goecks J, Nekrutenko A, Taylor J, Galaxy T: Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 2010, 11: R86-10.1186/gb-2010-11-8-r86.
Blankenberg D, Von Kuster G, Coraor N, Ananda G, Lazarus R, Mangan M, Nekrutenko A, Taylor J: Galaxy: a web-based genome analysis tool for experimentalists. Curr Protoc Mol Biol. 2010, Chapter 19: Unit 19.10.1-19.10.21.
Giardine B, Riemer C, Hardison RC, Burhans R, Elnitski L, Shah P, Zhang Y, Blankenberg D, Albert I, Taylor J, Miller W, Kent WJ, Nekrutenko A: Galaxy: a platform for interactive large-scale genome analysis. Genome Res. 2005, 15: 1451-1455. 10.1101/gr.4086505.
Cartagenia NV, Leuven, Belgium, [http://www.cartagenia.com]
Sincan M, Simeonov DR, Adams D, Markello TC, Pierson TM, Toro C, Gahl WA, Boerkoel CF: VAR-MD: a tool to analyze whole exome-genome variants in small human pedigrees with mendelian inheritance. Hum Mutat. 2012, 33: 593-598. 10.1002/humu.22034.
Yao J, Zhang KX, Kramer M, Pellegrini M, McCombie WR: FamAnn: an automated variant annotation pipeline to facilitate target discovery for family-based sequencing studies. Bioinformatics. 2014, 30: 1175-1176. 10.1093/bioinformatics/btt749.
Lee IH, Lee K, Hsing M, Choe Y, Park JH, Kim SH, Bohn JM, Neu MB, Hwang KB, Green RC, Kohane IS, Kong SW: Prioritizing disease-linked variants, genes, and pathways with an interactive whole-genome analysis pipeline. Hum Mutat. 2014, 35: 537-547. 10.1002/humu.22520.
Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM: A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. 2012, 6: 80-92. 10.4161/fly.19695.
Bai Y, Cavalcoli J: SNPAAMapper: An efficient genome-wide SNP variant analysis pipeline for next-generation sequencing data. Bioinformation. 2013, 9: 870-872. 10.6026/97320630009870.
Ng SB, Turner EH, Robertson PD, Flygare SD, Bigham AW, Lee C, Shaffer T, Wong M, Bhattacharjee A, Eichler EE, Bamshad M, Nickerson DA, Shendure J: Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 2009, 461: 272-276. 10.1038/nature08250.
Wang K, Li M, Hakonarson H: ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38: e164-10.1093/nar/gkq603.
Sifrim A, Van Houdt JK, Tranchevent LC, Nowakowska B, Sakai R, Pavlopoulos GA, Devriendt K, Vermeesch JR, Moreau Y, Aerts J: Annotate-it: a Swiss-knife approach to annotation, analysis and interpretation of single nucleotide variation in human disease. Genome Med. 2012, 4: 73-10.1186/gm374.
Zhang L, Zhang J, Yang J, Ying D, Lau YL, Yang W: PriVar: a toolkit for prioritizing SNVs and indels from next-generation sequencing data. Bioinformatics. 2013, 29: 124-125. 10.1093/bioinformatics/bts627.
Coutant S, Cabot C, Lefebvre A, Leonard M, Prieur-Gaston E, Campion D, Lecroq T, Dauchel H: EVA: Exome Variation Analyzer, an efficient and versatile tool for filtering strategies in medical genomics. BMC Bioinform. 2012, 13: S9-10.1186/1471-2105-13-S14-S9.
D'Antonio M, D'Onorio De Meo P, Paoletti D, Elmi B, Pallocca M, Sanna N, Picardi E, Pesole G, Castrignano T: WEP: a high-performance analysis pipeline for whole-exome data. BMC Bioinform. 2013, 14: S11-10.1186/1471-2105-14-S7-S11.
Helsmoortel C, Vulto-van Silfhout AT, Coe BP, Vandeweyer G, Rooms L, van den Ende J, Schuurs-Hoeijmakers JH, Marcelis CL, Willemsen MH, Vissers LE, Yntema HG, Bakshi M, Wilson M, Witherspoon KT, Malmgren H, Nordgren A, Anneren G, Fichera M, Bosco P, Romano C, de Vries BB, Kleefstra T, Kooy RF, Eichler EE, Van der Aa N: A SWI/SNF-related autism syndrome caused by de novo mutations in ADNP. Nat Genet. 2014, 46: 380-384. 10.1038/ng.2899.
VCF_To_VariantDB Galaxy Tool, [http://toolshed.g2.bx.psu.edu/view/geert-vandeweyer/vcf_to_variantdb]
Thorvaldsdottir H, Robinson JT, Mesirov JP: Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013, 14: 178-192. 10.1093/bib/bbs017.
Robinson PN, Mundlos S: The human phenotype ontology. Clin Genet. 2010, 77: 525-534. 10.1111/j.1399-0004.2010.01436.x.
Choi Y, Sims GE, Murphy S, Miller JR, Chan AP: Predicting the functional effect of amino acid substitutions and indels. PLoS One. 2012, 7: e46688-10.1371/journal.pone.0046688.
Schwarz JM, Rodelsperger C, Schuelke M, Seelow D: MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods. 2010, 7: 575-576. 10.1038/nmeth0810-575.
Ng PC, Henikoff S: SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31: 3812-3814. 10.1093/nar/gkg509.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G: Gene ontology: tool for the unification of biology. Gene Ontol Consort Nat Genet. 2000, 25: 25-29. 10.1038/75556.
Landrum MJ, Lee JM, Riley GR, Jang W, Rubinstein WS, Church DM, Maglott DR: ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014, 42: D980-D985. 10.1093/nar/gkt1113.
Exome Variant Server, [http://evs.gs.washington.edu/EVS/]
Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, Gibbs RA, Hurles ME, McVean GA: A map of human genome variation from population-scale sequencing. Nature. 2010, 467: 1061-1073. 10.1038/nature09534.
Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K: dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001, 29: 308-311. 10.1093/nar/29.1.308.
Liu X, Jian X, Boerwinkle E: dbNSFP: a lightweight database of human nonsynonymous SNPs and their functional predictions. Hum Mutat. 2011, 32: 894-899. 10.1002/humu.21517.
Davydov EV, Goode DL, Sirota M, Cooper GM, Sidow A, Batzoglou S: Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput Biol. 2010, 6: e1001025-10.1371/journal.pcbi.1001025.
Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J: A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014, 46: 310-315. 10.1038/ng.2892.
Fujita PA, Rhead B, Zweig AS, Hinrichs AS, Karolchik D, Cline MS, Goldman M, Barber GP, Clawson H, Coelho A, Diekhans M, Dreszer TR, Giardine BM, Harte RA, Hillman-Jackson J, Hsu F, Kirkup V, Kuhn RM, Learned K, Li CH, Meyer LR, Pohl A, Raney BJ, Rosenbloom KR, Smith KE, Haussler D, Kent WJ: The UCSC Genome Browser database: update 2011. Nucleic Acids Res. 2011, 39: D876-D882. 10.1093/nar/gkq963.
Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, Getz G: Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013, 31: 213-219. 10.1038/nbt.2514.
Sie AS, Prins JB, van Zelst-Stams WA, Veltman JA, Feenstra I, Hoogerbrugge N: Patient experiences with gene panels based on exome sequencing in clinical diagnostics: high acceptance and low distress.Clin Genet 2014, doi:10.1111/cge.12433.,
Kassahn KS, Scott HS, Caramins MC: Integrating massively parallel sequencing into diagnostic workflows and managing the annotation and clinical interpretation challenge. Hum Mutat. 2014, 35: 413-423. 10.1002/humu.22525.
Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A: Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 2010, 20: 110-121. 10.1101/gr.097857.109.
Grantham R: Amino acid difference formula to help explain protein evolution. Science. 1974, 185: 862-10.1126/science.185.4154.862.
Adzhubei I, Jordan DM, Sunyaev SR: Predicting Functional Effect of Human Missense Mutations Using PolyPhen-2. Curr Protoc Hum Genet. 2013, Chapter 7: Unit 7.20-
Teer JK, Green ED, Mullikin JC, Biesecker LG: VarSifter: visualizing and analyzing exome-scale sequence variation data on a desktop computer. Bioinformatics. 2012, 28: 599-600. 10.1093/bioinformatics/btr711.
Borra VM, Steenackers E, de Freitas F, Van Hul E, Glass I, Van Hul W: Localization of the gene for X-linked calvarial hyperostosis to chromosome Xq27.3-Xqter. Bone. 2014, 58: 67-71. 10.1016/j.bone.2013.10.011.
Gillis E, Kempers M, Salemink S, Timmermans J, Cheriex EC, Bekkers SC, Fransen E, De Die-Smulders CE, Loeys BL, Laer LV: An FBN1 deep intronic mutation in a familial case of Marfan syndrome: an explanation for genetically unsolved cases?. Hum Mutat. 2014, 35: 571-574. 10.1002/humu.22540.
Helsmoortel C, Vandeweyer G, Ordoukhanian P, Van Nieuwerburgh F, Van der Aa N, Kooy RF: Challenges and opportunities in the investigation of unexplained intellectual disability using family based whole exome sequencing.Clin Genet 2014, doi:2010.1111/cge.12470.,
Li MX, Gui HS, Kwan JS, Bao SY, Sham PC: A comprehensive framework for prioritizing variants in exome sequencing studies of Mendelian diseases. Nucleic Acids Res. 2012, 40: e53-10.1093/nar/gkr1257.
Santoni FA, Makrythanasis P, Nikolaev S, Guipponi M, Robyr D, Bottani A, Antonarakis SE: Simultaneous identification and prioritization of variants in familial, de novo, and somatic genetic disorders with VariantMaster. Genome Res. 2014, 24: 349-355. 10.1101/gr.163832.113.
Aleman A, Garcia-Garcia F, Salavert F, Medina I, Dopazo J: A web-based interactive framework to assist in the prioritization of disease candidate genes in whole-exome sequencing studies. Nucleic Acids Res. 2014, 42: W88-W93. 10.1093/nar/gku407.
Na YJ, Cho Y, Kim JH: AnsNGS: an annotation system to sequence variations of next generation sequencing data for disease-related phenotypes. Healthcare Inform Res. 2013, 19: 50-55. 10.4258/hir.2013.19.1.50.
Acknowledgements
This work was supported by the Belgian National Fund for Scientific Research-Flanders (FWO), IWT/TBM (grant 110700) and the Fondation Leducq.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Electronic supplementary material
13073_2014_74_MOESM1_ESM.tiff
Additional file 1: Database layout of VariantDB. The variant table holds unique IDs and information on genomic position and allelic composition of each observed variant. Annotations are stored in separate tables linked on the variant_id. To increase performance, ClinVar and Gene Ontology are stored as summary representations of the full releases. User authentication is based on email and password combinations. Data access is controlled on the project level, with access definitions for variant-level, sample-level and phenotype-level editing. (TIFF 566 KB)
13073_2014_74_MOESM2_ESM.tiff
Additional file 2: Input form of the VariantDB galaxy integration. Supported VCF sources are GATK Unified Genotyper, GATK Haplotype Caller, GATK MuTect and Sam Tools VarScan. Private VariantDB servers can be added in the tool configuration. (TIFF 307 KB)
13073_2014_74_MOESM3_ESM.tiff
Additional file 3: Alternative output format of VariantDB. In this format, all selected annotations are presented on a single line per variant. Annotations are grouped if they represent multiple entries for the same variant (for example, alternative transcripts, multiple entries in ClinVar). (TIFF 251 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Vandeweyer, G., Van Laer, L., Loeys, B. et al. VariantDB: a flexible annotation and filtering portal for next generation sequencing data. Genome Med 6, 74 (2014). https://doi.org/10.1186/s13073-014-0074-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13073-014-0074-6