
Abstract
Triple negative breast cancer (TNBC), which is typically lack of expression of estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2), represents the most aggressive and mortal subtype of breast cancer. Currently, only a few treatment options are available for TNBC due to the absence of molecular targets, which underscores the need for develo** novel therapeutic and preventive approaches for this disease. Recent evidence from clinical trials and preclinical studies has demonstrated a pivotal role of signal transducer and activator of transcription 3 (STAT3) in the initiation, progression, metastasis, and immune evasion of TNBC. STAT3 is overexpressed and constitutively activated in TNBC cells and contributes to cell survival, proliferation, cell cycle progression, anti-apoptosis, migration, invasion, angiogenesis, chemoresistance, immunosuppression, and stem cells self-renewal and differentiation by regulating the expression of its downstream target genes. STAT3 small molecule inhibitors have been developed and shown excellent anticancer activities in in vitro and in vivo models of TNBC. This review discusses the recent advances in the understanding of STAT3, with a focus on STAT3’s oncogenic role in TNBC. The current targeting strategies and representative small molecule inhibitors of STAT3 are highlighted. We also propose potential strategies that can be further examined for develo** more specific and effective inhibitors for TNBC prevention and therapy.
Similar content being viewed by others

Background
Triple negative breast cancer (TNBC) is the most aggressive form of breast cancer and accounts for much higher recurrence and metastasis rates [1]. Due to the absence of the expression of estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2), TNBC is unresponsive to endocrine and HER2-targeted therapies, which results in the high mortality of patients with this disease [1]. When patients are diagnosed with TNBC at the early stage, combination chemotherapy (anthracyclines, taxanes, platinum salts, etc.) with or without radiotherapy is used as standard non-surgical therapy and has shown some efficacy in patients with both primary and metastatic diseases [2]. Because of the inter- and the intratumoral heterogeneities of TNBC, the intrinsic chemoresistance as well as severe side effects are often observed and lead to limited success in the clinic [3, 4]. Targeted therapies (e.g., poly (ADP-ribose) polymerase (PARP) inhibitors and epidermal growth factor receptor (EGFR) inhibitors) and immunotherapies have also shown some promise in preliminary clinical studies, but further investigations are critically needed [5,6,7]. More recently, many efforts have been made to identify targetable molecules for treating TNBC via genomic profiling and several critical alternations have been discovered, including the overexpression and aberrant activation of signal transducer and activator of transcription 3 (STAT3) [8, 9]. The emerging data suggest that STAT3 may be a potential molecular target and biomarker for TNBC.
The STAT family of transcription factors is comprised of seven members with high structural and functional similarity, including STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b, and STAT6 [10, 11]. All STAT proteins consist of an amino acid domain (NH2), a coiled-coil domain (CCD) for binding with interactive proteins, a DNA binding domain (DBD), a linker domain, a SRC homology 2 (SH2) domain for phosphorylation and dimerization, and a C-terminal transactivation domain (TAD) [11]. Most of these domains are highly conserved among STAT proteins and only TAD is divergent and mainly contributes to their structure diversity [12]. STAT3 was initially discovered to bind to DNA in response to interleukin-6 (IL-6) and epidermal growth factor (EGF) in 1994 [13, 14]. Over the past decades, STAT3 has become one of the most investigated oncogenic transcription factors and is highly associated with cancer initiation, progression, metastasis, chemoresistance, and immune evasion [15, 16]. The recent evidence from both preclinical and clinical studies have demonstrated that STAT3 plays a critical role in TNBC and STAT3 inhibitors have shown efficacy in inhibiting TNBC tumor growth and metastasis.
Considering that there is an unmet medical need for TNBC treatment and innovative therapeutic agents are urgently required, an in-depth understanding of the roles of STAT3 in TNBC will facilitate the development of STAT3-targeted therapeutics and pave the way for a novel TNBC treatment approach. In this review, we focus on the recent findings related to STAT3’s role in TNBC as well as STAT3 inhibitors and current targeting strategies. We also discuss other potential strategies for develo** new STAT3 inhibitors for TNBC treatment.
The STAT3 signaling pathway
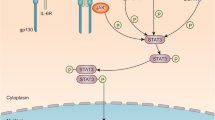
The classical STAT3 signaling pathway that is activated through the binding of cytokines or growth factors to their corresponding cell surface receptors has been extensively reviewed [16,17,18]. Here, we present a brief overview of the STAT3 signaling pathway, nonreceptor tyrosine kinases of STAT3, and its intrinsic inhibitors and coactivators, which are depicted in Fig. 1. Briefly, the overexpressed cytokine receptors, e.g., interleukin-6 receptor (IL-6R) and interleukin-10 receptor (IL-10R) and the hyperactive growth factor receptors, e.g., epidermal growth factor receptor (EGFR), fibroblast growth factor receptor (FGFR) and insulin-like growth factor receptor (IGFR) always trigger the tyrosine phosphorylation cascade through the binding of ligands to these receptors, leading to the aberrant activation of STAT3 and the transcription of its downstream target genes [17]. Once the ligands bind to their receptors on the cell surface, these receptors further form dimers and successively recruit glycoprotein 130 (gp130) and Janus kinases (JAKs), thus phosphorylating and activating JAKs [19]. Conversely, the cytoplasmic tyrosine residues of these receptors are phosphorylated by the activated JAKs and then interact with the SH2 domain of STAT3, resulting in STAT3 phosphorylation at Tyr705 by JAKs [16]. In addition, STAT3 can be phosphorylated and activated by several nonreceptor tyrosine kinases, e.g., Src and Abl [20]. The phosphorylated STAT3 (pSTAT3) further forms a homodimer through interaction between their phosphorylated Tyr705 site and SH2 domain, triggering the dissociation of STAT3 dimers from the cell surface receptors and its translocation from cytoplasm to the nucleus [21, 22]. With the help of a variety of coactivator proteins, including NCOA/SRC1a, apurinic/apyrimidinic endonuclease-1/redox factor-1 (APE/Ref-1), and CREB-binding protein (CBP)/p300, the nuclear STAT3 binds to specific DNA sequences and activates the transcription of genes that regulate various phenotypes of cancer cells [17, 18].

The STAT3 signaling pathway in cancer cells. Under normal physiological conditions, STAT3 activation is strictly controlled by the endogenous inhibitors, including the protein inhibitor of activated STAT (PIAS), the suppressor of cytokine signaling (SOCS), and several protein tyrosine phosphatases (PTPs). Once the upstream cytokines (e.g., IL-6) or growth factors (e.g., EGF, FGF, and VEGF) bind to cell surface receptors, STAT3 is phosphorylated and activated by JAK or Src. The nonreceptor tyrosine kinases (e.g., Src and Abl) also phosphorylate STAT3. The phosphorylated STAT3 undergoes dimerization and translocates from cytoplasm into the nucleus. The activated STAT3 further binds to DNA and its coactivators (e.g., NCOA, APE, and CBP) and induces the transcription of its downstream target genes
STAT3 is also highly expressed in some normal tissues and organs, including the bone marrow, peripheral nervous system, and digestive tract and plays a physiological role [23,24,25]. In the normal physiological conditions, STAT3 phosphorylation and activation are tightly controlled by several intrinsic inhibitors, including protein tyrosine phosphatases (PTPs), the suppressors of cytokine signaling (SOCS), and the protein inhibitor of activated STAT (PIAS) [26]. The Src homology domain-containing tyrosine phosphatases 1/2 (SHP-1/2) directly interact and dephosphorylate JAK and STAT3, resulting in their inactivation [27, 28]. The nuclear PTPs, including TC45 and T-cell protein-tyrosine phosphatase (TC-PTP) induce the inactivation of STAT3 through its dephosphorylation and translocation from nucleus to the cytoplasm [29, 30]. Other PTPs, such as PTP1B and PTPeC have also been reported to regulate STAT3 dephosphorylation and inactivation [31]. Moreover, SOCS directly interacts with JAK and STAT3 and inhibits their phosphorylation and activation via forming a negative feedback loop with JAK-STAT3 signaling pathway [32]. PIAS inhibits the binding of nuclear STAT3 to DNA and induces STAT3 dephosphorylation via protein tyrosine phosphatase receptor T (PTPRT), leading to the reduced expression of its downstream target genes [33]. In addition, the stability of STAT3 protein is also regulated by the ubiquitin-proteasome system via the ubiquitin ligase TRAF6 (tumor necrosis factor receptor-associated factor 6) [34]. Recent studies have also reported that miR-544 directly targets the 3′-untranslated region (UTR) on STAT3 mRNA, thus down-regulating STAT3 expression in TNBC cells [35]. Due to the presence of these endogenous inhibitors, STAT3 is strictly governed to exert its physiological functions in normal cells [36]. Herein, both direct inhibition of STAT3 and activation of the endogenous inhibitors may be considered as potential STAT3-inhibiting strategies for develo** novel cancer therapeutics.
The STAT3 signaling pathway in triple negative breast cancer
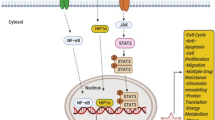
The oncogenic potential of STAT3 has been widely recognized through its involvement in regulating the expression of genes related to cancer cell proliferation, anti-apoptosis, migration, invasion, angiogenesis, chemoresistance, immune suppression, stem cell self-renewal and maintenance, and autophagy (as shown in Fig. 2) [17, 18]. Importantly, STAT3 is overexpressed and constitutively activated in TNBC, which is highly related to TNBC initiation, progression, metastasis, resistance to chemotherapy, and the poor survival outcomes [8]. STAT3 is not only capable of eliciting the expression of cancer-related genes, but also physically interacts and functionally cooperates with other oncogenic transcription factors, e.g., GLI1, promoting the aggressiveness of TNBC [8]. A recent study has also found a reduction of the gene associated with retinoic-interferon-induced mortality 19 (GRIM-19), an intrinsic inhibitor of STAT3 transcription accompanied by STAT3 overexpression in TNBC [37]. In addition, TCPTP, including two splice variants TC45 and TC48 are down-regulated in TNBC cells in vitro and in vivo, which also contributes to the activation of STAT3 signaling [38]. Indeed, STAT3 has also been found to localize in the mitochondria, where it is termed mitoSTAT3 and regulates the mitochondrial functions, including electron transport chain, ATP synthesis, calcium homeostasis, and reactive oxygen species (ROS) accumulation [39, 40]. Moreover, mitoSTAT3 has been shown to promote breast cancer cell growth, in which the phosphorylation of Serine 727 plays a critical role [41].

Activation of STAT3 signaling promotes growth, metastasis, chemoresistance, immune escape, and stemness in TNBC. One the upstream regulators are activated, STAT3 is phosphorylated, dimerized, and translocated into the nucleus, where it activates the transcription of the target genes that regulate cell proliferation, anti-apoptosis, migration, invasion, angiogenesis, chemoresistance, immune escape, stem cell phenotypes, and autophagy
A recent study has shown that acetylated STAT3 is highly elevated in TNBC, causing the methylation and inactivation of tumor-suppressor gene promoters [

Inhibiting STAT3 signaling at multiple levels for cancer therapy. Currently, the majority of STAT3 inhibitors have been developed through (1) targeting the upstream regulators of STAT3, (2) binding to the SH2 domain of STAT3 and inhibiting its activation, (3) inhibiting STAT3 phosphorylation or acetylation, or (4) blocking STAT3-DNA binding. Other potential strategies, such as (5) inhibiting the binding of STAT3 with its co-activators, (6) modulating the binding of STAT3 with other interactive proteins, and (7) promoting STAT3 ubiquitination and proteasomal degradation may also be evaluated for develo** novel STAT3 inhibitors
Target upstream regulators of STAT3
The majority of STAT3 inhibitors have been identified to target the upstream regulators of STAT3 signaling. STAT3 activation is often initiated through the binding of cytokines and growth factors to their corresponding cell surface receptors. Therefore, small molecules and natural products that are able to inhibit IL-6 secretion and production, e.g., carfilzomib [87], manuka honey [88], bazedoxifene [89, 90], and Ganoderma lucidum extract [91] or suppress EGFR expression and phosphorylation, e.g., deguelin [92], picrasidine G [93], cantharidin [94], and silibinin [95] have shown significant inhibitory effects on STAT3 signaling as well as the expression of its downstream target genes in TNBC cell lines. In addition, arsenic trioxide (ATO) was reported to inhibit IL-6-mediated STAT3 activation, consequently reducing the expression of VEGF and suppressing angiogenesis [96]. Further studies have demonstrated that ATO blocks the interaction between enhancer of zeste homolog 2 (EZH2) and NF-κB p65, herein suppressing the activity of NF-κB and reducing the expression of IL-6. All these indirect STAT3 inhibitors have exhibited potent in vitro and in vivo anti-TNBC activities (Table 1). However, most of them have also been found to inhibit other signaling pathways that are triggered by ligand-cell surface receptor binding in cancer cells, indicating a low level of specificity in targeting the STAT3 signaling pathway.
As discussed earlier, several protein tyrosine kinases, such as JAK2 contribute to STAT3 phosphorylation and activation in both receptor-dependent and/or receptor-independent manners. JAK2 inhibitors, including silibinin [97] and ganoderic acid A [98] were found to inhibit TNBC cell viability, migration, and invasion and induce apoptosis in vitro through inhibiting the JAK2/STAT3 signaling pathway. However, their in vivo efficacy still needs further investigation. Targeting the intrinsic STAT3 inhibitors, such as PTPs and SOCS have been considered as a potential strategy for repressing STAT3 signaling pathway. Several natural and synthetic compounds were identified to activate one of the STAT3 PTPs, SHP-1. Among them, nintedanib and SC-78 significantly increase SHP-1 activity without affecting its expression [99, 100], while 1,2,3,4,6-penta-O-galloyl-beta-D-glucose (PGG) and SC-2001 largely induce the expression of SHP-1 [101, 102]. All these SHP-1 activators were also shown to inhibit STAT3 phosphorylation and the expression of its downstream target genes, thus suppressing TNBC cell growth and migration and inducing apoptosis in vitro and in vivo [99,100,101,102]. In addition, isolinderalactone was reported to increase SOCS3 expression and then enhance SOCS3-mediated STAT3 dephosphorylation and inactivation [103].
As one of the major client proteins of heat shock protein 90 (HSP90), STAT3 can be degraded through inhibiting HSP90. Two deguelin-derived HSP90 inhibitors, termed compound 57 and L80 have been observed to inhibit STAT3 expression and phosphorylation by interacting with the C-terminal ATP-binding pocket of HSP90 and blocking its function [104, 105]. Both compounds have also exerted their anticancer activities in TNBC models in vitro and in vivo [104, 105]. Moreover, nor-wogonin was found to inhibit the expression of transforming growth factor β-activated kinase 1 (TAK1), therefore dephosphorylating STAT3 without affecting its total expression level [106]. The dopamine receptor D2 (DRD2)-targeting drug thioridazine inhibits TNBC cell self-renewal through reducing DRD2-mediated STAT3 activation [107]. Due to the highly conserved structures among STAT family members, targeting the upstream regulators always results in the wide-spectrum inhibition of all STAT proteins, causing off-target effects. Therefore, directly targeting STAT3 and/or inhibiting its functions may be more promising strategies for develo** safe and effective anticancer therapeutics.
Directly bind to STAT3 and inhibit its activation
Due to advances in the understanding of the structural biology of STAT3, small molecule inhibitors have been developed to directly bind to STAT3 and inhibit its activity. Currently, many small molecule inhibitors have been designed to target the SH2 domain and block its phosphorylation, dimerization, and nuclear translocation. Several STAT3-binding small molecule inhibitors that are under preclinical and clinical investigations have shown excellent efficacy in TNBC cells in vitro and in vivo.
Recently, a dual-luciferase assay-based screening of 1563 compounds for STAT3 inhibitors was performed, leading to the identification of Bt354 [108]. Further studies have shown that Bt354 inhibits STAT3 phosphorylation and nuclear translocation, which may be attributed to the binding of this compound to the SH2 domain of STAT3. Bt354 did not cause significant changes in the expression of STAT3 upstream regulators JAK2 and Src, indicating a specific targeting effect on STAT3 [108]. Moreover, this small molecule inhibitor also suppresses the viability of TNBC cells with constitutively activated STAT3, induces the G2/M phase arrest and late apoptosis, and impairs cell migration in vitro and represses the growth of TNBC xenograft tumors in vivo [108]. Additionally, several natural products, including osthole [109], arctigenin [110], and alantolactone [111] have also been shown to directly bind to the SH2 domain of STAT3, inhibit its phosphorylation and activation, and suppress the growth and metastasis of TNBC in vitro and in vivo. Cryptotanshinone is a well-documented natural product inhibitor of STAT3, which also binds to the SH2 domain and inhibits the phosphorylation and dimerization of STAT3 [112]. KYZ3, a synthetic derivative of cryptotanshinone has recently been developed and shown to exert anticancer activity in TNBC cells in vitro and in vivo through binding to and inhibiting STAT3 activation [113]. However, none of these compounds have been evaluated for their binding affinity to STAT3. Their selectivity among STAT3 and other STAT family members is yet to be determined.
Inhibit STAT3 phosphorylation or acetylation
Except for the STAT3-binding small molecule inhibitors that we discussed above, a number of natural products and their derivatives were found to inhibit STAT3 phosphorylation and/or nuclear translocation without affecting the upstream regulators. Sesquiterpene lactones, which are enriched in the hexane fraction from Inula helenium L. have been shown to suppress tumor growth in vitro and in vivo by inhibiting STAT3 phosphorylation and decreasing the expression of the downstream target genes, including cyclin D1, c-Myc, and Bcl-2 [114]. Another crude extract from the fruits of Rhus coriaria was also discovered to inhibit angiogenesis, tumor growth and metastasis in TNBC models in vitro and in vivo by repressing STAT3 phosphorylation and STAT3-mediated VEGF expression [115]. Moreover, several natural compounds and derivatives, including schisandrin B [116], eupalinolide J [117], galiellalactone analogs 16 and 17 [118], and ursolic acid derivative FZU-03,010 [119] have shown in vitro and in vivo efficacy in TNBC models through inhibition of STAT3 phosphorylation and/or nuclear translocation. None of them have been investigated for the binding ability with STAT3. Considering that these compounds did not show any significant effects on STAT3 regulators and interactive proteins, further studies for examining the potential binding between STAT3 and these compounds would provide important information regarding their underlying molecular mechanisms.
Of note, several approved drugs have shown potent inhibitory effects on pSTAT3 and may be repositioned as anticancer drugs. Niclosamide, an FDA-approved anthelmintic drug was identified as a potent STAT3 inhibitor. A recent study demonstrated that niclosamide not only inhibits TNBC cell viability but also sensitizes TNBC cells to ionizing irradiation (IR) by blocking IR-induced STAT3 phosphorylation and activation [
