
Abstract
Background
Chronic pain is naturally aversive and often distressing for patients. Pain co** and self-regulatory skills have been shown to effectively reduce pain-related distress and other symptoms. In this trial, the primary goal is to pilot test the comparative efficacy of a single-session videoconference-delivered group pain education class to a waitlist control among patients with chronic pain.
Methods
Our study is a randomized clinical trial pilot testing the superiority of our 2-h single-session videoconference-delivered group pain education class against a waitlist control. We will enroll 120 adult patients with mixed etiology chronic pain and randomize 1:1 to one of the two study arms. We hypothesize superiority for the pain education class for bolstering pain and symptom management. Team researchers masked to treatment assignment will assess the outcomes up to 3 months post-treatment.
Discussion
This study aims to test the utility of a single-session videoconference-delivered group pain education class to improve self-regulation of pain and pain-related outcomes. Findings from our project have the potential to significantly reduce barriers to effective psychological treatment for pain, optimizing the delivery of increasingly vital online and remote-delivered intervention options.
Trial registration
ClinicalTrials.govNCT04546685. Registered on 04 September 2020.
Similar content being viewed by others


Background
Chronic pain is a problem of considerable public health importance that is estimated to impact approximately 100 million Americans and cost society up to $635 billion annually [1]. Chronic pain is particularly challenging to treat given the limited efficacy of existing pharmacological treatments [2,3,4] and the multisession nature of current psychological treatments, which are difficult to integrate into healthcare settings and thereby perpetuating medical utilization, disparities, and disease. To date, the USA lacks scalable behavioral medicine for pain, underscoring the need for solutions that are accessible, low-cost, and low-burden.
Despite the documented benefits of cognitive behavioral therapy (CBT) for chronic pain [3, 5], it remains limited by multiple obstacles including treatment time, healthcare costs, travel burden, lack of services for patients in remote or rural areas, and lack of available trained therapists and services [6,7,8,9]. Given these barriers, as well as the rapidly expanding need for alternatives to face-to-face encounters due to the COVID-19 pandemic [10], there is an urgent need for brief online and remotely delivered psychological interventions. Online and remotely delivered interventions have been demonstrated to be effective in treating a variety of physical and mental health issues, with Internet-based CBT classified as a well-established treatment for depression, panic disorder, and social phobia [11]. Online CBT has also been found to be effective in treating psychological issues related to living with chronic illnesses, including irritable bowel syndrome, tinnitus, and chronic pain [12], with small-to-moderate reported effect sizes (d range 0.04 to 1.23, mean 0.60) in chronic pain [11]. Internet- and remote-delivered CBT is also effective in reducing pain catastrophizing and improving patient attitudes toward chronic pain [13]. Other online interventions for patients with pain include the use of compassionate mind training [14], social media-based online community intervention [15], pain self-management [16], and online hypnosis [17]. Although showing promising results, these were multisession interventions ranging from 20 days to 12 weeks, which can be costly, time-consuming, and burdensome for patients.
Increasingly, the field is trending toward a population health approach that uses ultra-brief and targeted behavioral treatments that have the greatest reach at the lowest cost and burden. Single-session interventions (SSIs) have shown promise and efficacy in diverse populations and health conditions such as serious mental illness [18], anxiety and conduct disorder in youth [19], acute insomnia [20], heavy alcohol consumption in college students [21], trauma and adversity [22, 23], postsurgical pain [24], and general chronic pain [25,26,27,28,29]. Several online SSIs have been conducted as well, with demonstrated feasibility and patient acceptance of online-delivered SSIs for multiple sclerosis pain [30], disordered gambling [31], and adolescent mental health [32,33,34]. Data suggest that SSIs are only slightly less effective than longer course treatments requiring up to 16 h of treatment time [29, 35]. Effective SSIs, especially if delivered digitally, would eliminate many barriers to care such as lack of available trained therapists, treatment cost and burden, and insurance limits.
Our team has developed and tested a 2-h skills-based SSI (“Empowered Relief”), which was shown to reduce pain-specific distress and improve self-regulation at a 4-week follow-up in a cohort of 57 mixed etiology chronic pain patients receiving treatment at a tertiary referral, multidisciplinary chronic pain clinic [25]. Importantly, in a 3-arm randomized controlled trial, Empowered Relief was non-inferior to an 8-week cognitive behavioral therapy and superior to a health education class for reducing pain-specific distress (i.e., catastrophizing) and improving multiple secondary outcomes at 3 months post-treatment in individuals with chronic low back pain. For the first time, the current study seeks to test the impact of a group-based single-session intervention “Empowered Relief” that is delivered online on bolstering pain and symptom management in adults with mixed etiology chronic pain. Participants who are randomized in the waitlist or usual care do not receive the study intervention and will be instructed to continue with the care they would normally receive as part of their ongoing clinical care. Upon completion of the 3-month study, participants in the waitlist will be invited to enroll in “Empowered Relief.”
Specific aims
Our specific aim and three corresponding hypotheses are outlined below.
-
1.
We will conduct a randomized controlled trial comparing the preliminary effects of a single-session videoconference-delivered pain education class compared to a waitlist control (i.e., usual care) condition.
-
(a)
Hypothesis 1: The single-session intervention (Empowered Relief (ER)) will lead to greater reductions in pain catastrophizing compared to a usual care condition.
-
(b)
Hypothesis 2: ER will lead to greater reductions in pain bothersomeness and sleep disturbance compared to a usual care condition.
-
(c)
Hypothesis 3: ER will lead to greater reductions in pain intensity, anxiety, depression, and physical function compared to a usual care condition.
-
(a)
Methods/design
Overview
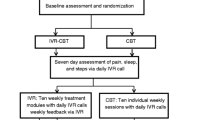
We are piloting a randomized clinical trial in which individuals with a chronic pain condition are randomly assigned to one of two arms: a single-session videoconference-delivered group pain education class or a waitlist control (WLC) receiving usual care (Figs. 1 and 2). Participants will be followed for 3 months after treatment. Participants will be assessed via an online screening form, a phone screening, enrollment survey, 2-week follow-up, and at 1, 2, and 3 months post-treatment. Team statisticians blinded to participant treatment assignment will assess the outcomes 2 weeks after treatment and after 1, 2, and 3 months. The primary outcome is pain catastrophizing 1 month post-treatment. Secondary outcomes include reductions in pain bothersomeness and PROMIS sleep disturbance at 1 month post-treatment. Tertiary outcomes include reductions in pain intensity, PROMIS anxiety, PROMIS depression, and PROMIS physical function. The protocol for this trial has been approved by the Stanford Institutional Review Board (IRB). All participants will be required to give informed consent to a trained study team member prior to enrollment in the study.

Flowchart of the trial protocol

The schedule of enrollment, interventions, and assessments
Study sample and setting
Participants for this trial will be recruited through targeted emails and advertisements at Stanford’s pain management clinics, in addition to the Stanford Systems Neuroscience and Pain Lab (SNAPL) database. All advertisements will direct interested individuals to an online screening form that assesses for initial eligibility. The study will enroll 120 adults (age >18 years) with a chronic non-cancer pain condition and who meet the study criteria (Table 1). The sample size accounts for expected attrition, and eligibility will be assessed by the research staff.
Inclusion and exclusion criteria
Tables 1 and 2 list the inclusion and exclusion criteria, respectively, as well as the rationale for each criterion and the sources where each criterion will be assessed. Additionally, we require that the participants be willing and available to participate in the full treatment session and able to respond to the enrollment survey (3–6 days before the class date) and post-treatment (2 weeks and 1, 2, and 3 months) follow-up questionnaires.
Recruitment procedures
Because the study intervention is group-based and delivered via videoconference, we are recruiting participants in 2–3 large cohorts consisting of 30 participants per class cohort (minimum of 20 participants, maximum of 30 participants per cohort) for both study arms.
Interested individuals deemed initially eligible by the online screening will be further screened over the phone. Eligible individuals will then be invited to enroll in the study and consented with a research staff over the phone, after which they provide an electronic signature to the consent form emailed to them. Participants are randomized following eligibility confirmation and informed consent procedures. Then, participants complete the enrollment survey, which includes information related to their chronic pain and psychosocial well-being.
Randomization
Enrolled participants will be randomized to one of two study arms: Empowered Relief (ER) and waitlist control (WLC; usual care). An automated program in REDCap will randomly assign a participant to a study arm when enrolled and will ensure blinded randomization, as well as equal numbers in both arms at the end of data collection.
Blinding
Participants cannot and will not be blinded to the study arm they are randomized to. A clinical psychologist who will be trained in the intervention and has no involvement in the data analysis will deliver the intervention. The study coordinator will be responsible for handling the randomization process through REDCap but will remain blinded to the randomization scheme. All data given to the statistician will be blinded. All research data will be kept separate from identifiers and linked using a participant number. Only the principal investigator will have access to the file linking names and participant numbers, and the file will be stored behind password-protected and fire-walled systems. The team will have access to the final unidentified dataset.
Study arms
Empowered Relief consists of a single-session 2-h videoconference-delivered group class. After the ER class, participants will receive home-based resources that facilitate ongoing self-regulation and pain self-management. Participants randomized to the waitlist control arm do not receive the study intervention and will be instructed to continue with the medical care they would normally receive. Upon completion of the 3-month study, participants in the waitlist control will be invited to enroll in ER.
Single-session videoconference-delivered skills-based pain class (Empowered Relief)
Our group developed the single-session intervention (SSI) for pain management in 2013 with the goal of rapidly equip** patients with skills to self-regulate pain-specific distress. Pilot data revealed significantly reduced pain-specific distress—as indexed by reductions in pain catastrophizing—1 month post-treatment, regardless of comorbid depression and anxiety [25]. The SSI (Empowered Relief) was also the subject of a NIH-funded randomized controlled trial in chronic low back pain [29]; the findings showed that Empowered Relief was non-inferior to an 8-week cognitive behavioral therapy and superior to a health education class for reducing pain catastrophizing and improving multiple secondary outcomes at 3 months post-treatment.
For the present study, for the first time, we seek to pilot test the impact of “Empowered Relief,” a group-based SSI that is delivered online, on bolstering pain and symptom management in adults with mixed etiology chronic pain. A clinical psychologist trained in delivering the 2-h intervention will administer the class via the Zoom platform to groups of enrolled participants. The class is delivered by PowerPoint presentation and includes mind-body pain science, the importance of self-regulation in the context of pain and stress, and evidence-based skills that target pain-specific distress and enhance pain control. Participants will be guided in develo** their own self-treatment plan and acquiring the skills necessary to decrease pain and stress-related physiological hyperarousal and to enhance regulation of cognition and emotion within the context of pain. At the end of the class, participants will (1) develop a self-tailored personalized plan to target pain-specific distress, (2) receive a 20-min guided relaxation response audio file, and (3) obtain an electronic copy of the didactic class content. Cohort effects are expected to be minimal due to the single session nature of the class, the highly structured and manualized nature of the class, and the minimal participant interaction via a videoconference platform.
Usual care (waitlist control) arm
The control condition will be a usual care comparator, which was selected because clinical trial methodologists [36, 37] argue that waitlist or usual care controls are appropriate for early-phase, proof-of-principle trials where the goal is to promote innovation. Participants in the usual care group will not receive a study intervention and will be instructed by a research study coordinator to continue with the care they would normally receive as part of their ongoing clinical care. Upon completion of all study measures, participants in the usual care condition will be given the opportunity to participate in the intervention arm so as to ensure equality in study participation.
Class platform
All treatment sessions will occur via the Zoom platform that is password-protected and hosted within the fire-walled Stanford University School of Medicine and Stanford HealthCare systems.
Instructors
For the ER treatment group, all instructors will be doctoral-level clinical psychologists trained in the treatment of chronic pain.
Training and monitoring of instructors
ER instructors will be trained in the study protocol for their classes prior to administering treatment. Existing treatment manuals as well as highly structured and standardized class content will assure treatment fidelity. A research coordinator, serving as a fidelity rater, will directly observe the ER classes. Cohort effects are likely to be minimal due to the single-session format and relatively minimal participant interaction.
Measures
Demographic data, pain characteristics, and physical and psychosocial health measures will be collected at enrollment. Pain catastrophizing will be assessed using the Pain Catastrophizing Scale [38]. Pain bothersomeness will be assessed by a single-item question that is commonly used in chronic low back pain research [39, 40]. PROMIS measures will be used to assess sleep disturbance, pain intensity, pain interference, physical function, depression, and anxiety using short forms [41]. Our group has applied the NIH PROMIS measures in multiple nationally funded clinical pain treatment trials and other studies [42,43,44] (Table 3).
Baseline (pre-treatment) assessment
Three to 6 days pre-treatment, patients will complete an online baseline survey including demographic information, pain condition and characteristics, pain bothersomeness, pain catastrophizing, and the PROMIS measures. Participants will not be asked to repeat demographic information again, but these measures will be identical to those assessed post-treatment.
Post-treatment assessment
One week post-treatment, patients in the ER arm will complete a brief questionnaire assessing patient satisfaction with the intervention, in addition to their frequency of skills use and audio file use. At 2 weeks and 1, 2, and 3 months post-treatment, all participants will complete a set of questionnaires identical to those administered at baseline. The primary study endpoint is 1 month post-treatment. Participants may receive up to $50 for study completion.
Primary outcome measure
Our primary outcome measure is pain-specific distress as indexed by pain catastrophizing scores at 1 month post-treatment. Pain catastrophizing will be assessed using the Pain Catastrophizing Scale (PCS), and a total score will be generated using the sum of the 13-items of the PCS. The PCS has valid and reliable psychometrics [38].
Secondary outcome measures
Our secondary outcome is pain bothersomeness, whereby participants rate their chronic pain bothersomeness during the previous 7 days on a 0 to 10 numeric rating scale anchored by 0 (not at all bothersome) and 10 (extremely bothersome) that is commonly used in CLBP research [39, 40]. PROMIS sleep disturbance will be assessed with the NIH PROMIS sleep disturbance short form.
Tertiary outcome measures
NIH PROMIS measures [41] will be administered to assess pain intensity, anxiety, depression, and physical function. These measures have been successfully applied to pain research [45,46,47,48].
Data collection, quality control, and confidentiality
The online assessments completed by participants will be gathered securely in a REDCap database. No questionnaires will be collected on paper. Additionally, the members of the team will be trained to use and complete case report forms (CRFs), how to review them for completeness, and how to maintain participant confidentiality. The patient flow will be reported according to the Consolidated Standards of Reporting Trials (CONSORT) guidelines [49].
Protection of human participants and assessment of safety
Protection of human participants
The Stanford University Institutional Review Board (IRB) has approved this study.
Safety monitoring
This trial will not be monitored by an independent Data and Safety Monitoring Board (DSMB). However, two researchers composed of a physician and a clinical psychologist with knowledge in the treatment of chronic pain conditions will oversee the project. In addition, we have a dedicated research regulatory manager who will provide oversight for all regulatory processes. The team will meet twice a year or on an as-needed basis and will make relevant safety decisions regarding reported participant cases.
Adverse experiences
The treatments in this study are not anticipated to pose any risks to participants. However, a study coordinator will review enrolled patient records periodically to monitor for adverse events. In the case of an adverse event, an adverse event case report form will be completed. These will be discussed in monthly team meetings and reported to the IRB annually. Serious adverse events will be reported to the Stanford IRB and NIH. The PI and study committee will evaluate all serious adverse events within 24 h after the study team becomes aware of the incident. All study-related adverse events will be included in the annual report to the NIH, and serious adverse events will be reported within 2 weeks.
Stop** rules
The trial will be stopped if (1) either treatment intervention is associated with adverse effects that calls into question the safety of the intervention, (2) there is difficulty in study recruitment or retention that will impact the ability to evaluate study endpoints, (3) new information becomes available during the trial that indicates the need to stop the trial, or (4) other unforeseen situations occur that would warrant stop** the study.
Statistical issues
Sample size and detectable differences
We chose our sample size to ensure adequate power to detect treatment effects on the primary outcome (i.e., pain catastrophizing). The project will enroll 120 participants (ages >18 years) with a diagnosis of chronic non-cancer pain (>3 months more than half the time).
To compare the main effect of ER class on pain catastrophizing scores against the WLC condition, we will plan to enroll 120 participants and have 116 completers (58 per group). The proposed sample size accounts for 4% attrition in each study arm. This is lower than the attrition rate we observed in our Empowered Relief trial [29] of 14.9% or of that seen in pain CBT literature of 18–25% [50, 51], but we believe that the digital format and single-session nature of the intervention will be less burdensome and lead to lower attrition rates. We hope to achieve 80% power to detect medium-large treatment effects on the primary outcome (i.e., pain catastrophizing).
Statistical analyses
Primary analyses
We will use an intent-to-treat approach in all analyses (i.e., the assessment of individuals will be analyzed by a randomized group regardless of participation in the intervention). By doing so, we protect against any confounds that arise as a result of subject dropout.
The main effect of ER on pain catastrophizing will be compared against the WLC using a 2-sample t test. Our primary endpoint is pain catastrophizing at 1 month post-treatment. We will also compare the proportion of success rate, defined as ≥30% reduction in pain catastrophizing for a clinically significant treatment response [52].
Secondary objectives
To test the secondary and tertiary aims that the ER class will have greater reductions in pain bothersomeness, sleep disruption, pain intensity, anxiety, depression, and physical function compared to WLC, our primary endpoint will be considered at 1 month post-treatment, and its within-subject difference from baseline will be calculated. The mean difference in the ER group will be compared against the WLC arm using the two-sample t test. Similarly, we will also compare the proportion of success rate, defined as ≥30% reduction in our outcomes for a clinically significant treatment response [52].
Discussion
In this trial, we will seek to determine whether a group-based single-session intervention (SSI) that is delivered online is an effective treatment option for persons with chronic pain. In 2019, the US Health and Human Services cited “Empowered Relief” as a promising scalable behavioral pain treatment [53], and for the first time, this study aims to test whether a videoconference-delivered version of the class may similarly effectively and efficiently reduce the burden of chronic pain and improve symptom management. Importantly, it addresses the rapidly expanding need for alternatives to face-to-face encounters due to the COVID-19 pandemic. Finally, the study will identify a proportion of patients who achieve a meaningful reduction in a number of pain-related indices in response to this online single-session intervention. This will facilitate the future application of the digital version of the class across a variety of settings, such as in primary care or in pre-surgical populations, and possibly across chronic health conditions with a primary pain complaint.
Availability of data and materials
Data will be available on ClinicalTrials.gov (NCT04546685).
References
Institute of Medicine (US) Committee. Relieving pain in America: a blueprint for transforming prevention, care, education, and research. Washington, DC: National Academies Press; 2011.
Finnerup NB, Attal N, Haroutounian S, McNicol E, Baron R, Dworkin RH, et al. Pharmacotherapy for neuropathic pain in adults: a systematic review and meta-analysis. Lancet Neurol. 2015;14(2):162–73. https://doi.org/10.1016/S1474-4422(14)70251-0.
Morley S, Eccleston C, Williams A. Systematic review and meta-analysis of randomized controlled trials of cognitive behaviour therapy and behaviour therapy for chronic pain in adults, excluding headache. Pain. 1999;80(1-2):1–13. https://doi.org/10.1016/S0304-3959(98)00255-3.
Chou R, Huffman LH. Medications for acute and chronic low back pain: a review of the evidence for an American Pain Society/American College of Physicians clinical practice guideline. Ann Intern Med. 2007;147(7):505–14. https://doi.org/10.7326/0003-4819-147-7-200710020-00008.
Mariano TY, Urman RD, Hutchison CA, Jamison RN, Edwards RR. Cognitive behavioral therapy (CBT) for subacute low back pain: a systematic review. Curr Pain Headache Rep. 2018;22(3):15. https://doi.org/10.1007/s11916-018-0669-5.
Bennett-Levy J, Perry H. The promise of online cognitive behavioural therapy training for rural and remote mental health professionals. Australas Psychiatry. 2009;17(sup1):S121–S4.
Cartreine JA, Ahern DK, Locke SE. A roadmap to computer-based psychotherapy in the United States. Harv Rev Psychiatry. 2010;18(2):80–95. https://doi.org/10.3109/10673221003707702.
Darnall BD, Scheman J, Davin S, Burns JW, Murphy JL, Wilson AC, et al. Pain psychology: a global needs assessment and national call to action. Pain Med. 2016;17(2):250–63. https://doi.org/10.1093/pm/pnv095.
Griffiths KM, Carron-Arthur B, Parsons A, Reid R. Effectiveness of programs for reducing the stigma associated with mental disorders. A meta-analysis of randomized controlled trials. World Psychiatry. 2014;13(2):161–75. https://doi.org/10.1002/wps.20129.
Eccleston C, Blyth FM, Dear BF, Fisher EA, Keefe FJ, Lynch ME, et al. Managing patients with chronic pain during the COVID-19 outbreak: considerations for the rapid introduction of remotely supported (eHealth) pain management services. Pain. 2020;161(5):889–93. https://doi.org/10.1097/j.pain.0000000000001885.
Hedman E, Ljótsson B, Lindefors N. Cognitive behavior therapy via the Internet: a systematic review of applications, clinical efficacy and cost–effectiveness. Expert Rev Pharmacoecon Outcomes Res. 2012;12(6):745–64. https://doi.org/10.1586/erp.12.67.
Beatty L, Lambert S. A systematic review of internet-based self-help therapeutic interventions to improve distress and disease-control among adults with chronic health conditions. Clin Psychol Rev. 2013;33(4):609–22. https://doi.org/10.1016/j.cpr.2013.03.004.
Garg S, Garg D, Turin TC, Chowdhury MFU. Web-based interventions for chronic back pain: a systematic review. J Med Internet Res. 2016;18(7):e139. https://doi.org/10.2196/jmir.4932.
Dhokia M, Elander J, Clements K, Gilbert P. A randomized-controlled pilot trial of an online compassionate mind training intervention to help people with chronic pain avoid analgesic misuse. Psychol Addict Behav. 2020;34(7):726–33. https://doi.org/10.1037/adb0000579.
Young SD, Koussa M, Lee S-J, Perez H, Gill N, Gelberg L, et al. Feasibility of a social media/online community support group intervention among chronic pain patients on opioid therapy. J Addict Dis. 2018;37(1-2):96–101. https://doi.org/10.1080/10550887.2018.1557992.
Wilson M, Finlay M, Orr M, Barbosa-Leiker C, Sherazi N, Roberts MLA, et al. Engagement in online pain self-management improves pain in adults on medication-assisted behavioral treatment for opioid use disorders. Addict Behav. 2018;86:130–7. https://doi.org/10.1016/j.addbeh.2018.04.019.
Flynn N. Effect of an online hypnosis intervention in reducing migraine symptoms: a randomized controlled trial. Int J Clin Exp Hypn. 2019;67(3):313–35. https://doi.org/10.1080/00207144.2019.1612674.
Depp CA, Perivoliotis D, Holden J, Dorr J, Granholm EL. Single-session mobile-augmented intervention in serious mental illness: a three-arm randomized controlled trial. Schizophr Bull. 2019;45(4):752–62. https://doi.org/10.1093/schbul/sby135.
Schleider JL, Weisz JR. Little treatments, promising effects? Meta-analysis of single-session interventions for youth psychiatric problems. J Am Acad Child Adolesc Psychiatry. 2017;56(2):107–15. https://doi.org/10.1016/j.jaac.2016.11.007.
Ellis JG, Cushing T, Germain A. Treating acute insomnia: a randomized controlled trial of a “single-shot” of cognitive behavioral therapy for insomnia. Sleep. 2015;38(6):971–8.
Samson JE, Tanner-Smith EE. Single-session alcohol interventions for heavy drinking college students: a systematic review and meta-analysis. J Stud Alcohol. 2015;76(4):530–43. https://doi.org/10.15288/jsad.2015.76.530.
Ziadni MS, Carty JN, Doherty HK, Porcerelli JH, Rapport LJ, Schubiner H, et al. A life-stress, emotional awareness, and expression interview for primary care patients with medically unexplained symptoms: a randomized controlled trial. Health Psychol. 2018;37(3):282–90. https://doi.org/10.1037/hea0000566.
Carty JN, Ziadni MS, Holmes HJ, Tomakowsky J, Peters K, Schubiner H, et al. The effects of a life stress emotional awareness and expression interview for women with chronic urogenital pain: a randomized controlled trial. Pain Med. 2019;20(7):1321–9. https://doi.org/10.1093/pm/pny182.
Darnall BD, Ziadni MS, Krishnamurthy P, Flood P, Heathcote LC, Mackey IG, et al. “My surgical success”: effect of a digital behavioral pain medicine intervention on time to opioid cessation after breast cancer surgery—a pilot randomized controlled clinical trial. Pain Med. 2019;20(11):2228–37. https://doi.org/10.1093/pm/pnz094.
Darnall BD, Sturgeon JA, Kao M-C, Hah JM, Mackey SC. From catastrophizing to recovery: a pilot study of a single-session treatment for pain catastrophizing. J Pain Res. 2014;7:219.
Darnall BD, Ziadni MS, Roy A, Kao M-C, Sturgeon JA, Cook KF, et al. Comparative efficacy and mechanisms of a single-session pain psychology class in chronic low back pain: study protocol for a randomized controlled trial. Trials. 2018;19(1):1–15.
ClinicalTrials.gov [Internet]. Virtual single-session “Empowered Relief” group intervention for chronic pain. Bethesda (MD): National Library of Medicine (US). Identifier NCT04546685. September 14, 2020 [cited October 5, 2020] Available from: https://clinicaltrials.gov/ct2/show/NCT04546685?id=NCT04546685&draw=2&rank=1.
Ziadni MS, Chen AL, Winslow T, Mackey SC, Darnall BD. Efficacy and mechanisms of a single-session behavioral medicine class among patients with chronic pain taking prescription opioids: study protocol for a randomized controlled trial. Trials. 2020;21(1):1–12.
Darnall BD, Roy A, Kao MC, Ziadni M, You DS, Cook KF, et al. Comparison of a single-session pain management skills class (“Empowered Relief”) vs. cognitive behavioral therapy or health education for chronic low back pain: a non-inferiority and combined superiority randomized trial. Accepted.
Alschuler KN, Altman JK, Ehde DM. Feasibility and acceptability of a single-session, videoconference-delivered group intervention for pain in multiple sclerosis. Rehabil Psychol. 2021;66(1):22–30. https://doi.org/10.1037/rep0000360.
Rodda SN, Lubman D, Jackson A, Dowling NA. Improved outcomes following a single session web-based intervention for problem gambling. J Gambl Stud. 2017;33(1):283–99. https://doi.org/10.1007/s10899-016-9638-2.
Schleider JL, Dobias M, Sung J, Mumper E, Mullarkey MC. Acceptability and utility of an open-access, online single-session intervention platform for adolescent mental health. JMIR Ment Health. 2020;7(6):e20513. https://doi.org/10.2196/20513.
Wasil AR, Park SJ, Gillespie S, Shingleton R, Shinde S, Natu S, et al. Harnessing single-session interventions to improve adolescent mental health and well-being in India: development, adaptation, and pilot testing of online single-session interventions in Indian secondary schools. Asian J Psychiatr. 2020;50:101980.
Cardamone-Breen MC, Jorm AF, Lawrence KA, Rapee RM, Mackinnon AJ, Yap MBH. A single-session, web-based parenting intervention to prevent adolescent depression and anxiety disorders: randomized controlled trial. J Med Internet Res. 2018;20(4):e148. https://doi.org/10.2196/jmir.9499.
Schleider JL, Dobias ML, Sung JY, Mullarkey MC. Future directions in single-session youth mental health interventions. J Clin Child Adolesc Psychol. 2020;49(2):264–78. https://doi.org/10.1080/15374416.2019.1683852.
Hart T, Fann JR, Novack TA. The dilemma of the control condition in experience-based cognitive and behavioural treatment research. Neuropsychol Rehabil. 2008;18(1):1–21. https://doi.org/10.1080/09602010601082359.
Mohr DC, Spring B, Freedland KE, Beckner V, Arean P, Hollon SD, et al. The selection and design of control conditions for randomized controlled trials of psychological interventions. Psychother Psychosom. 2009;78(5):275–84. https://doi.org/10.1159/000228248.
Sullivan MJ, Bishop SR, Pivik J. The pain catastrophizing scale: development and validation. Psychol Assess. 1995;7(4):524–32. https://doi.org/10.1037/1040-3590.7.4.524.
Cherkin DC, Sherman KJ, Balderson BH, Cook AJ, Anderson ML, Hawkes RJ, et al. Effect of mindfulness-based stress reduction vs cognitive behavioral therapy or usual care on back pain and functional limitations in adults with chronic low back pain: a randomized clinical trial. Jama. 2016;315(12):1240–9. https://doi.org/10.1001/jama.2016.2323.
Turner JA, Anderson ML, Balderson BH, Cook AJ, Sherman KJ, Cherkin DC. Mindfulness-based stress reduction and cognitive-behavioral therapy for chronic low back pain: similar effects on mindfulness, catastrophizing, self-efficacy, and acceptance in a randomized controlled trial. Pain. 2016;157(11):2434–44. https://doi.org/10.1097/j.pain.0000000000000635.
Cella D, Riley W, Stone A, Rothrock N, Reeve B, Yount S, et al. The Patient-Reported Outcomes Measurement Information System (PROMIS) developed and tested its first wave of adult self-reported health outcome item banks: 2005–2008. J Clin Epidemiol. 2010;63(11):1179–94. https://doi.org/10.1016/j.jclinepi.2010.04.011.
Darnall BD, Sturgeon JA, Cook KF, Taub CJ, Roy A, Burns JW, et al. Development and validation of a daily pain catastrophizing scale. J Pain. 2017;18(9):1139–49. https://doi.org/10.1016/j.jpain.2017.05.003.
Sturgeon JA, Hah JM, Sharifzadeh Y, Middleton SK, Rico T, Johnson KA, et al. Predictors of daily pain medication use in individuals with recurrent back pain. J Behav Med. 2018;25(2):252–8.
Ziadni M, Sturgeon J, Darnall B. The relationship between negative metacognitive thoughts, pain catastrophizing and adjustment to chronic pain. Eur J Pain. 2018;22(4):756–62. https://doi.org/10.1002/ejp.1160.
Mojtabai R. National trends in long-term use of prescription opioids. Pharmacoepidemiol Drug Saf. 2018;27(5):526–34. https://doi.org/10.1002/pds.4278.
Edlund MJ, Martin BC, Fan M-Y, Devries A, Braden JB, Sullivan MD. Risks for opioid abuse and dependence among recipients of chronic opioid therapy: results from the TROUP study. Drug Alcohol Depend. 2010;112(1-2):90–8. https://doi.org/10.1016/j.drugalcdep.2010.05.017.
Compton WM, Volkow ND. Major increases in opioid analgesic abuse in the United States: concerns and strategies. Drug Alcohol Depend. 2006;81(2):103–7. https://doi.org/10.1016/j.drugalcdep.2005.05.009.
Boscarino JA, Rukstalis M, Hoffman SN, Han JJ, Erlich PM, Gerhard GS, et al. Risk factors for drug dependence among out-patients on opioid therapy in a large US health-care system. Addiction. 2010;105(10):1776–82. https://doi.org/10.1111/j.1360-0443.2010.03052.x.
Boutron I, Moher D, Altman DG, Schulz KF, Ravaud P. Extending the CONSORT statement to randomized trials of nonpharmacologic treatment: explanation and elaboration. Ann Intern Med. 2008;148(4):295–309. https://doi.org/10.7326/0003-4819-148-4-200802190-00008.
Thorn BE, Pence LB, Ward LC, Kilgo G, Clements KL, Cross TH, et al. A randomized clinical trial of targeted cognitive behavioral treatment to reduce catastrophizing in chronic headache sufferers. J Pain. 2007;8(12):938–49. https://doi.org/10.1016/j.jpain.2007.06.010.
Glombiewski JA, Hartwich-Tersek J, Rief W. Attrition in cognitive-behavioral treatment of chronic back pain. Clin J Pain. 2010;26(7):593–601. https://doi.org/10.1097/AJP.0b013e3181e37611.
Dworkin RH, Turk DC, McDermott MP, Peirce-Sandner S, Burke LB, Cowan P, et al. Interpreting the clinical importance of group differences in chronic pain clinical trials: IMMPACT recommendations. Pain. 2009;146(3):238–44. https://doi.org/10.1016/j.pain.2009.08.019.
U.S. Department of Health & Human Services. Pain Management Best Practices Inter-Agency Task Force Report: updates, gaps, inconsistencies, and recommendations 2019. Available from: U.S. Department of Health and Human Services website: https://www.hhs.gov/ash/advisory-committees/pain/reports/index.html.
Acknowledgements
The authors thank the National Institute of Health for providing funding (NIH K23DA047473).
Sponsors
the investigator was sponsored by the NIH National Institute on Drug Abuse (NIH K23DA047473) who approved the study design, but played no part in the data collection, management, analysis, and interpretation of the data; writing of the report; and decision to submit the report for publication.
Trial status
NCT04546685, recruitment began on 09/28/2020. The expected date when recruitment will be completed is 03/2021. IRB (protocol #31191) was initially approved on 09/25/2020.
Funding
NIH K23DA047473 (MSZ). The design and methodology of this clinical trial were approved by the NIDA Office of Clinical and Regulatory Affairs.
Author information
Authors and Affiliations
Contributions
MSZ and BDD conceived the trial. BDD created the single-session skills-based pain management intervention. MSZ, BDD, and LGC refined the protocol and selected the measures. MZ and BDD developed the plans for the statistical analyses. MSZ, BDD, SRA, and LGC drafted the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was approved by the Stanford’s Institutional Review Board (protocol # #31191) 3000 El-Camino Real, Five Palo Alto Square, 4th Floor, Palo Alto, CA 94306. An informed consent will be obtained from all participants in the study.
Consent for publication
Not applicable.
Competing interests
All authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Findings from this study will be submitted for publication in peer-reviewed journals regardless of the study outcome, and an accessible summary of findings will be produced for participants and members of the public. We do not plan to use professional writers in future publications. The funding agency has participated in the design of the study and ensuring there is a proper data monitoring process.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ziadni, M.S., Anderson, S.R., Gonzalez-Castro, L. et al. Comparative efficacy of a single-session “Empowered Relief” videoconference-delivered group intervention for chronic pain: study protocol for a randomized controlled trial. Trials 22, 358 (2021). https://doi.org/10.1186/s13063-021-05303-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13063-021-05303-8