
Abstract
The lack of effective therapies that slow the progression of Alzheimer’s disease (AD) and related tauopathies highlights the need for a more comprehensive understanding of the fundamental cellular mechanisms underlying these diseases. Model organisms, including yeast, worms, and flies, provide simple systems with which to investigate the mechanisms of disease. The evolutionary conservation of cellular pathways regulating proteostasis and stress response in these organisms facilitates the study of genetic factors that contribute to, or protect against, neurodegeneration. Here, we review genetic modifiers of neurodegeneration and related cellular pathways identified in the budding yeast Saccharomyces cerevisiae, the nematode Caenorhabditis elegans, and the fruit fly Drosophila melanogaster, focusing on models of AD and related tauopathies. We further address the potential of simple model systems to better understand the fundamental mechanisms that lead to AD and other neurodegenerative disorders.
Similar content being viewed by others


Background
Alzheimer’s disease (AD) is the most common form of dementia. It is characterized by the accumulation of amyloid-beta in senile plaques and hyperphosphorylated Tau in neurofibrillary tangles (NFTs) [22]. Mutations in the amyloid precursor protein (APP) or the proteins that cleave it, the presenillins, PSEN1 and PSEN2 (as part of the gamma secretase), have been identified in familial AD [63, 277]. Large-scale RNAi screens have been performed with both models, with only one gene, the HSP70 family member hsp-1, found in both screens [154, 165].
Similar to other models, expression of AD-related proteins in flies results in aggregation and impaired cellular functions. Expressing hAPP in flies, together with the cleaving enzyme BACE and presenilins, in photoreceptor cells generates amyloid plaques and leads to neurodegeneration [110]. In addition, ubiquitous expression of these genes caused ectopic wing vein formation and a shortened lifespan. Amyloid deposition and neurodegeneration were also observed when human Aβ42 was expressed in the fly brain [84, 138]. Interestingly, expressing Aβ40 causes age-dependent learning defects but no obvious neurodegeneration, consistent with observations in mammalian systems where Aβ40 is less fibrillogenic and toxic than Aβ42 [139, 216].
Tau-based models of disease
Tau is a highly soluble protein that binds tubulins and promotes the assembly and stabilization of microtubules. In the human brain, six major Tau isoforms are generated by alternative splicing [104]. Isoforms have one (1N), two (2N) or no amino-terminal inserts (0N) and differ in the exclusion or inclusion of exon 10, resulting in a protein with either 3 (3R) or 4 (4R) microtubule-binding regions. In a normal adult brain, the ratio of 3R to 4R isoforms is approximately equal [124]. Both 3R and 4R isoforms are found in the AD brain but the ratio of the two may change during the course of the disease [124]. The phosphorylation status of Tau also changes in AD. Hyperphosphorylation of Tau precedes its aggregation into NFTs [3]. Although MAPT mutations are not a cause of AD, they are causal in other tauopathies including frontotemporal dementia (FTD) and Pick’s disease [141].
Expression of FTD-associated Tau variants (V337M, V301L, R406W) has been used to model AD in both flies and worms [2, 107, 204, 245]. Expression of these Tau variants in C. elegans induces disease-associated pathologies, including phosphorylation of Tau at disease-relevant sites, accumulation of insoluble Tau aggregates, synaptic loss, decreased neuronal function and neurodegeneration [23, 167, 209]. In studies comparing wild-type and mutant Tau, disease-associated mutations produced more severe phenotypes [167]. Transgenic flies expressing mutant human Tau show increased Tau phosphorylation and disease-associated Tau conformations, reduced lifespan, and vacuolization and degeneration of cortical cells [243, 274, 320, 321]. In C. elegans, impaired locomotion occurs before insoluble Tau aggregates are detected, suggesting that neurodegeneration in this model is not a general effect of aggregated protein [167]. Increased Tau phosphorylation and age-related neurodegeneration were observed in a Drosophila model in the absence of neurofibrillary tangles [320, 321]. Overall, expressing AD-related proteins in simple models recapitulates many, but not all, features of the disease (Fig. 1) and provides insight into the progression of the disease.

Modeling Alzheimer's disease in simple model systems. Expressing AD-related proteins Tau and Amyloid beta, in simple model systems, including S. cerevisiae, C. elegans, and Drosophila melanogaster recapitulates many disease-associated phenotypes
Large-scale screens in yeast, worms, and fruit flies have identified many genetic modifiers of amyloid-beta and Tau toxicity (Table 1) [17, 30, 42, 93, 154, 156, 165, 255, 274, 275, 285, 297]. Here we summarize these modifiers and compare findings across species. Many of these modifier genes can be linked to cellular pathways and processes associated with AD, such as protein trafficking and localization, cell cycle, metabolic processes, gene expression, and stress response (Fig. 2).

Pathways enriched amoung genetic modifiers of AD models. Human homologs of genetic modifiers were analyzed using ShinyGO v0.77 (Ge et al. 2020). Nodes represent the top 30 enriched GO terms. Node size is scaled to the number of genes. Darker nodes are more significantly enriched. Edge thickness is proportional to the number of overlap** genes in each category
While extensive validation of many of these modifiers is lacking, modifiers identified in different studies could reveal important regulators of neurodegeneration and identify processes that are important for maintaining neuronal health. We performed functional annotation of modifiers from each species using online tools including FunSpec, GO Slim Mapper, Wormcat, Flybase, and GLAD [20, 120, 130, 135, 257] (Tables S6-S11). We also identified human homologs of these genes (Tables S1-S5). Homologs of AD-associated genes reported from genome-wide association studies (GWAS) were identified by comparing our list to the > 900 loci collected in the Alzheimer’s Disease Variant Portal (ADVP) [168]. In Drosophila, 60 genes (6%) identified as modifiers had potential human orthologs with some association to AD. Of the 176 modifiers identified in C. elegans, 23 genes had no predicted human ortholog and seven genes (4%) had orthologs listed in the ADVP database (Supplementary tables).
AD-associated pathways in simple model systems
The largest collection of modifiers of Aβ toxicity was identified in yeast. Three separate Aβ modifier screens were carried out, two using loss-of-function approaches and one using an overexpression system [42, 93, 297]. Although over 1000 genes (~ 17% of the genome) were identified as modifiers, only 16 genes had the same effect in more than one screen and only two genes (XRN1 and SLA1) had the same effect in all three screens (Fig. 3A &B). Twelve of these genes had human orthologs, with two (PDE2 and SCD6) identified as potential risk genes for AD [1]. PDE2 is a phosphodiesterase that enhances Aβ toxicity in yeast. It is homologous to several human phosphodiesterases, including PDE9A, which regulates cGMP and functions in learning and long-term memory. Consistent with PDE9A promoting neurodegeneration, PDE9A inhibitors have had success in pre-clinical studies where they improve cognitive function in rats [305]. SCD6, an ortholog of the processing body assembly factor LSM14A, functions in RNA processing.

Enhancer and suppressor screens identify distinct and overlap** gene sets within, and between species. A Summary of three large-scale screens performed in S. cerevisiae. Human orthologs are shown on the right. Blue colour indicates that the gene function was protective, orange indicates gene function enhances toxicity. B Venn diagram showing overlap of genes recovered between yeast screens. C Physical interaction network of human orthologs of genes identified in C. elegans (pink) or D. melanogaster (blue) as modifiers of AD models. Network from Biogrid, 0.4 confidence, physical interaction subnetwork
The lack of overlap in yeast screens could reflect a limitation of the phenotype used to score toxicity in yeast. Because the yeast screens use growth as a readout, there is potential for false positives to arise when deletions affect growth. While mutants with growth defects are generally either removed from consideration or normalized in some way, there is the potential for Aβ to act as a sensitized background, producing synthetic interactions that are not specific to Aβ biology. Further analysis would be required to confirm the role of these homologs in AD-related processes.
Similar to what was observed in yeast, a comparison of the C. elegans and Drosophila screens showed little overlap. No overlap was found in Drosophila and C. elegans Aβ screens, and only one gene (fat-7/CG8630), a homolog of human SCD/SCD5, had the same effect in Tau screens across species. This may be attributed to differences in cell type expression or phenotypic output. In the two largest C. elegans Aβ screens, Aβ was expressed in different compartments (one neuronal and one muscular) and different phenotypic readouts were used. While these screens have little overlap between species on a gene-by-gene basis, they do overlap in the types of genes and processes that are recovered. Primarily, these genes can be categorized into some common functional groups already associated with neurodegeneration, including proteostasis, trafficking, cellular stress-related pathways including ER stress and oxidative stress, immune response, metabolism, cytoskeleton, and signaling (Fig. 2). We also found that physical interactions had been reported between some of the modifiers (Figs. 4 and 5). Additionally, we examined the reported protein interactions for the human orthologs of C. elegans and Drosophila modifiers and found that they could be connected in a larger network (Fig. 3C).

C. elegans interaction network. Physical subnetwork generated by String. Stars indicate genes with human orthologs with some evidence of AD involvement (Alzheimer's disease variant portal). Weight of edges indicates confidence score. Genes that did not interact with another gene in the network were removed

Drosophila interaction network. Physical subnetwork of modifier genes generated by String. Stars indicate genes with human orthologs with some evidence of AD involvement (Alzheimer's disease variant portal). Weight of edges indicates confidence score. Genes that did not interact with another gene in the network were removed
Dysregulation of protein folding machinery promotes AD
The misfolding and aggregating of proteins are countered by the action of molecular chaperones that support proteostasis by promoting proper folding or by promoting the destruction of aberrant proteins by the ubiquitin-proteosome or autophagy systems [261]. Although chaperones would generally be expected to play a positive role in protecting against neurodegeneration by promoting proteostasis, chaperones may also have negative effects by inadvertently stabilizing more toxic protein forms at the expence of less toxic ones [258].
Chaperones and co-chaperones were identified as modifiers of toxicity in large scale screens in yeast, Drosophila and C. elegans. Chaperones have been implicated in the response to protein aggregation associated with neurodegenerative diseases [258]. Increased expression of Heat shock proteins (HSPs) is observed in patients with AD and other neurodegenerative diseases [173]. Further, plasma Hsp70 levels are negatively correlated with cognitive performance in the elderly [278].
Many HSPs are protective in mammalian models of AD, with Hsp27, Hsp60, Hsp70, and Hsp90 decreasing protein aggregation and toxicity, or promoting neuroprotection [13, 16, 29, 133, 193, 195, 264]. Consistent with this, expression of the human Hsp70, either cytoplasmically or extracellularly, rescues memory decline in flies expressing Aβ42 [198] and Tau toxicity in C. elegans [209]. In a Drosophila Aβ model, a gain-of-function mutation in the Hsp70 co-chaperone Hsp110 extends lifespan [255]. High-throughput screens have identified a number of chaperones and co-chaperones that play protective roles in Aβ, Tau and other models of neurodegenerative disease [40].
The cochaperone CHIP that marks Hsp70 and Hsp90 substrates for degradation, ubiquitinates Tau for degradation [242]. As previously observed, CHIP interacts physically or mechanistically with a number of other modifiers of neurodegeneration [40]. In Drosophila, the deubiquitinase USP7 reduces Tau ubiquitination and promotes neurodegeneration, countering the activity of CHIP [156]. Knockdown of two E3 ubiquitin ligases that destabilize CHIP, RNF130 and RNF149, decreased neurodegeneration in Drosophila [156]. Knockdown of these genes in mice also reduced pathological Tau species and improved learning and memory in a tauopathy model [156]. Together, these studies suggest a conserved role for CHIP in preventing neurodegeneration.
Although it is likely that chaperones have wide-ranging effects on proteostasis, there is evidence to suggest that some chaperones act directly on Aβ or Tau. Analysis of C. elegans Aβ deposits identified six chaperones that co-immunoprecipitated with Aβ, including three alpha B-crystallin-related small heat shock protein HSP-16 s (HSP-16.1, HSP-16.2, HSP-16.48), two members of the Hsp70 family (C15H9.6 and F26D10.3), and a putative ortholog of a small glutamine-rich tetratricopeptide repeat-containing protein (SGT) (R05F9.10) [87]. Like Hsp70, overexpression of HSP-16.2 suppresses Aβ toxicity in C. elegans [88, 324]. Hsp90 physically interacts with Tau to aid in at least two functions (1) the interaction of Tau with microtubules and (2) the targeting of Tau for proteosomal degradation [150]. However, the unfolding of Tau mediated by Hsp90 also permits the formation of oligomers, which could promote toxicity [317].
In yeast (Ydj1), Drosophila (Droj2) and C. elegans (dnj-7), DNAJ proteins were identified in screens as enhancers of Aβ toxicity [42, 154, 254]. The yeast HSP40 family chaperone Ydj1, and its human ortholog DnaJA1, physically interact with Aβ and increase its accumulation in the mitochondria [254, 288]. This effect may be mediated through the ability of Ydj1 to delay Aβ fibrillization in favour of more toxic oligomers that are more easily transported into the mitochondria [254]. In contrast to its effects in the Aβ model, Droj2 downregulation enhanced Tau phenotype, suggesting different roles in regulating Aβ and Tau toxicity [156]. Although this requires further validation, it may reflect the fact that the effects in the Aβ model are independent of HSP70.
HSPA8/Hsc70 works in complex with a DNAJ protein DNAJB1, and Hsp110 as a disaggregase, which can aid in the clearance of amyloids [15, 72, 338]. Hsc70 and DNAJ proteins can also control the extracellular release of Tau [86]. In Drosophila, Hsc70Cb/Hsp110 and a co-chaperone Csp, were identified in two independent Tau-based screens, one using mutant and one using overexpression of wild-type Tau. In both cases, the expression of these proteins was detrimental [17, 156]. However, because Hsc70 proteins interact with a large number of proteins, it is difficult to identify the targets that are relevant to degeneration.
The role of ribosomal proteins in AD
Ribosome dysfunction is an early event in the development of AD [62]. Both pathological (mutant or wild-type but hyperphosphorylated) and non-pathological Tau can associate with ribosomal proteins (RPs), with a different complement of proteins interacting with each [114, 160, 206]. In vitro studies suggest that, for pathological Tau, this interaction is inhibitory for translation [206].
Translation may also be inhibited through the regulation of ribosomal subunits and translation initiation factors, including eIF2α, eIF3η, and eIF5 whose expression is altered in AD, in some cases early in the disease [126, 159]. Decreased synthesis of ribosomal proteins RPL23, RPLP0, RPL19, and RPS16 is also observed in mouse models of tauopathy [78].
Ribosomal subunits were identified as modifiers of Aβ toxicity in C. elegans and S. cerevisiae. Deletion or knockdown of the orthologs of human RPL8, RPS13, RPS16, and RPS19 suppressed Aβ toxicity in both yeast and worm models. However, decreased toxicity in response to a reduction of ribosomal protein expression is difficult to interpret. While these interactions may be relevant, it is also possible that they act by reducing the expression of the transgenes used to induce toxicity. Deciphering the role of ribosomal proteins therefore presents a challenge with these models.
While there is the potential for a reduced rate of translation to be protective in neurodegeneration, several ribosomal proteins have extra-ribosomal functions that may be relevant to neurodegeneration. Some ribosomal proteins interact with MDM2 preventing its interaction with, and degradation of p53. No MDM2 ortholog has been identified in yeast or C. elegans, suggesting this may not be a function of ribosomal proteins in these organisms. RPL9 knockdown is associated with Id-1/NF-κB signaling inactivation [10]. Enhanced NF-κB activation is observed in AD patients and is believed to contribute to disease pathology [147, 286], suggesting RPL9 reduction may be protective by reducing NF-kB signaling. Similarly, RPL11 inhibits PPARα activity, whose activation is neuroprotective [109, 322]. RPL26 enhances p53 translation [40, 289], whose expression is increased and plays a critical role in AD [37, 143, 207]. Defining how specific ribosomal protein genes regulate AD will prove challenging but may identify new therapeutic targets.
Protein homeostasis is required to prevent neurodegeneration
Impaired protein homeostasis is a characteristic of many neurodegenerative disorders, including AD [45, 329]. The excessive burden of protein misfolding triggers ER stress and activates the unfolded protein response (UPR), a conserved signaling pathway that increases ER folding capacity and inhibits new protein synthesis [128]. In the short term, activating the UPR increases the expression of ER chaperones and helps maintain protein homeostasis. However, prolonged UPR activation can provoke apoptosis [92]. Genome-wide expression analysis revealed that, as in mammalian models, Aβ induces ER stress and activates the UPR in yeast and C. elegans [41, 122].
In yeast, UPR response is mediated by the IRE1α branch of the UPRER that activates the transcription factor XBP1s; it is the only conserved branch in yeast. In a yeast model expressing Aβ42, compounds that inhibit UPR prevent apoptosis and confer a protective effect [58]. In C. elegans, knockdown of xbp-1 reduces Aβ aggregation and delays paralysis in the Aβ42 model [260], suggesting a negative impact of UPR activation in this model. However, in a C. elegans Tau model, loss of xbp-1 function exacerbated Tau toxicity and constitutive activation of XBP-1 promoted the clearance of misfolded Tau and attenuated Tau-related phenotypes [311]. These seemingly disparate findings may be distinguished by their effects on the long-term and short-term consequences of UPR activation or may reflect differences in the model used or the cell type expression of the transgenes.
Overall, UPR activation may either promote or prevent neurodegeneration, depending on the stages of the disease and specific branches of the pathway affected [127]. The UPR effectors PERK and downstream eIF2α are activated in human AD patients, where they co-localize with abnormal Tau protein [304]. Interestingly, two drugs that inhibit eIF2α activation, trazodone and dibenzoylmethane, are neuroprotective in mouse models of dementia [116].
Protein degradation pathways prevent the accumulation of misfolded protein
Proteostasis is maintained by the regulation of protein synthesis and degradation pathways. Misfolded or toxic proteins are prevented from accumulating by the two protein degradation systems, the ubiquitin-proteosome system (UPS) and autophagy [106, 210, 226]. In addition to its role in protein degradation, autophagy is involved in the extracellular release of Aβ and plaque formation [221, 222]. Dysregulation of the ubiquitin–proteasome system is observed in patient samples [226]. In neurodegenerative disorders, dysfunctions of protein degradation pathways have been identified as contributors to neurodegeneration [301, 302]. This has also been demonstrated in C. elegans, Drosophila, and yeast models of AD (Tables S11-S14). The C. elegans AIRAP/AIRAPL homolog AIP-1, a component of the proteasome 19S regulatory cap, plays an essential role in preventing AD phenotypes in worms [123]. Drosophila mir-9a enhances Tau-related phenotypes by repressing the UBE4B ubiquitin ligase that targets Tau for degradation [285]. Overexpression of Atg, the Drosophila ortholog of ULK1, a mediator of autophagy, suppressed Aβ toxicity [30]. It is likely that the C. elegans ortholog of this gene, unc-51, was not identified in RNAi screens designed to identify enhancers of neurodegeneration because RNAi clones that produce an uncoordinated phenotype on their own are generally excluded from consideration.
In many models of neurodegeneration, increasing the activity of protein degradation pathways can reduce neurodegeneration [236, 251]. For example, in Drosophila deficiency of S5b/PSMD5, the 26S proteasome regulatory subunit increases proteasome activity and reduces Tau rough eye phenotype [271]. Another AD-relevant protein, CD2AP, is vital for the UPS in a Drosophila AD model [229]. CD2AP mutation inhibits proteasome activity and synaptic vesicle recycling, which enhances Tau neurotoxicity in flies.
Mitochondrial dysfunction in AD
The central nervous system has high energy demands; although it represents 2% of the body’s weight, it consumes 20% of the total oxygen [149, 276]. This energy is provided by mitochondria, which are essential for ATP and amino acid production and maintaining calcium homeostasis [233, 256]. Mitochondrial dysfunction is a hallmark of AD. Defects in mitochondrial morphology, dynamics, trafficking, and mitophagy occur in AD [250, 283, 314]. Such dysfunction leads to increased ROS, decreased ATP production and altered ion homeostasis [36, 59, 212, 213, 279, 287]. These phenotypes are also observed in AD models, suggesting that the involvement of mitochondrial dysfunction is conserved across species.
Both Aβ and Tau models produce mitochondrial pathologies. Firstly, Aβ accumulates in the mitochondria of AD patients and in Aβ transgenic mouse, yeast, and Drosophila models (Anandatheerthavarada et al. 2003, [34, 43, 134, 194, 234, 288]). As previously discussed, DNAJ proteins have been implicated in the transport of Aβ into the mitochondria. In addition, a recent study in yeast showed that Aβ is specifically recognized by the mitochondrial translocase of outer mitochondrial membrane subunit 22 (TOMM22) and that Aβ transport into the mitochondria depends on the TOM complex [134]. Another component of the TOM complex, TOMM40 is a susceptibility gene for late-onset AD [112]. Overexpression of TOMM22 or TOMM40 increased mitochondrial Aβ in a human cell line and was accompanied by changes in mitochondrial morphology, mitochondrial damage and an increase in autophagosomes and autolysosomes [65].
Mitochondrial dysfunction may also occur when cells are unable to eliminate damaged mitochondria. In C. elegans, expression of disease-associated Tau (P301L) inhibits mitophagy [54]. For neuronal cells, the location of mitochondria is an added consideration. In Drosophila, axonal loss of mitochondria enhances neurodegeneration in a Tau-based model [140]. Moreover, expression of disease-associated mutant forms of Tau enhanced mitochondrial elongation in both Drosophila and mouse models. Increasing mitochondrial fission reduced mitochondrial length and neurotoxicity in Drosophila, suggesting that abnormal mitochondrial dynamics promote neurodegeneration [70].
In high throughput studies, many mitochondria-related genes were identified as modifiers of AD phenotypes. Notably, in C. elegans, RNAi-mediated knockdown of electron transport chain components, including the ATP synthase subunits atp-2 and atp-5 (complex V) and the complex I NADH ubiquinone oxidoreductase nuo-2/NDUFS3, and NADH dehydrogenase nuo-3/NDUFA6, suppressed paralysis in Aβ42 models, although the mechanisms were unclear [154]. One possibility is that partial knockdown generates a mild mitochondrial stress that induces a protective response. In both C. elegans and Drosophila, a slight decrease in the activity of the mitochondrial respiratory chain increases lifespan [49, 83, 117, 176]. A severe reduction of ETC function is lethal in C. elegans [300].
Regulation of oxidative stress in AD
Oxidative damage is an early event in AD development, contributing to toxic oligomer formation and disease development [125, 196, 205, 224, 235, 309]. Oxidative stress causes DNA damage [266], protein oxidation [14], lipid peroxidation [220], contributes to neuronal cell damage, and promotes apoptosis [187, 249].
As in AD, oxidative stress contributes to toxicity and neurodegeneration in model systems [67, 312]. High throughput screens conducted in yeast identified 25 Aβ modifiers involved in oxidative stress response (Tables S9, S10). Similarly, a high throughput genetic modifier screen in a Drosophila model of AD identified targets significantly enriched in oxidative stress-related genes [255]. This study also showed that overexpressing antioxidative genes, specifically genes encoding the iron-binding protein ferritin and H2O2 scavenger catalases suppressed Aβ toxicity. Furthermore, knocking down mitoferrin-1, a mitochondrial iron transporter, reduced ROS and extended lifespan in C. elegans AD models, indicating its critical role in regulating mitochondrial iron metabolism in AD [136]. Interestingly, a mild increase in ROS can be neuroprotective by the formation of glial lipid droplets that transfer peroxidized lipids from neurons to glia, where homologs of AD-risk genes ABCA1, ABCA7, VLDLR, VPS26, VPS35, AP2A, PICALM, and CD2AP are required in Drosophila [214].
The association between oxidative stress and Tau phosphorylation is controversial. Treatment of H2O2 leads to decreased Tau phosphorylation in rat hippocampal and SH-SY5Y human neuroblastoma cells [331], but chronic oxidative stress through inhibition of glutathione synthesis increased Tau phosphorylation in M17 neuroblastoma cells [284]. This increased phosphorylation is proposed to occur as a result of increased activity of JNK and p38 MAPK and decreased activity of PP2A [284]. Vanhelmont and colleagues reported that oxidative stress induces Tau aggregation in yeast but decreases Tau phosphorylation [308]. Similarly, in Drosophila, increased oxidative stress increases neurodegeneration, but not by increasing Tau phosphorylation [60]. Thus, Tau phosphorylation and oxidative stress may work in parallel to promote aggregation.
Cellular trafficking influences Aβ toxicity
Defects in cytoskeletal dynamics, vesicle trafficking and sorting systems are observed in AD [180]. Genes related to cellular trafficking were recovered in yeast and C. elegans screens. In yeast, Aβ expression impairs clathrin-mediated endocytosis [297]. Single nucleotide polymorphisms in phosphatidylinositol binding clathrin assembly protein (PICALM), an adapter protein that functions in clathrin-mediated endocytosis and autophagy, are associated with AD [119, 172, 327]. PICALM has been implicated in the trafficking and processing of APP, the turnover of Aβ, and as a modulator of glutamatergic signaling [294, 325, 331]. Overexpression of PICALM orthologs in C. elegans and Drosophila protect against neuronally expressed Aβ [238, 297, 330]. Surprisingly, PICALM increased Aβ toxicity in a yeast model expressing an ER-targeted Aβ42-GFP fusion protein [55] and in C. elegans, the knockdown of the PICALM ortholog unc-11 suppressed Aβ toxicity when Aβ42 was expressed in the muscle [215]. While these data are taken from large-scale screens that require additional validation, these seemingly contradictory findings may be the result of the many roles of PICALM and further investigation is needed to disentangle these effects.
Defects in ER-Golgi trafficking reduce Aβ toxicity in yeast, whereas mutations in genes involved in cytoskeleton, endocytosis, and the ESCRT machinery which function in vesicular trafficking, increase Aβ toxicity [93]. However, in C. elegans, endocytic gene depletion suppresses necrotic neurodegeneration [299]. In addition, a large-scale RNAi screen in C. elegans identified several genes involved in ER to Golgi trafficking, including copd-1, sly-1, syx-5, sec-12, sec-23, and rab-1, that when knocked down suppress Aβ toxicity [154].
In both C. elegans and Drosophila screens, cytoskeletal proteins and regulators modified outcomes in Tau-based models (Tables S2, S3). These regulators comprise a range of proteins that bind or modify actin or microtubules.
Notably, genes involved in F-actin processing were identified in screens in both organisms. F-actin can associate with Tau [94], which may mechanistically explain how these proteins act as modifiers in Tau-based models. In general, F-actin-associated proteins promoted worse outcomes. This is consistent with the finding in Drosophila that overexpression of actin (Act5C) exacerbates toxicity resulting from overexpression of human Tau [94]. Likewise, knockdown of the C. elegans actins, act-1 and act-5, suppresses paralysis in an Aβ42 model [154].
Lipid metabolism influences neurodegeneration
Dysregulation of glucose and lipid metabolism have been implicated in the development of AD [185, 328]. In C. elegans, the knockdown of four genes involved in fatty acid biosynthesis, elo-4, acs-1, fat-6, and pod-2, suppressed paralysis in an Aβ model. Increased expression of the orthologs of these four genes ELOVL3, ACSF2, SCD5, and ACACA was reported in either mouse models or AD patients [8, 11, 184, 240], suggesting they may have conserved roles in AD. Similarly, decreased expression of the Δ9 desaturases fat-5 or fat-7 rescues neurodegeneration in a C. elegans model of Parkinson’s disease [201]. Together these data suggest that specific lipids may either promote or protect against neurodegeneration. Consistent with an important role for lipid metabolism in AD, Triggering Receptor Expressed on Myeloid Cells 2 (TREM2) and ATP-binding cassette transporter A7 (ABCA7), two genes with risk variants associated with AD function are required for lipid homeostasis [148, 282].
Cell signaling in AD
Many conserved cell signaling pathways can influence AD development [103, 129] including Wnt, MAPK, and TOR pathways [227, 313, 338].
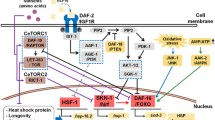
Wnt signaling pathway components were identified in both C. elegans and Drosophila screens [165, 275]. GSK-3β phosphorylates Tau at several disease-relevant sites [118, 189, 191], but also antagonizes the Wnt pathway, both functions may be relevant to neurodegeneration. In C. elegans, knocking down gsk-3 enhances Tau-related neurodegeneration. In Drosophila, downstream mediators of the Wnt pathway have also been identified as modifiers. Overexpression of the Drosophila β-catenin exacerbates neurodegeneration in a Tau model [142].
Gain-of-function mutations in the fly TAOK1 homolog enhanced disease phenotype in a Drosophila model of tauopathy [274]. The thousand-and-one kinases (TAOKs) belong to the MAP3K family. TAOKs have many targets, including proteins in the p38 and Hippo pathways [81]. Furthermore, a TAOK inhibitor reduces Tau phosphorylation in mice and induced pluripotent stem cell-derived neurons from frontotemporal lobar degeneration (FTLD) patients [99].
Tau is heavily phosphorylated in AD, and these modifications are believed to contribute to the disease. Many kinases that phosphorylate Tau have been identified, and many are conserved in C. elegans and Drosophila. Further, when human Tau is expressed in Drosophila or C. elegans, it is phosphorylated at disease-relevant sites [165, 248]. While there is limited overlap at the single gene level, orthologs of kinases that phosphorylate human hTau were identified as modifiers in both C. elegans (TTBK, TAOK, GSK-3) and Drosophila (CaMKII, MARK, TAOK, GSK-3) (Fig. 6, Supplementary Tables 3, 4). The ability of these proteins to phosphorylate Tau may be conserved across organisms. Consistent with this idea, many phosphorylation sites, including SKXGS sites and several proline-directed serines, are conserved in Drosophila and C. elegans Tau proteins. Nevertheless, there are likely species-specific ways in which Tau is modified and regulated. Intriguingly, in Drosophila, the activity of these genes correlates with increased phosphorylation having a negative impact. By contrast, In C. elegans, knockdown of the orthologs of GSK-3β, TAOK and TTBK enhanced toxicity, contrary to what is expected based on their abilities to phosphorylate Tau.

Tau related kinases recovered in C. elegans and Drosophila AD modifier screens. Fill colour indicates species, line colour indicates effect on neurodegeneration. Nodes are connected by phosphorylation or signaling events. Nodes with 2-colour outlines indicate genes where findings between studies are incongruent
Downregulation of the focal adhesion kinase (Fak), the PTK2B homolog, suppressed Tau toxicity in a fly model [66]. PTK2B co-localized with hyperphosphorylated Tau in AD patient brain samples, suggesting that it may directly phosphorylate Tau. PTK2B was also implicated in Aβ regulation, but the mechanisms involved remain elusive, as two different transgenic mouse models found seemingly opposing roles for the protein, with PTK2B deletion or overexpression were both shown to be protective [101, 263].
Challenges and limits to the use of model systems in Alzheimer’s research
While simple model organisms provide many advantages in discovery research, differences in the biology of humans and model organisms can provide a challenge in modelling certain aspects of the disease. For example, neuropsychiatric symptoms in AD cannot be modelled in simple organisms. Specific anatomical and biological differences between humans and model organisms may also limit the study of some aspects of the disease. Drosophila and C. elegans do not accumulate neurofibrillary tangles [320, 321]. Further, microglia play an important role in the progression of AD, contributing to phagocytosis and inflammation [208] but C. elegans and Drosophila do not have an obvious equivalent cell type. Although it is possible that some roles of the microglia are filled by other cells, some genes that play important roles in AD are highly expressed in microglia. Many proteins believed to influence AD through their activity in the microglia, including TREM2, CLU and CD33, do not have orthologs in Drosophila or C. elegans.
Differences in the biology of humans and model organisms can be an advantage and a disadvantage. In the case of neurofibrillary tangles, the observation that neurodegeneration occurs in the absence of neurofibrillary tangle formation was an important demonstration that toxic forms of Tau precede neurofibrillary tangle formation [320]. Similarly, the absence of an adaptive immune system facilitates the examination of processes independent of inflammation.
The use of overexpression models may be a limitation in that expression is generally much higher than what would be observed in AD. Also, the ability of transgenes to promote rapid degeneration may not model all aspects of a disease that progresses more slowly. These effects may bias which genes are recovered as modifiers. Differences in levels of expression of different transgenes could explain the limited overlap between screens where the same protein is being expressed. The timing and location of gene expression could also explain differences between screens. Expression of these transgenes during development could induce developmental effects that predispose animals to more rapid aging or decreased stress response. Moreover, simple overexpression models cannot capture subtleties in the production of Aβ or Tau. Aβ overexpression models typically use the Aβ42 peptide alone and therefore do not integrate the regulation of APP cleavage or the potential influence of other forms of Aβ. Similarly, Tau-based models typically overexpress one of the six Tau isoforms normally found in the human brain. The specific isoform chosen, as well as the absence of multiple isoforms, may affect which modifiers are isolated in a given screen.
The design of individual models and screens affects which genes are recovered and which are not. An advantage of Drosophila and yeast is the ability to easily perform both gain-of-function and loss-of-function screens, whereas resources for large-scale gain-of-function screens in C. elegans are lacking. Modifiers identified by overexpression in Drosophila may not be identified in C. elegans because their impact is not obvious in knockdown experiments. For amyloid and Tau-based screens, the cell type chosen for expression may also impact which genes are identified as modifiers. In C. elegans, early screens used a muscle-expressed Aβ, Tau models on the other hand, used neuronal expression.
Species-specific gene function may explain why some gene modifiers do not translate directly to human biology and genes that act as modifiers in humans may not function as such in model organisms. There are several reasons a human risk gene might not be uncovered as a modifier in a model organism. Large-scale screens in these organisms often rely on knockdown or knockout approaches, while disease-associated alleles may produce more subtle effects, for example, an amino acid substitution that produces a specific functional alteration. Species-specific expansions of gene families may create redundancy that masks the function of individual genes. Functions specific to either humans or the model system may also explain the limited overlap in screens. In some cases, an ortholog to a human disease gene may not exist, for example, APOE, the strongest risk factor for late-onset AD [183], does not have a direct ortholog in Drosophila or C. elegans. In other cases, the functional ortholog may not have been identified. In human disease-associated genes identified in GWAS studies and genes identified in genetic screens, there is overlap in protein families that is not captured when analyzing on a gene by gene basis. For example, DNAJ family proteins have been identified in GWAS studies and as modifiers in all three organisms examined, but they do not overlap on a gene-by-gene basis using predicted orthologs.
Summary
The study of human AD samples has provided a wealth of information; however, it remains a challenge to decipher cause from consequence using only these samples. Model organisms allow rapid hypothesis testing and unbiased genetic screening that contribute to the discovery of AD-related cellular processes and signaling pathways. Integration of data across different models can be a powerful approach to understanding the biology of neurodegeneration.
AD modifiers identified from high throughput screens and targeted studies can be classified into functional categories relevant to neurodegeneration. Major pathways involving Aβ modifiers identified in all three model systems examined include transcription and translation-related processes, stress response and chaperones, and protein trafficking. When comparing Tau modifiers found in C. elegans and Drosophila, transcription and translation-related processes, stress response and chaperones, cytoskeleton-related pathways, and metabolism are shown to play critical roles in regulating abnormal Tau expression. These evolutionarily conserved pathways reveal fundamental mechanisms of AD and other neurodegenerative disorders.
Caution should be taken in interpreting negative findings from large-scale screens. These screens are generally designed for ease of screening and can be biased toward dramatic effects, while missing more subtle ones. In addition, whether a gene was effectively interrogated in a given screen depends on whether it is present in a deletion set or RNAi library, whether loss of function is lethal or produces a phenotype that excludes it from consideration, and whether it is expressed in the cell type and at the age examined. For example, in C. elegans, knockdowns that produce a movement defect, or incoordination in a wild-type background, are generally excluded from analysis when a screen measures the enhancement of movement defects. Additionally, the presence of paralogs or other redundantly functioning genes can hide the involvement of some genes in neurodegeneration. Species-specific gene duplications could therefore result in a gene being recovered in a screen in one organism, but not in another.
Caution should also be exercised in interpreting positive results from large-scale screens without additional validation. While we have highlighted some findings from these screens, many require additional validation, including the analysis of mutants and more direct analysis of neurons, rather than a phenotypic proxy. Determining whether effects observed are cell autonomous or non-cell autonomous can also clarify the role of a specific modifier in neurodegeneration. While the ability to perform screens in whole animals is an advantage, it is important to consider that modifiers recovered may not function cell-autonomously.
Some genetic modifiers identified in model organisms do not have obvious human orthologs and their functions remain unknown. These genes may represent species-specific signaling or these genes may have human orthologs that cannot be identified on the basis of sequence homology. Nevertheless, their activities may be related to processes that also influence neurodegeneration in humans.
The use of simple model systems to study AD and related tauopathies has revealed important cellular mechanisms of neurodegeneration and provides powerful tools for discovering therapeutic targets and strategies to combat these diseases. In combination with cell lines, animal models, and clinical studies, simple model organisms can provide insights into disease mechanisms and aid in the development of effective treatments for AD and other neurodegenerative disorders. In fact, studies that combine different models can be very powerful (Kim et al., 2019). These studies leverage the conservation of processes between animals to identify robust modifiers.
References
Abraham R, Moskvina V, Sims R, Hollingworth P, Morgan A, Georgieva L, et al. A genome-wide association study for late-onset Alzheimer’s disease using DNA pooling. BMC Med Genomics. 2008;1:44.
Alexander AG, Marfil V, Li C. Use of Caenorhabditis elegans as a model to study Alzheimer’s disease and other neurodegenerative diseases. Front Genet. 2014;5:279.
Alonso A, Zaidi T, Novak M, Grundke-Iqbal I, Iqbal K. Hyperphosphorylation Induces Self-Assembly of Tau into Tangles of Paired Helical Filaments/Straight Filaments. Proc Natl Acad Sci USA. 2001;98(12):6923–8.
Alvarez J, Alvarez-Illera P, Santo-Domingo J, Fonteriz RI, Montero M. Modeling Alzheimer’s Disease in Caenorhabditis elegans. Biomedicines. 2022; 10(2).
Annunziata I, Patterson A, Helton D, Hu H, Moshiach S, et al. Lysosomal NEU1 deficiency affects amyloid precursor protein levels and amyloid-β secretion via deregulated lysosomal exocytosis. Nat Commun. 2013;4:2734.
Antonova Simona V, et al. Chaperonin CCT checkpoint function in basal transcription factor TFIID assembly. Nat Struct Mol Biol. 2018;25(12):1119–27.
Arber C, Toombs J, Lovejoy C, Ryan NS, Paterson RW, Willumsen N, Gkanatsiou E, et al. Familial Alzheimer’s disease patient-derived neurons reveal distinct mutation-specific effects on amyloid beta. Mol Psychiatry. 2020;25(11):2919–31.
Astarita G, Jung K-M, Vasilevko V, Dipatrizio NV, Martin SK, et al. Elevated stearoyl-CoA desaturase in brains of patients with Alzheimer’s disease. PLoS One. 2011;6(10):e24777.
Bagriantsev S, Liebman S. Modulation of Abeta42 low-n oligomerization using a novel yeast reporter system. BMC Biol. 2006;4:32.
Baik IH, Jo G-H, Seo D, Ko MJ, Cho CH, et al. Knockdown of RPL9 expression inhibits colorectal carcinoma growth via the inactivation of Id-1/NF-κB signaling axis. Int J Oncol. 2016;49(5):1953–62.
Bao W-D, Pang P, Zhou X-T, Hu F, **ong W, et al. Loss of ferroportin induces memory impairment by promoting ferroptosis in Alzheimer’s disease. Cell Death Differ. 2021;28(5):1548–62.
Bardai FH, Ordonez DG, Bailey RM, Hamm M, Lewis J, Feany MB. Lrrk promotes tau neurotoxicity through dysregulation of actin and mitochondrial dynamics. PLoS Biol. 2018;16(12):e2006265.
Baughman HER, Clouser AF, Klevit RE, Nath A. HspB1 and Hsc70 chaperones engage distinct tau species and have different inhibitory effects on amyloid formation. J Biol Chem. 2018;293(8):2687–700.
Berlett BS, Stadtman ER. Protein oxidation in aging, disease, and oxidative stress. J Biol Chem. 1997;272(33):20313–6.
Beton JG, Monistrol J, Wentink A, Johnston EC, Roberts AJ, Bukau BG, Hoogenboom BW, Saibil HR. Cooperative amyloid fibre binding and disassembly by the Hsp70 disaggregase. EMBO J. 2022;41(16):e110410.
Björkdahl C, Sjögren MJ, Zhou X, Concha H, Avila J, et al. Small heat shock proteins Hsp27 or alphaB-crystallin and the protein components of neurofibrillary tangles: tau and neurofilaments. J Neurosci Res. 2008;86(6):1343–52.
Blard O, Feuillette S, Bou J, Chaumette B, Frébourg T, et al. Cytoskeleton proteins are modulators of mutant tau-induced neurodegeneration in Drosophila. Hum Mol Genet. 2007;16(5):555–66.
Bloom GS. Amyloid-β and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 2014;71(4):505–8.
Bonet-Costa V, Pomatto LC-D, Davies KJA. The proteasome and oxidative stress in alzheimer’s disease. Antioxid Redox Signal. 2016;25(16):886–901.
Boyle EI, Weng S, Gollub J, ** H, Botstein D, et al. GO::TermFinder–open source software for accessing Gene Ontology information and finding significantly enriched Gene Ontology terms associated with a list of genes. Bioinformatics. 2004;20(18):3710–5.
Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K. Staging of Alzheimer Disease-Associated Neurofibrillary Pathology Using Paraffin Sections and Immunocytochemistry. Acta Neuropathol. 2006;112(4):389–404.
Breijyeh Z, Karaman R. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules (Basel, Switzerland). 2020;25(24)5789. https://doi.org/10.3390/molecules25245789.
Brandt R, Gergou A, Wacker I, Fath T, Hutter H. A Caenorhabditis elegans model of tau hyperphosphorylation: induction of developmental defects by transgenic overexpression of Alzheimer’s disease-like modified tau. Neurobiol Aging. 2009;30(1):22–33.
Burdick D, Soreghan B, Kwon M, Kosmoski J, Knauer M, Henschen A, Yates J, Cotman C, Glabe C. Assembly and Aggregation Properties of Synthetic Alzheimer’s A4/Beta Amyloid Peptide Analogs. J Biol Chem. 1992;267(1):546–54.
Burnouf S, Grönke S, Augustin H, Dols J, Gorsky MK, Werner J, Kerr F, Alic N, Martinez P, Partridge L. Deletion of endogenous Tau proteins is not detrimental in Drosophila. Sci Rep. 2016;6:23102.
Cacho-Valadez B, Muñoz-Lobato F, Pedrajas JR, Cabello J, Fierro-González JC, et al. The characterization of the Caenorhabditis elegans mitochondrial thioredoxin system uncovers an unexpected protective role of thioredoxin reductase 2 in β-amyloid peptide toxicity. Antioxid Redox Signal. 2012;16(12):1384–400.
Caine J, Sankovich S, Antony H, Waddington L, Macreadie P, et al. Alzheimer’s Abeta fused to green fluorescent protein induces growth stress and a heat shock response. FEMS Yeast Res. 2007;7(8):1230–6.
Caldeira GL, Ferreira IL, Rego AC. Impaired transcription in Alzheimer’s disease: key role in mitochondrial dysfunction and oxidative stress. J Alzheimers Dis. 2013;34(1):115–31.
Campanella C, Pace A, Caruso Bavisotto C, Marzullo P, Marino Gammazza A, et al. Heat shock proteins in alzheimer’s disease: role and targeting. Int J Mol Sci. 2018;19(9):2603. https://doi.org/10.3390/ijms19092603.
Cao W, Song H-J, Gangi T, Kelkar A, Antani I, et al. Identification of novel genes that modify phenotypes induced by Alzheimer’s beta-amyloid overexpression in Drosophila. Genetics. 2008;178(3):1457–71.
Cao W, Zheng H. Peripheral immune system in aging and Alzheimer’s disease. Mol Neurodegener. 2018;13(1):51.
Carmine-Simmen K, Proctor T, Tschäpe J, Poeck B, Triphan T, et al. Neurotoxic effects induced by the Drosophila amyloid-beta peptide suggest a conserved toxic function. Neurobiol Dis. 2009;33(2):274–81.
Casas-Tinto S, Zhang Y, Sanchez-Garcia J, Gomez-Velazquez M, Rincon-Limas DE, Fernandez-Funez P. The ER stress factor XBP1s prevents amyloid-beta neurotoxicity. Hum Mol Genet. 2011;20(11):2144–60.
Caspersen C, Wang N, Yao J, Sosunov A, Chen X, et al. Mitochondrial Abeta: a potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. FASEB J. 2005;19(14):2040–1.
Cassar M, Kretzschmar D. Analysis of amyloid precursor protein function in drosophila melanogaster. Front Mol Neurosci. 2016;9:61.
Cha M-Y, Cho HJ, Kim C, Jung YO, Kang MJ, et al. Mitochondrial ATP synthase activity is impaired by suppressed O-GlcNAcylation in Alzheimer’s disease. Hum Mol Genet. 2015;24(22):6492–504.
Chang JR, Ghafouri M, Mukerjee R, Bagashev A, Chabrashvili T, Sawaya BE. Role of p53 in neurodegenerative diseases. Neurodegener Dis. 2012;9(2):68–80.
Chanu SI, Sarkar S. Targeted downregulation of dMyc suppresses pathogenesis of human neuronal tauopathies in drosophila by limiting heterochromatin relaxation and tau hyperphosphorylation. Mol Neurobiol. 2017;54(4):2706–19.
Chatterjee S, Ambegaokar SS, Jackson GR, Mudher A. Insulin-mediated changes in tau hyperphosphorylation and autophagy in a drosophila model of tauopathy and neuroblastoma cells. Front Neurosci. 2019;13:801.
Chen J, Guo K, Kastan MB. Interactions of nucleolin and ribosomal protein L26 (RPL26) in translational control of human p53 mRNA. J Biol Chem. 2012;287(20):16467–76.
Chen X, Bisschops MMM, Agarwal NR, Ji B, Shanmugavel KP, Petranovic D. Interplay of energetics and ER stress exacerbates Alzheimer’s Amyloid-β (Aβ) toxicity in yeast. Front Mol Neurosci. 2017;10:232.
Chen X, Ji B, Hao X, Li X, Eisele F, et al. FMN reduces Amyloid-β toxicity in yeast by regulating redox status and cellular metabolism. Nat Commun. 2020;11(1):867.
Chen X, Petranovic D. Amyloid-β peptide-induced cytotoxicity and mitochondrial dysfunction in yeast. FEMS Yeast Res. 2015;15(6):fov61.
Chen X-F, Zhang Y, Xu H, Bu G. Transcriptional regulation and its misregulation in Alzheimer’s disease. Mol Brain. 2013;6(1):1–9.
Cheng J, North BJ, Zhang T, Dai X, Tao K, et al. The emerging roles of protein homeostasis-governing pathways in Alzheimer’s disease. Aging Cell. 2018;17(5):e12801.
Chew YL, Fan X, Götz J, Nicholas HR. PTL-1 regulates neuronal integrity and lifespan in C. elegans. J Cell Sci. 2013;126(Pt 9):2079–91.
Chikka MR, Anbalagan C, Dvorak K, Dombeck K, Prahlad V. The Mitochondria-Regulated Immune Pathway Activated in the C. elegans Intestine Is Neuroprotective. Cell Rep. 2016;16(9):2399–414.
Conte D, MacNeil LT, Walhout AJM, Mello CC. RNA Interference in Caenorhabditis elegans. Curr Protoc Mol Biol. 2015;109:26.3.1-26.3.30.
Copeland JM, Cho J, Lo T Jr, Hur JH, Bahadorani S, Arabyan T, Rabie J, Soh J, Walker DW. Extension of Drosophila life span by RNAi of the mitochondrial respiratory chain. Curr Biol. 2009;19(19):1591–8.
Costanzo M, Baryshnikova A, Bellay J, Kim Y, Spear ED, Sevier CS, et al. The genetic landscape of a cell. Science. 2010;327:425–31.
Crowther DC, Kinghorn KJ, Miranda E, Page R, Curry JA, et al. Intraneuronal Abeta, non-amyloid aggregates and neurodegeneration in a Drosophila model of Alzheimer’s disease. Neuroscience. 2005;132(1):123–35.
Cui YH, Le Y, Zhang X, Gong W, Abe K, et al. Up-regulation of FPR2, a chemotactic receptor for amyloid beta 1–42 (A beta 42), in murine microglial cells by TNF alpha. Neurobiol Dis. 2002;10(3):366–77.
Culetto E, Sattelle DB. A role for Caenorhabditis elegans in understanding the function and interactions of human disease genes. Hum Mol Genet. 2000;9:869–77.
Cummins, N., Tweedie, A., Zuryn, S., Bertran-Gonzalez, J., & Götz, J. Disease-associated tau impairs mitophagy by inhibiting Parkin translocation to mitochondria. EMBO J. 2019; 38(3). https://doi.org/10.15252/embj.201899360.
D’Angelo F, Vignaud H, Di Martino J, Salin B, Devin A, et al. A yeast model for amyloid-β aggregation exemplifies the role of membrane trafficking and PICALM in cytotoxicity. Dis Model Mech. 2013;6(1):206–16.
De Mena L, Chhangani D, Fernandez-Funez P, Rincon-Limas DE. secHsp70 as a tool to approach amyloid-β42 and other extracellular amyloids. Fly (Austin). 2017;11(3):179–84.
De Vos A, Bynens T, Rosseels J, Coun C, Ring J, et al. The peptidyl prolyl cis/trans isomerase Pin1/Ess1 inhibits phosphorylation and toxicity of tau in a yeast model for Alzheimer’s disease. AIMS Mol Sci. 2015;2(2):144–60.
Derf A, Mudududdla R, Bharate SB, Chaudhuri B. Inhibitors of Aβ42-induced endoplasmic reticular unfolded protein response (UPRER), in yeast, also rescue yeast cells from Aβ42-mediated apoptosis. Eur J Pharm Sci. 2019;128:118–27.
Devi L, Prabhu BM, Galati DF, Avadhani NG, Anandatheerthavarada HK. Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. J Neurosci. 2006;26(35):9057–68.
Dias-Santagata D, Fulga TA, Duttaroy A, Feany MB. Oxidative stress mediates tau-induced neurodegeneration in Drosophila. J Clin Invest. 2007;117(1):236–45.
Dickson JR, Yoon H, Frosch MP, Hyman BT. Cytoplasmic mislocalization of RNA polymerase II subunit RPB1 in alzheimer disease is linked to pathologic tau. J Neuropathol Exp Neurol. 2021;80(6):530–40.
Ding Q, Markesbery WR, Chen Q, Li F, Keller JN. Ribosome dysfunction is an early event in Alzheimer’s disease. J Neurosci. 2005;25(40):9171–5.
Dosanjh LE, Brown MK, Rao G, Link CD, Luo Y. Behavioral phenoty** of a transgenic Caenorhabditis elegans expressing neuronal amyloid-beta. J Alzheimers Dis. 2010;19(2):681–90.
Dostal V, Link CD. Assaying β-amyloid toxicity using a transgenic C. elegans model. J Vis Exp. 2010;9(44):e2252.
Dou Y, Tan Y. Presequence protease reverses mitochondria-specific amyloid-β-induced mitophagy to protect mitochondria. FASEB J. 2023;37:e22890.
Dourlen P, Fernandez-Gomez FJ, Dupont C, Grenier-Boley B, Bellenguez C, et al. Functional screening of Alzheimer risk loci identifies PTK2B as an in vivo modulator and early marker of Tau pathology. Mol Psychiatry. 2017;22(6):874–83.
Drake J, Link CD, Butterfield DA. Oxidative stress precedes fibrillar deposition of Alzheimer’s disease amyloid beta-peptide (1–42) in a transgenic Caenorhabditis elegans model. Neurobiol Aging. 2003;24(3):415–20.
Drewes G, Trinczek B, Illenberger S, Biernat J, Schmitt-Ulms G, et al. Microtubule-associated protein/microtubule affinity-regulating kinase (p110mark). A novel protein kinase that regulates tau-microtubule interactions and dynamic instability by phosphorylation at the Alzheimer-specific site serine 262. J Biol Chem. 1995;270(13):7679–88.
Drummond E, Wisniewski T. Alzheimer’s disease: experimental models and reality. Acta Neuropathol. 2017;133:155–75.
DuBoff B, Götz J, Feany MB. Tau promotes neurodegeneration via DRP1 mislocalization in vivo. Neuron. 2012;75:618–32.
Dubey H, Gulati K, Ray A. Recent studies on cellular and molecular mechanisms in Alzheimer’s disease: focus on epigenetic factors and histone deacetylase. Rev Neurosci. 2018;29(3):241–60.
Duennwald ML, Echeverria A, Shorter J. Small heat shock proteins potentiate amyloid dissolution by protein disaggregases from yeast and humans. PLoS Biol. 2012;10(6):e1001346.
Dugan JM, deWit C, McConlogue L, Maltese WA. The Ras-related GTP-binding protein, Rab1B, regulates early steps in exocytic transport and processing of beta-amyloid precursor protein. J Biol Chem. 1995;270(18):10982–9.
EL Zhou T, Koh S, Chuang M, Sharma R, et al. An antimicrobial peptide and its neuronal receptor regulate dendrite degeneration in aging and infection. Neuron. 2018;97(1):125-138.e5.
Eid R, Sheibani S, Gharib N, Lapointe JF, Horowitz A, Vali H, Mandato CA, Greenwood MT. Human ribosomal protein L9 is a Bax suppressor that promotes cell survival in yeast. FEMS Yeast Res. 2014;14(3):495–507.
Emmons SW, Yemini E, Zimmer M. Methods for analyzing neuronal structure and activity in Caenorhabditis elegans. Genetics. 2021;218(4):iyab072.
Esposito M, Sherr GL. Epigenetic modifications in alzheimer’s neuropathology and therapeutics. Front Neurosci. 2019;13:476.
Evans HT, Benetatos J, van Roijen M, Bodea L-G, Götz J. Decreased synthesis of ribosomal proteins in tauopathy revealed by non-canonical amino acid labelling. EMBO J. 2019;38(13):e101174.
Evans HT, Taylor D, Kneynsberg A, Bodea L-G, Götz J. Altered ribosomal function and protein synthesis caused by tau. Acta Neuropathol Commun. 2021;9(1):110.
Ewald CY, Raps DA, Li C. APL-1, the Alzheimer’s Amyloid precursor protein in Caenorhabditis elegans, modulates multiple metabolic pathways throughout development. Genetics. 2012;191(2):493–507.
Fang C-Y, Lai T-C, Hsiao M, Chang Y-C. The diverse roles of TAO kinases in health and diseases. Int J Mol Sci. 2020;21(20):7463.
Fay DS, Fluet A, Johnson CJ, Link CD. In vivo aggregation of beta-amyloid peptide variants. J Neurochem. 1998;71(4):1616–25.
Feng J, Bussière F, Hekimi S. Mitochondrial electron transport is a key determinant of life span in Caenorhabditis elegans. Dev Cell. 2001;1(5):633–44.
Finelli A, Kelkar A, Song H-J, Yang H, Konsolaki M. A model for studying Alzheimer’s Abeta42-induced toxicity in Drosophila melanogaster. Mol Cell Neurosci. 2004;26(3):365–75.
Florez-McClure ML, Hohsfield LA, Fonte G, Bealor MT, Link CD. Decreased insulin-receptor signaling promotes the autophagic degradation of beta-amyloid peptide in C. elegans. Autophagy. 2007;3(6):569–80.
Fontaine SN, Zheng D, Sabbagh JJ, Martin MD, Chaput D, Darling A, Trotter JH, Stothert AR, Nordhues BA, Lussier A, Baker J, Shelton L, Kahn M, Blair LJ, Stevens SM Jr, Dickey CA. DnaJ/Hsc70 chaperone complexes control the extracellular release of neurodegenerative-associated proteins. EMBO J. 2016;35(14):1537–49.
Fonte V, Kapulkin WJ, Taft A, Fluet A, Friedman D, Link CD. Interaction of intracellular beta amyloid peptide with chaperone proteins. Proc Natl Acad Sci USA. 2002;99(14):9439–44.
Fonte V, Kipp DR, Yerg J, Merin D, Forrestal M, et al. Suppression of in vivo beta-amyloid peptide toxicity by overexpression of the HSP-16.2 small chaperone protein. J Biol Chem. 2008;283(2):784–91.
Fortini ME, Skupski MP, Boguski MS, Hariharan IK. A survey of human disease gene counterparts in the Drosophila genome. J Cell Biol. 2000;150:F23–30.
França MB, Lima KC, Eleutherio ECA. Oxidative stress and amyloid toxicity: insights from yeast. J Cell Biochem. 2017;118(6):1442–52.
Freeman MR. Drosophila central nervous system glia. Cold Spring Harb Perspect Biol. 2015;7(11):a020552.
Fribley A, Zhang K, Kaufman RJ. Regulation of apoptosis by the unfolded protein response. Methods Mol Biol. 2009;559:191–204.
Fruhmann G, Marchal C, Vignaud H, Verduyckt M, Talarek N, et al. The Impact of ESCRT on Aβ1-42 Induced Membrane Lesions in a Yeast Model for Alzheimer’s Disease. Front Mol Neurosci. 2018;11:406.
Fulga TA, Elson-Schwab I, Khurana V, Steinhilb ML, Spires TL, et al. Abnormal bundling and accumulation of F-actin mediates tau-induced neuronal degeneration in vivo. Nat Cell Biol. 2007;9(2):139–48.
Gagliano SA, Pouget JG, Hardy J, Knight J, Barnes MR, et al. Genomics implicates adaptive and innate immunity in Alzheimer’s and Parkinson’s diseases. Ann Clin Transl Neurol. 2016;3(12):924–33.
Garcia-Esparcia P, Sideris-Lampretsas G, Hernandez-Ortega K, Grau-Rivera O, Sklaviadis T, et al. Altered mechanisms of protein synthesis in frontal cortex in Alzheimer disease and a mouse model. Am J Neurodegener Dis. 2017;6(2):15–25.
Garrido-Maraver J, Loh SHY, Martins LM. Forcing contacts between mitochondria and the endoplasmic reticulum extends lifespan in a Drosophila model of Alzheimer’s disease. Biol Open. 2020;9(1):bio047530.
GBD 2016 Neurology Collaborators. Global, regional, and national burden of neurological disorders, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019;18(5):459–80.
Giacomini C, Koo C-Y, Yankova N, Tavares IA, Wray S, et al. A new TAO kinase inhibitor reduces tau phosphorylation at sites associated with neurodegeneration in human tauopathies. Acta Neuropathol Commun. 2018;6(1):37.
Giaever G, Chu AM, Ni L, Connelly C, Riles L, et al. Functional profiling of the Saccharomyces cerevisiae genome. Nature. 2002;418(6896):387–91.
Giralt A, de Pins B, Cifuentes-Díaz C, López-Molina L, Farah AT, et al. PTK2B/Pyk2 overexpression improves a mouse model of Alzheimer’s disease. Exp Neurol. 2018;307:62–73.
Godini R, Pocock R, Fallahi H. Caenorhabditis elegans hub genes that respond to amyloid beta are homologs of genes involved in human Alzheimer’s disease. PLoS One. 2019;14(7):e0219486.
Godoy JA, Rios JA, Zolezzi JM, Braidy N, Inestrosa NC. Signaling pathway cross talk in Alzheimer’s disease. Cell Commun Signal. 2014;12:23.
Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA. Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron. 1989;3:519–26.
Goffeau A, Barrell BG, Bussey H, Davis RW, Dujon B, et al. Life with 6000 genes. Science. 1996;274(5287):546, 563–7.
Goldberg AL. Protein degradation and protection against misfolded or damaged proteins. Nature. 2003;426(6968):895–9.
Götz J, Gladbach A, Pennanen L, van Eersel J, Schild A, David D, Ittner LM. Animal models reveal role for tau phosphorylation in human disease. Biochem Biophys Acta. 2010;1802:860–71.
Götz JJ, Götz J. Experimental Models of Tauopathy - From Mechanisms to Therapies. Adv Exp Med Biol. 2019;1184:381–91.
Gray JP, Davis JW, Gopinathan L, Leas TL, Nugent CA, Vanden Heuvel JP. The ribosomal protein rpL11 associates with and inhibits the transcriptional activity of peroxisome proliferator-activated receptor-alpha. Toxicol Sci. 2006;89(2):535–46.
Greeve I, Kretzschmar D, Tschäpe J-A, Beyn A, Brellinger C, et al. Age-dependent neurodegeneration and Alzheimer-amyloid plaque formation in transgenic Drosophila. J Neurosci. 2004;24(16):3899–906.
Grune T, Jung T, Merker K, Davies KJA. Decreased proteolysis caused by protein aggregates, inclusion bodies, plaques, lipofuscin, ceroid, and “aggresomes” during oxidative stress, aging, and disease. Int J Biochem Cell Biol. 2004;36(12):2519–30.
Grupe A, Williams J. Evidence for novel susceptibility genes for late-onset Alzheimer’s disease from a genome-wide association study of putative functional variants. Hum Mol Gen. 2007;16(8):865–73.
Gu Q, Cuevas E, Raymick J, Kanungo J, Sarkar S. Downregulation of 14-3-3 Proteins in Alzheimer’s Disease. Mol Neurobiol. 2020;57(1):32–40.
Gunawardana CG, Mehrabian M, Wang X, Mueller I, Lubambo IB, Jonkman JEN, Wang H, Schmitt-Ulms G. The Human Tau Interactome: Binding to the Ribonucleoproteome, and Impaired Binding of the Proline-to-Leucine Mutant at Position 301 (P301L) to Chaperones and the Proteasome. Mol Cell Proteomics. 2015;14(11):3000–14.
Guthrie CR, Schellenberg GD, Kraemer BC. SUT-2 potentiates tau-induced neurotoxicity in Caenorhabditis elegans. Hum Mol Genet. 2009;18(10):1825–38.
Halliday M, Radford H, Zents KAM, Molloy C, Moreno JA, et al. Repurposed drugs targeting eIF2α-P-mediated translational repression prevent neurodegeneration in mice. Brain. 2017;140(6):1768–83.
Hamilton B, Dong Y, Shindo M, Liu W, Odell I, Ruvkun G, et al. A systematic RNAi screen for longevity genes in C. elegans. Genes Dev. 2005;19:1544–55.
Hanger DP, Hughes K, Woodgett JR, Brion JP, Anderton BH. Glycogen synthase kinase-3 induces Alzheimer’s disease-like phosphorylation of tau: generation of paired helical filament epitopes and neuronal localisation of the kinase. Neurosci Lett. 1992;147:58–62.
Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, Pahwa JS, Moskvina V, Dowzell K, Williams A, Jones N, Thomas C, Stretton A, Morgan AR, Lovestone S, Powell J, Proitsi P, Lupton MK, Brayne C, Williams J. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet. 2009;41(10):1088–93.
Harris MA, Clark J, Ireland A, Lomax J, Ashburner M, et al. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004;32(Database issue):D258-61.
Hassan A, Scott H, Hill M. Regulation of microglial transcription factor MEF2C by Alzheimer’s disease‐relevant stimuli. Alzheimers Dement. 2021;17(S3):05748.
Hassan WM, Dostal V, Huemann BN, Yerg JE, Link CD. Identifying Aβ-specific pathogenic mechanisms using a nematode model of Alzheimer’s disease. Neurobiol Aging. 2015;36(2):857–66.
Hassan WM, Merin DA, Fonte V, Link CD. AIP-1 ameliorates beta-amyloid peptide toxicity in a Caenorhabditis elegans Alzheimer’s disease model. Hum Mol Genet. 2009;18(15):2739–47.
Hefti MM, Farrell K, Kim S, Bowles KR, Fowkes ME, Raj T, Crary JF. High-resolution temporal and regional map** of MAPT expression and splicing in human brain development. PLoS One. 2018;13:e0195771.
Hensley K, Hall N, Subramaniam R, Cole P, Harris M, et al. Brain regional correspondence between Alzheimer’s disease histopathology and biomarkers of protein oxidation. J Neurochem. 1995;65(5):2146–56.
Hernández-Ortega K, Garcia-Esparcia P, Gil L, Lucas JJ, Ferrer I. Altered machinery of protein synthesis in alzheimer’s: from the nucleolus to the ribosome. Brain Pathol. 2016;26(5):593–605.
Hetz C, Mollereau B. Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat Rev Neurosci. 2014;15(4):233–49.
Hetz C, Zhang K, Kaufman RJ. Mechanisms, regulation and functions of the unfolded protein response. Nat Rev Mol Cell Biol. 2020;21(8):421–38.
Ho GJ, Drego R, Hakimian E, Masliah E. Mechanisms of cell signaling and inflammation in Alzheimer’s disease. Curr Drug Targets Inflamm Allergy. 2005;4(2):247–56.
Holdorf AD, Higgins DP, Hart AC, Boag PR, Pazour GJ, et al. WormCat: an online tool for annotation and visualization of caenorhabditis elegans genome-scale data. Genetics. 2020;214(2):279–94.
Hoozemans JJM, Veerhuis R, Van Haastert ES, Rozemuller JM, Baas F, et al. The unfolded protein response is activated in Alzheimer’s disease. Acta Neuropathol. 2005;110(2):165–72.
Hornsten A, Lieberthal J, Fadia S, Malins R, Ha L, et al. APL-1, a Caenorhabditis elegans protein related to the human beta-amyloid precursor protein, is essential for viability. Proc Natl Acad Sci USA. 2007;104(6):1971–6.
Hoshino T, Murao N, Namba T, Takehara M, Adachi H, et al. Suppression of Alzheimer’s disease-related phenotypes by expression of heat shock protein 70 in mice. J Neurosci. 2011;31(14):5225–34.
Hu W, Wang Z, Zheng H. Mitochondrial accumulation of amyloid β (Aβ) peptides requires TOMM22 as a main Aβ receptor in yeast. J Biol Chem. 2018;293(33):12681–9.
Hu Y, Comjean A, Perkins LA, Perrimon N, Mohr SE. GLAD: an online database of gene list annotation for drosophila. J Genomics. 2015;3:75–81.
Huang J, Chen S, Hu L, Niu H, Sun Q, et al. Mitoferrin-1 is Involved in the Progression of Alzheimer’s Disease Through Targeting Mitochondrial Iron Metabolism in a Caenorhabditis elegans Model of Alzheimer’s Disease. Neuroscience. 2018;385:90–101.
Husseman JW, Hallows JL, Bregman DB, Leverenz JB, Nochlin D, et al. Hyperphosphorylation of RNA polymerase II and reduced neuronal RNA levels precede neurofibrillary tangles in Alzheimer disease. J Neuropathol Exp Neurol. 2001;60(12):1219–32.
Iijima K, Iijima-Ando K. Drosophila models of Alzheimer’s amyloidosis: the challenge of dissecting the complex mechanisms of toxicity of amyloid-beta 42. J Alzheimers Dis. 2008;15(4):523–40.
Iijima K, Liu H-P, Chiang A-S, Hearn SA, Konsolaki M, Zhong Y. Dissecting the pathological effects of human Abeta40 and Abeta42 in Drosophila: a potential model for Alzheimer’s disease. Proc Natl Acad Sci USA. 2004;101(17):6623–8.
Iijima-Ando K, Sekiya M, Maruko-Otake A, Ohtake Y, Suzuki E, Lu B, Iijima KM. Loss of axonal mitochondria promotes tau-mediated neurodegeneration and Alzheimer’s disease-related tau phosphorylation via PAR-1. PLoS Genet. 2012;8(8):e1002918.
Ingram EM, Spillantini MG. Tau gene mutations: dissecting the pathogenesis of FTDP-17. Trends Mol Med. 2002;8:555–62.
Jackson GR, Wiedau-Pazos M, Sang T-K, Wagle N, Brown CA, et al. Human wild-type tau interacts with wingless pathway components and produces neurofibrillary pathology in Drosophila. Neuron. 2002;34(4):509–19.
Jazvinšćak Jembrek M, Slade N, Hof PR, Šimić G. The interactions of p53 with tau and Aß as potential therapeutic targets for Alzheimer’s disease. Prog Neurobiol. 2018;168:104–27.
Jeon Y, Lee JH, Choi B, Won S-Y, Cho KS. Genetic dissection of alzheimer’s disease using drosophila models. Int J Mol Sci. 2020;21(3):884.
Jiang Y, Di Gregorio SE, Duennwald ML, Lajoie P. Polyglutamine toxicity in yeast uncovers phenotypic variations between different fluorescent protein fusions. Traffic. 2017;18(1):58–70.
Jones MW, Errington ML, French PJ, Fine A, Bliss TV, et al. A requirement for the immediate early gene Zif268 in the expression of late LTP and long-term memories. Nat Neurosci. 2001;4(3):289–96.
Jones SV, Kounatidis I. Nuclear Factor-Kappa B and Alzheimer Disease, unifying genetic and environmental risk factors from cell to humans. Front Immunol. 2017;8:1805.
Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med. 2013;368:107–16.
Kann O, Kovács R. Mitochondria and neuronal activity. Am J Physiol Cell Physiol. 2007;292(2):C641–57.
Karagöz GE, Duarte AMS, Akoury E, Ippel H, Biernat J, Morán Luengo T, et al. Hsp90-Tau complex reveals molecular basis for specificity in chaperone action. Cell. 2014;156:963–74.
Karch CM, Goate AM. Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol Psychiat. 2015;77:43–51.
Kawai M, Cras P, Richey P, Tabaton M, Lowery DE, et al. Subcellular localization of amyloid precursor protein in senile plaques of Alzheimer’s disease. Am J Pathol. 1992;140(4):947–58.
Kelleher RJ, Shen J. Presenilin-1 mutations and Alzheimer’s disease. Proc Natl Acad Sci USA. 2017;114(4):629–33.
Khabirova E, Moloney A, Marciniak SJ, Williams J, Lomas DA, et al. The TRiC/CCT chaperone is implicated in Alzheimer’s disease based on patient GWAS and an RNAi screen in Aβ-expressing Caenorhabditis elegans. PLoS One. 2014;9(7):e102985.
Kheiri G, Dolatshahi M, Rahmani F, Rezaei N. Role of p38/MAPKs in Alzheimer’s disease: implications for amyloid beta toxicity targeted therapy. Rev Neurosci. 2018;30(1):9–30.
Kim J, de Haro M, Al-Ramahi I, Garaicoechea LL, Jeong H-H, Sonn JY, Zoghbi HY. Evolutionarily conserved regulators of tau identify targets for new therapies. Neuron. 2023;111:824-838.e7.
Kinney JW, Bemiller SM, Murtishaw AS, Leisgang AM, Salazar AM, Lamb BT. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimers Dement (N Y). 2018;4:575–90.
Kocahan S, Doğan Z. Mechanisms of Alzheimer’s Disease pathogenesis and prevention: the brain, neural pathology, N-methyl-D-aspartate receptors, tau protein and other risk factors. Clin Psychopharmacol Neurosci. 2017;15(1):1–8.
Kong W, Mou X, Liu Q, Chen Z, Vanderburg CR, et al. Independent component analysis of Alzheimer’s DNA microarray gene expression data. Mol Neurodegener. 2009;4:5.
Koren SA, Hamm MJ, Meier SE, Weiss BE, Nation GK, Chishti EA, Arango JP, Chen J, Zhu H, Blalock EM, Abisambra JF. Tau drives translational selectivity by interacting with ribosomal proteins. Acta Neuropathol. 2019;137(4):571–83.
Kouli A, Torsney KM, Kuan W-L. 2018. Parkinson’s disease: etiology, neuropathology, and pathogenesis. In Parkinson’s Disease: Pathogenesis and Clinical Aspects, eds. TB Stoker, JC Greenland. Brisbane (AU): Codon Publications
Kovacs GG. Tauopathies. Handb Clin Neurol. 2017;145:355–68.
Kow RL, Sikkema C, Wheeler JM, Wilkinson CW, Kraemer BC. DOPA decarboxylase modulates tau toxicity. Biol Psychiatry. 2018;83(5):438–46.
Kow RL, Strovas TJ, McMillan PJ, Jacobi AM, Behlke MA, et al. Distinct Poly(A) nucleases have differential impact on sut-2 dependent tauopathy phenotypes. Neurobiol Dis. 2021;147:105148.
Kraemer BC, Burgess JK, Chen JH, Thomas JH, Schellenberg GD. Molecular pathways that influence human tau-induced pathology in Caenorhabditis elegans. Hum Mol Genet. 2006;15(9):1483–96.
Kraemer BC, Schellenberg GD. SUT-1 enables tau-induced neurotoxicity in C. elegans. Hum Mol Genet. 2007;16(16):1959–71.
Kraemer BC, Zhang B, Leverenz JB, Thomas JH, Trojanowski JQ, Schellenberg GD. Neurodegeneration and defective neurotransmission in a Caenorhabditis elegans model of tauopathy. Proc Natl Acad Sci USA. 2003;100(17):9980–5.
Kuksa PP, Liu C-L, Fu W, Qu L, Zhao Y, Katanic Z, Leung YY. Alzheimer’s disease variant portal: A catalog of genetic findings for alzheimer’s disease. J Alzheimer’s Dis. 2022;86:461–77.
Kwak SS, Washicosky KJ, Brand E, von Maydell D, Aronson J, Kim S, Kim DY. Amyloid-β42/40 ratio drives tau pathology in 3D human neural cell culture models of Alzheimer’s disease. Nat Commun. 2020;11:1377.
Lackie RE, Maciejewski A, Ostapchenko VG, Marques-Lopes J, Choy W-Y, et al. The hsp70/hsp90 chaperone machinery in neurodegenerative diseases. Front Neurosci. 2017;11:25.
Lai CH, Chou CY, Ch’ang LY, Liu CS, Lin W. Identification of novel human genes evolutionarily conserved in Caenorhabditis elegans by comparative proteomics. Genome Res. 2000;10:703–13.
Lambert JC, Zelenika D, Hiltunen M, Chouraki V, Combarros O, Bullido MJ, Tognoni G, Fiévet N, Boland A, Arosio B, Coto E, Del Zompo M, Mateo I, Frank-Garcia A, Helisalmi S, Porcellini E, Pilotto A, Forti P, Ferri R, Amouyel P. Evidence of the association of BIN1 and PICALM with the AD risk in contrasting European populations. Neurobiol Aging. 2011;32(4):756.e11-e756.e15. https://doi.org/10.1016/j.neurobiolaging.2010.11.022.
Leak RK. Heat shock proteins in neurodegenerative disorders and aging. J Cell Commun Signal. 2014;8(4):293–310.
Lee JK, Kim N-J. Recent advances in the inhibition of p38 MAPK as a potential strategy for the treatment of alzheimer’s disease. Molecules. 2017;22(8):1287.
Lee K-S, Huh S, Lee S, Wu Z, Kim A-K, et al. Altered ER-mitochondria contact impacts mitochondria calcium homeostasis and contributes to neurodegeneration in vivo in disease models. Proc Natl Acad Sci USA. 2018;115(38):E8844–53.
Lee SS, Lee RYN, Fraser AG, Kamath RS, Ahringer J, Ruvkun G. A systematic RNAi screen identifies a critical role for mitochondria in C. elegans longevity. Nat Genet. 2003;33(1):40–8.
Leng F, Edison P. Neuroinflammation and microglial activation in Alzheimer disease: where do we go from here? Nat Rev Neurol. 2021;17:157–72.
Li X-L, Hu N, Tan M-S, Yu J-T, Tan L. Behavioral and psychological symptoms in Alzheimer’s disease. Biomed Res Int. 2014;2014:927804.
Licastro F, Pedrini S, Caputo L, Annoni G, Davis LJ, et al. Increased plasma levels of interleukin-1, interleukin-6 and alpha-1-antichymotrypsin in patients with Alzheimer’s disease: peripheral inflammation or signals from the brain? J Neuroimmunol. 2000;103(1):97–102.
Limone A, Veneruso I, D’Argenio V, Sarnataro D. Endosomal trafficking and related genetic underpinnings as a hub in Alzheimer’s disease. J Cell Physiol. 2022;237(10):3803–15.
Ling D, Magallanes M, Salvaterra PM. Accumulation of amyloid-like Aβ1–42 in AEL (autophagy-endosomal-lysosomal) vesicles: potential implications for plaque biogenesis. ASN Neuro. 2014;6(2):AN20130044.
Link CD. Expression of human beta-amyloid peptide in transgenic Caenorhabditis elegans. Proc Natl Acad Sci USA. 1995;92(20):9368–72.
Liu C-C, Liu C-C, Kanekiyo T, Xu H, Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol. 2013;9:106–18.
Liu L, Wu Q, Zhong W, Chen Y, Zhang W, et al. Microarray Analysis of Differential Gene Expression in Alzheimer’s Disease Identifies Potential Biomarkers with Diagnostic Value. Med Sci Monit. 2020;26:e919249.
Liu Q, Zhang J. Lipid metabolism in Alzheimer’s disease. Neurosci Bull. 2014;30(2):331–45.
Loewen CA, Feany MB. The unfolded protein response protects from tau neurotoxicity in vivo. PLoS One. 2010;5(9):e13084.
Loh KP, Huang SH, De Silva R, Tan BKH, Zhu YZ. Oxidative stress: apoptosis in neuronal injury. Curr Alzheimer Res. 2006;3(4):327–37.
Lotz M, Ebert S, Esselmann H, Iliev AI, Prinz M, et al. Amyloid beta peptide 1–40 enhances the action of Toll-like receptor-2 and -4 agonists but antagonizes Toll-like receptor-9-induced inflammation in primary mouse microglial cell cultures. J Neurochem. 2005;94(2):289–98.
Lovestone S, Reynolds CH, Latimer D, Davis DR, Anderton BH, Gallo JM, et al. Alzheimer’s disease-like phosphorylation of the microtubule-associated protein tau by glycogen synthase kinase-3 in transfected mammalian cells. Curr Biol. 1994;4:1077–86.
Lu KP, Zhou XZ. The prolyl isomerase PIN1: a pivotal new twist in phosphorylation signalling and disease. Nat Rev Mol Cell Biol. 2007;8(11):904–16.
Lucas JJ, Hernández F, Gómez-Ramos P, Morán MA, Hen R, Avila J. Decreased nuclear beta-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3beta conditional transgenic mice. EMBO J. 2001;20:27–39.
Lye SH, Chtarbanova S. Drosophila as a model to study brain innate immunity in health and disease. Int J Mol Sci. 2018;19(12):3922.
Magrané J, Smith RC, Walsh K, Querfurth HW. Heat shock protein 70 participates in the neuroprotective response to intracellularly expressed beta-amyloid in neurons. J Neurosci. 2004;24(7):1700–6.
Manczak M, Anekonda TS, Henson E, Park BS, Quinn J, Reddy PH. Mitochondria are a direct site of A beta accumulation in Alzheimer’s disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum Mol Genet. 2006;15(9):1437–49.
Mangione MR, Vilasi S, Marino C, Librizzi F, Canale C, et al. Hsp60, amateur chaperone in amyloid-beta fibrillogenesis. Biochim Biophys Acta. 2016;1860(11):2474–83.
Marcus DL, Thomas C, Rodriguez C, Simberkoff K, Tsai JS, et al. Increased peroxidation and reduced antioxidant enzyme activity in Alzheimer’s disease. Exp Neurol. 1998;150(1):40–4.
Martínez-Menárguez JÁ, Martínez-Alonso E, Cara-Esteban M, Tomás M. Focus on the small gtpase rab1: A key player in the pathogenesis of parkinson’s disease. Int J Mol Sci. 2021;22(21):12087.
Martín-Peña A, Rincón-Limas DE, Fernandez-Fúnez P. Engineered Hsp70 chaperones prevent Aβ42-induced memory impairments in a Drosophila model of Alzheimer’s disease. Sci Rep. 2018;8(1):9915.
Mast N, Saadane A, Valencia-Olvera A, Constans J, Maxfield E, et al. Cholesterol-metabolizing enzyme cytochrome P450 46A1 as a pharmacologic target for Alzheimer’s disease. Neuropharmacology. 2017;123:465–76.
Matlack KES, Tardiff DF, Narayan P, Hamamichi S, Caldwell KA, et al. Clioquinol promotes the degradation of metal-dependent amyloid-β (Aβ) oligomers to restore endocytosis and ameliorate Aβ toxicity. Proc Natl Acad Sci USA. 2014;111(11):4013–8.
Maulik M, Mitra S, Basmayor AM, Lu B, Taylor BE, Bult-Ito A. Genetic Silencing of Fatty Acid Desaturases Modulates α-Synuclein Toxicity and Neuronal Loss in Parkinson-Like Models of C. elegans. Front Aging Neurosci. 2019;11:207.
Mazzulli JR, Zunke F, Isacson O, Studer L, Krainc D. α-Synuclein-induced lysosomal dysfunction occurs through disruptions in protein trafficking in human midbrain synucleinopathy models. Proc Natl Acad Sci USA. 2016;113(7):1931–6.
McColgan P, Tabrizi SJ. Huntington’s disease: a clinical review. Eur J Neurol. 2018;25(1):24–34.
Mcdonald J, Dhakal S, Macreadie I. Yeast contributions to Alzheimer’s Disease. J Human Clin Gen. 2020;2(2):1–19.
Mecocci P, MacGarvey U, Beal MF. Oxidative damage to mitochondrial DNA is increased in Alzheimer’s disease. Ann Neurol. 1994;36(5):747–51.
Meier S, Bell M, Lyons DN, Rodriguez-Rivera J, Ingram A, Fontaine SN, Mechas E, Chen J, Wolozin B, LeVine H 3rd, Zhu H, Abisambra JF. Pathological Tau Promotes Neuronal Damage by Impairing Ribosomal Function and Decreasing Protein Synthesis. J Neurosci. 2016;36(3):1001–7.
Merlo P, Frost B, Peng S, Yang YJ, Park PJ, Feany M. p53 prevents neurodegeneration by regulating synaptic genes. Proc Natl Acad Sci USA. 2014;111(50):18055–60.
Miao J, Ma H, Yang Y, Liao Y, Lin C, Zheng J, et al. Microglia in Alzheimer’s disease: pathogenesis, mechanisms, and therapeutic potentials. Front Aging Neurosci. 2023;15:1201982.
Miyasaka T, Ding Z, Gengyo-Ando K, Oue M, Yamaguchi H, et al. Progressive neurodegeneration in C. elegans model of tauopathy. Neurobiol Dis. 2005;20(2):372–83.
Mizushima N. Autophagy: process and function. Genes Dev. 2007;21(22):2861–73.
Moltedo O, Remondelli P, Amodio G. The Mitochondria-endoplasmic reticulum contacts and their critical role in aging and age-associated diseases. Front Cell Dev Biol. 2019;7:172.
Moreira PI, Carvalho C, Zhu X, Smith MA, Perry G. Mitochondrial dysfunction is a trigger of Alzheimer’s disease pathophysiology. Biochim Biophys Acta. 2010;1802(1):2–10.
Mosconi L, de Leon M, Murray J E L, Lu J, et al. Reduced mitochondria cytochrome oxidase activity in adult children of mothers with Alzheimer’s disease. J Alzheimers Dis. 2011;27(3):483–90.
Moulton MJ, Barish S, Ralhan I, Chang J, Goodman LD, Harland JG, Bellen HJ. Neuronal ROS-induced glial lipid droplet formation is altered by loss of Alzheimer’s disease-associated genes. Proc Natl Acad Sci U SA. 2021;118(52):e2112095118.
Mukherjee S, Russell JC, Carr DT, Burgess JD, Allen M, et al. Systems biology approach to late-onset Alzheimer’s disease genome-wide association study identifies novel candidate genes validated using brain expression data and Caenorhabditis elegans experiments. Alzheimers Dement. 2017;13(10):1133–42.
Murphy MP, LeVine H. Alzheimer’s disease and the amyloid-beta peptide. J Alzheimers Dis. 2010;19(1):311–23.
Nangia V, O’Connell J, Chopra K, Qing Y, Reppert C, et al. Genetic reduction of tyramine β hydroxylase suppresses Tau toxicity in a Drosophila model of tauopathy. Neurosci Lett. 2021;755:135937.
Nassari S, Del Olmo T, Jean S. Rabs in signaling and embryonic development. Int J Mol Sci. 2020;21(3):1064.
Navarro-Mabarak C, Camacho-Carranza R, Espinosa-Aguirre JJ. Cytochrome P450 in the central nervous system as a therapeutic target in neurodegenerative diseases. Drug Metab Rev. 2018;50(2):95–108.
Niki E. Lipid peroxidation products as oxidative stress biomarkers. BioFactors. 2008;34(2):171–80.
Nilsson P, Loganathan K, Sekiguchi M, Matsuba Y, Hui K, et al. Aβ secretion and plaque formation depend on autophagy. Cell Rep. 2013;5(1):61–9.
Nilsson P, Saido TC. Dual roles for autophagy: degradation and secretion of Alzheimer’s disease Aβ peptide. BioEssays. 2014;36(6):570–8.
Nisha, Sarkar S. Downregulation of glob1 suppresses pathogenesis of human neuronal tauopathies in Drosophila by regulating tau phosphorylation and ROS generation. Neurochem Int. 2021;146:105040.
Nunomura A, Perry G, Aliev G, Hirai K, Takeda A, et al. Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol. 2001;60(8):759–67.
O’Brien KP, Westerlund I, Sonnhammer ELL. OrthoDisease: a database of human disease orthologs. Hum Mutat. 2004;24:112–9.
Oddo S. The ubiquitin-proteasome system in Alzheimer’s disease. J Cell Mol Med. 2008;12(2):363–73.
Oddo S. The role of mTOR signaling in Alzheimer disease. Front Biosci (Schol Ed). 2012;4:941–52.
Ojala J, Alafuzoff I, Herukka S-K, van Groen T, Tanila H, Pirttilä T. Expression of interleukin-18 is increased in the brains of Alzheimer’s disease patients. Neurobiol Aging. 2009;30(2):198–209.
Ojelade SA, Lee TV, Giagtzoglou N, Yu L, Ugur B, et al. cindr, the Drosophila Homolog of the CD2AP Alzheimer’s Disease Risk Gene, Is Required for Synaptic Transmission and Proteostasis. Cell Rep. 2019;28(7):1799-1813.e5.
Oka M, Fujisaki N, Maruko-Otake A, Ohtake Y, Shimizu S, et al. Ca2+/calmodulin-dependent protein kinase II promotes neurodegeneration caused by tau phosphorylated at Ser262/356 in a transgenic Drosophila model of tauopathy. J Biochem. 2017;162(5):335–42.
Omata Y, Lim Y-M, Akao Y, Tsuda L. Age-induced reduction of autophagy-related gene expression is associated with onset of Alzheimer’s disease. Am J Neurodegener Dis. 2014;3(3):134–42.
Orr ME, Sullivan AC, Frost B. A brief overview of tauopathy: causes, consequences, and therapeutic strategies. Trends Pharmacol Sci. 2017;38(7):637–48.
Osellame LD, Blacker TS, Duchen MR. Cellular and molecular mechanisms of mitochondrial function. Best Pract Res Clin Endocrinol Metab. 2012;26(6):711–23.
Pagani L, Eckert A. Amyloid-Beta interaction with mitochondria. Int J Alzheimers Dis. 2011;2011:925050.
Palmer AM, Burns MA. Selective increase in lipid peroxidation in the inferior temporal cortex in Alzheimer’s disease. Brain Res. 1994;645(1–2):338–42.
Papaevgeniou N, Sakellari M, Jha S, Tavernarakis N, Holmberg CI, et al. 18α-Glycyrrhetinic Acid Proteasome Activator Decelerates Aging and Alzheimer’s Disease Progression in Caenorhabditis elegans and Neuronal Cultures. Antioxid Redox Signal. 2016;25(16):855–69.
Park MH, Park KH, Choi BJ, Han WH, Yoon HJ, et al. Discovery of a dual-action small molecule that improves neuropathological features of Alzheimer’s disease mice. Proc Natl Acad Sci USA. 2022;119(3):e2115082119.
Park S-K, Ratia K, Ba M, Valencik M, Liebman SW. Inhibition of Aβ42 oligomerization in yeast by a PICALM ortholog and certain FDA approved drugs. Microb Cell. 2016;3(2):53–64.
Park SY, Seo J, Chun YS. Targeted Downregulation of kdm4a Ameliorates Tau-engendered Defects in Drosophila melanogaster. J Korean Med Sci. 2019;34(33):e225.
Peña-Bautista C, Álvarez-Sánchez L, Cañada-Martínez AJ, Baquero M, Cháfer-Pericás C. Epigenomics and lipidomics integration in Alzheimer disease: pathways involved in early stages. Biomedicines. 2021;9(12):1812.
Penserga T, Kudumala SR, Poulos R, Godenschwege TA. A Role for Drosophila Amyloid Precursor Protein in Retrograde Trafficking of L1-Type Cell Adhesion Molecule Neuroglian. Front Cell Neurosci. 2019;13:322.
Petrucelli L, Dickson D, Kehoe K, Taylor J, Snyder H, Grover A, Hutton M. CHIP and Hsp70 regulate tau ubiquitination, degradation and aggregation. Hum Mol Genet. 2004;13:703–14.
Povellato G, Tuxworth RI, Hanger DP, Tear G. Modification of the Drosophila model of in vivo Tau toxicity reveals protective phosphorylation by GSK3β. Biol Open. 2014;3(1):1–11.
Prokop S, Miller KR, Heppner FL. Microglia actions in Alzheimer’s disease. Acta Neuropathol. 2013;126(4):461–77.
Prüßing K, Voigt A, Schulz JB. Drosophila melanogaster as a model organism for Alzheimer’s disease. Mol Neurodegener. 2013;8:35.
Qin X, Wang Y, Paudel HK. Inhibition of Early Growth Response 1 in the Hippocampus Alleviates Neuropathology and Improves Cognition in an Alzheimer Model with Plaques and Tangles. Am J Pathol. 2017;187(8):1828–47.
Qu Z, Sun J, Zhang W, Yu J, Zhuang C. Transcription factor NRF2 as a promising therapeutic target for Alzheimer’s disease. Free Radic Biol Med. 2020;159:87–102.
Qureshi HY, Han D, MacDonald R, Paudel HK. Overexpression of 14-3-3z promotes tau phosphorylation at Ser262 and accelerates proteosomal degradation of synaptophysin in rat primary hippocampal neurons. PLoS One. 2013;8(12):e84615.
Radi E, Formichi P, Battisti C, Federico A. Apoptosis and oxidative stress in neurodegenerative diseases. J Alzheimers Dis. 2014;42(Suppl 3):S125–52.
Reddy PH. Abnormal tau, mitochondrial dysfunction, impaired axonal transport of mitochondria, and synaptic deprivation in Alzheimer’s disease. Brain Res. 2011;1415:136–48.
Regitz C, Dußling LM, Wenzel U. Amyloid-beta (Aβ1-42)-induced paralysis in Caenorhabditis elegans is inhibited by the polyphenol quercetin through activation of protein degradation pathways. Mol Nutr Food Res. 2014;58(10):1931–40.
Reisberg B, Ferris SH, de Leon MJ, Kluger A, Franssen E, Borenstein J, et al. The stage specific temporal course of Alzheimer’s disease: functional and behavioral concomitants based upon cross-sectional and longitudinal observation. Prog Clin Biol Res. 1989;317:23–41.
Rimal S, Li Y, Vartak R, Geng J, Tantray I, et al. Inefficient quality control of ribosome stalling during APP synthesis generates CAT-tailed species that precipitate hallmarks of Alzheimer’s disease. Acta Neuropathol Commun. 2021;9(1):169.
Ring J, Tadic J, Ristic S, Poglitsch M, Bergmann M, Radic N, Mossmann D, Liang Y, Maglione M, Jerkovic A, Hajiraissi R, Hanke M, Küttner V, Wolinski H, Zimmermann A, Domuz Trifunović L, Mikolasch L, Moretti DN, Broeskamp F, Madeo F. The HSP40 chaperone Ydj1 drives amyloid beta 42 toxicity. EMBO Mol Med. 2022;14(5):e13952.
Rival T, Page RM, Chandraratna DS, Sendall TJ, Ryder E, et al. Fenton chemistry and oxidative stress mediate the toxicity of the beta-amyloid peptide in a Drosophila model of Alzheimer’s disease. Eur J Neurosci. 2009;29(7):1335–47.
Rizzuto R, Bernardi P, Pozzan T. Mitochondria as all-round players of the calcium game. J Physiol (Lond). 2000;529(Pt 1):37–47.
Robinson MD, Grigull J, Mohammad N, Hughes TR. FunSpec: a web-based cluster interpreter for yeast. BMC Bioinformatics. 2002;3:35.
Rutledge BS, Choy W-Y, Duennwald ML. Folding or holding?-Hsp70 and Hsp90 chaperoning of misfolded proteins in neurodegenerative disease. J Biol Chem. 2022;298:101905.
Ryan KC, Ashkavand Z, Norman KR. The role of mitochondrial calcium homeostasis in Alzheimer’s and related diseases. Int J Mol Sci. 2020;21(23):9153.
Safra M, Ben-Hamo S, Kenyon C, Henis-Korenblit S. The ire-1 ER stress-response pathway is required for normal secretory-protein metabolism in C elegans. J Cell Sci. 2013;126(Pt 18):4136–46.
Saibil H. Chaperone machines for protein folding, unfolding and disaggregation. Nat Rev Mol Cell Biol. 2013;14(10):630–42.
Saido TC. Metabolism of amyloid β peptide and pathogenesis of Alzheimer’s disease. Proc Jpn Acad Ser B Phys Biol Sci. 2013;89(7):321–39.
Salazar SV, Cox TO, Lee S, Brody AH, Chyung AS, et al. Alzheimer’s Disease Risk Factor Pyk2 Mediates Amyloid-β-Induced Synaptic Dysfunction and Loss. J Neurosci. 2019;39(4):758–72.
Salminen A, Ojala J, Kaarniranta K, Hiltunen M, Soininen H. Hsp90 regulates tau pathology through co-chaperone complexes in Alzheimer’s disease. Prog Neurobiol. 2011;93(1):99–110.
Salminen A, Ojala J, Kauppinen A, Kaarniranta K, Suuronen T. Inflammation in Alzheimer’s disease: amyloid-beta oligomers trigger innate immunity defence via pattern recognition receptors. Prog Neurobiol. 2009;87(3):181–94.
Salmon TB, Evert BA, Song B, Doetsch PW. Biological consequences of oxidative stress-induced DNA damage in Saccharomyces cerevisiae. Nucleic Acids Res. 2004;32(12):3712–23.
Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, Younkin S. Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat Med. 1996;2:864–70.
Schwikowski B, Uetz P, Fields S. A network of protein-protein interactions in yeast. Nat Biotechnol. 2000;18(12):1257–61.
Seynnaeve D, Vecchio MD, Fruhmann G, Verelst J, Cools M, et al. Recent Insights on Alzheimer’s Disease Originating from Yeast Models. Int J Mol Sci. 2018;19(7):1947.
Sharma P, Srivastava P, Seth A, Tripathi PN, Banerjee AG, Shrivastava SK. Comprehensive review of mechanisms of pathogenesis involved in Alzheimer’s disease and potential therapeutic strategies. Prog Neurobiol. 2019;174:53–89.
Shim SM, Lee WJ, Kim Y, Chang JW, Song S, Jung Y-K. Role of S5b/PSMD5 in proteasome inhibition caused by TNF-α/NFκB in higher eukaryotes. Cell Rep. 2012;2(3):603–15.
Shukla S, Tekwani BL. Histone deacetylases inhibitors in neurodegenerative diseases, neuroprotection and neuronal differentiation. Front Pharmacol. 2020;11:537.
Shulman JM, Chipendo P, Chibnik LB, Aubin C, Tran D, et al. Functional screening of Alzheimer pathology genome-wide association signals in Drosophila. Am J Hum Genet. 2011;88(2):232–8.
Shulman JM, Feany MB. Genetic modifiers of tauopathy in Drosophila. Genetics. 2003;165(3):1233–42.
Shulman JM, Imboywa S, Giagtzoglou N, Powers MP, Hu Y, et al. Functional screening in Drosophila identifies Alzheimer’s disease susceptibility genes and implicates Tau-mediated mechanisms. Hum Mol Genet. 2014;23(4):870–7.
Silver I, Erecińska M. Oxygen and ion concentrations in normoxic and hypoxic brain cells. Adv Exp Med Biol. 1998;454:7–16.
Sinnige T, Ciryam P, Casford S, Dobson CM, de Bono M, Vendruscolo M. Expression of the amyloid-β peptide in a single pair of C. elegans sensory neurons modulates the associated behavioural response. PLoS One. 2019;14(5):e0217746.
Son SJ, Lee KS, Chung JH, Chang KJ, Roh HW, et al. Increased plasma levels of heat shock protein 70 associated with subsequent clinical conversion to mild cognitive impairment in cognitively healthy elderly. PLoS One. 2015;10(3):e0119180.
Sorbi S, Bird ED, Blass JP. Decreased pyruvate dehydrogenase complex activity in Huntington and Alzheimer brain. Ann Neurol. 1983;13(1):72–8.
Sparvero LJ, Patz S, Brodsky JL, Coughlan CM. Proteomic analysis of the amyloid precursor protein fragment C99: expression in yeast. Anal Biochem. 2007;370(2):162–70.
Spindler SR, Hartenstein V. The Drosophila neural lineages: a model system to study brain development and circuitry. Dev Genes Evol. 2010;220(1–2):1–10.
Steinberg S, Stefansson H, Jonsson T, Johannsdottir H, Ingason A, Helgason H, et al. Loss-of-function variants in ABCA7 confer risk of Alzheimer’s disease. Nat Genet. 2015;47:445–7.
Stokin GB, Goldstein LSB. Axonal transport and Alzheimer’s disease. Annu Rev Biochem. 2006;75:607–27.
Su B, Wang X, Lee H-G, Tabaton M, Perry G, et al. Chronic oxidative stress causes increased tau phosphorylation in M17 neuroblastoma cells. Neurosci Lett. 2010;468(3):267–71.
Subramanian M, Hyeon SJ, Das T, Suh YS, Kim YK, et al. UBE4B, a microRNA-9 target gene, promotes autophagy-mediated Tau degradation. Nat Commun. 2021;12(1):3291.
Sun E, Motolani A, Campos L, Lu T. The Pivotal Role of NF-kB in the Pathogenesis and Therapeutics of Alzheimer’s Disease. Int J Mol Sci. 2022;23(16):8972.
Swerdlow RH. Mitochondria and mitochondrial cascades in alzheimer’s disease. J Alzheimers Dis. 2018;62(3):1403–16.
Tadic J, Ring J, Jerkovic A, Ristic S, Maglione M, Dengjel J, Sigrist SJ, Eisenberg T. A pathological role of the Hsp40 protein Ydj1/DnaJA1 in models of Alzheimer’s disease [Review of A pathological role of the Hsp40 protein Ydj1/DnaJA1 in models of Alzheimer’s disease]. Cell Stress Chaperones. 2022;6(5):61–4.
Takagi M, Absalon MJ, McLure KG, Kastan MB. Regulation of p53 translation and induction after DNA damage by ribosomal protein L26 and nucleolin. Cell. 2005;123(1):49–63.
Tan L, Schedl P, Song H-J, Garza D, Konsolaki M. The Toll–>NFkappaB signaling pathway mediates the neuropathological effects of the human Alzheimer’s Abeta42 polypeptide in Drosophila. PLoS One. 2008;3(12):e3966.
Tarassov K, Messier V, Landry CR, Radinovic S, Serna Molina MM, Shames I, et al. An in vivo map of the yeast protein interactome. Science. 2008;320:1465–70.
Terai K, Matsuo A, McGeer PL. Enhancement of immunoreactivity for NF-kappa B in the hippocampal formation and cerebral cortex of Alzheimer’s disease. Brain Res. 1996;735(1):159–68.
Thomas PD, Ebert D, Muruganujan A, Mushayahama T, Albou L-P, Mi H. PANTHER: Making genome-scale phylogenetics accessible to all. Protein Sci. 2022;31:8–22.
Thomas RS, Henson A, Gerrish A, Jones L, Williams J, Kidd EJ. Decreasing the expression of PICALM reduces endocytosis and the activity of β-secretase: implications for Alzheimer’s disease. BMC Neurosci. 2016;17(1):50.
Timm T, Matenia D, Li XY, Griesshaber B, Mandelkow EM. Signaling from MARK to tau: regulation, cytoskeletal crosstalk, and pathological phosphorylation. Neurodegener Dis. 2006;3(4–5):207–17.
Tonoki A, Kuranaga E, Tomioka T, Hamazaki J, Murata S, et al. Genetic evidence linking age-dependent attenuation of the 26S proteasome with the aging process. Mol Cell Biol. 2009;29(4):1095–106.
Treusch S, Hamamichi S, Goodman JL, Matlack KES, Chung CY, et al. Functional links between Aβ toxicity, endocytic trafficking, and Alzheimer’s disease risk factors in yeast. Science. 2011;334(6060):1241–5.
Troemel ER, Chu SW, Reinke V, Lee SS, Ausubel FM, Kim DH. p38 MAPK regulates expression of immune response genes and contributes to longevity in C. elegans. PLoS Genet. 2006;2(11):e183.
Troulinaki K, Tavernarakis N. Endocytosis and intracellular trafficking contribute to necrotic neurodegeneration in C. elegans. EMBO J. 2012;31(3):654–66.
Tsang WY, Lemire BD. Mitochondrial ATP synthase controls larval development cell nonautonomously in Caenorhabditis elegans. Dev Dynamics. 2003;226(4):719–26.
Tseng BP, Green KN, Chan JL, Blurton-Jones M, LaFerla FM. Abeta inhibits the proteasome and enhances amyloid and tau accumulation. Neurobiol Aging. 2008;29(11):1607–18.
Uddin MS, Stachowiak A, Mamun AA, Tzvetkov NT, Takeda S, et al. Autophagy and alzheimer’s disease: from molecular mechanisms to therapeutic implications. Front Aging Neurosci. 2018;10:04.
Ulamec SM, Brockwell DJ, Radford SE. Looking beyond the core: the role of flanking regions in the aggregation of amyloidogenic peptides and proteins. Front Neurosci. 2020;14:611285.
Unterberger U, Höftberger R, Gelpi E, Flicker H, Budka H, Voigtländer T. Endoplasmic reticulum stress features are prominent in Alzheimer disease but not in prion diseases in vivo. J Neuropathol Exp Neurol. 2006;65(4):348–57.
van der Staay FJ, Rutten K, Bärfacker L, Devry J, Erb C, Heckroth H, Hendrix M. The novel selective PDE9 inhibitor BAY 73–6691 improves learning and memory in rodents. Neuropharmacology. 2008;55:908–18.
van Heusden GPH, Steensma HY. Yeast 14-3-3 proteins. Yeast. 2006;23(3):159–71.
van Leeuwen FW, de Kleijn DP, van den Hurk HH, Neubauer A, Sonnemans MA, et al. Frameshift mutants of beta amyloid precursor protein and ubiquitin-B in Alzheimer’s and Down patients. Science. 1998;279(5348):242–7.
Vanhelmont T, Vandebroek T, De Vos A, Terwel D, Lemaire K, et al. Serine-409 phosphorylation and oxidative damage define aggregation of human protein tau in yeast. FEMS Yeast Res. 2010;10(8):992–1005.
Vergallo A, Giampietri L, Baldacci F, Volpi L, Chico L, et al. Oxidative stress assessment in alzheimer’s disease: a clinic setting study. Am J Alzheimers Dis Other Demen. 2018;33(1):35–41.
Vérièpe J, Fossouo L, Parker JA. Neurodegeneration in C. elegans models of ALS requires TIR-1/Sarm1 immune pathway activation in neurons. Nat Commun. 2015;6:7319.
Waldherr SM, Strovas TJ, Vadset TA, Liachko NF, Kraemer BC. Constitutive XBP-1s-mediated activation of the endoplasmic reticulum unfolded protein response protects against pathological tau. Nat Commun. 2019;10(1):4443.
Wan L, Nie G, Zhang J, Luo Y, Zhang P, et al. β-Amyloid peptide increases levels of iron content and oxidative stress in human cell and Caenorhabditis elegans models of Alzheimer disease. Free Radic Biol Med. 2011;50(1):122–9.
Wan W, **a S, Kalionis B, Liu L, Li Y. The role of Wnt signaling in the development of Alzheimer’s disease: a potential therapeutic target? Biomed Res Int. 2014;2014:301575.
Wang X, Su B, Fujioka H, Zhu X. Dynamin-like protein 1 reduction underlies mitochondrial morphology and distribution abnormalities in fibroblasts from sporadic Alzheimer’s disease patients. Am J Pathol. 2008;173(2):470–82.
Wang X, Wang W, Li L, Perry G, Lee H, Zhu X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim Biophys Acta. 2014;1842(8):1240–7.
Webster CP, Smith EF, Bauer CS, Moller A, Hautbergue GM, et al. The C9orf72 protein interacts with Rab1a and the ULK1 complex to regulate initiation of autophagy. EMBO J. 2016;35(15):1656–76.
Weickert S, Wawrzyniuk M, John LH, Rüdiger SGD, Drescher M. The mechanism of Hsp90-induced oligomerizaton of Tau. Sci Adv. 2020;6:eaax6999.
Wiese M, Antebi A, Zheng H. Intracellular trafficking and synaptic function of APL-1 in Caenorhabditis elegans. PLoS One. 2010;5(9):12790.
Wilson DM, Cookson MR, Van Den Bosch L, Zetterberg H, Holtzman DM, Dewachter I. Hallmarks of neurodegenerative diseases. Cell. 2023;186(4):693–714.
Wittmann CW, Wszolek MF, Shulman JM, Salvaterra PM, Lewis J, et al. Tauopathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science. 2001;293(5530):711–4.
Wójtowicz S, Strosznajder AK, Jeżyna M, Strosznajder JB. The novel role of PPAR alpha in the brain: promising target in therapy of alzheimer’s disease and other neurodegenerative disorders. Neurochem Res. 2020;45(5):972–88.
Woo J-AA, Liu T, Fang CC, Cazzaro S, Kee T, et al. Activated cofilin exacerbates tau pathology by impairing tau-mediated microtubule dynamics. Commun Biol. 2019;2:112.
Wu Y, Cao Z, Klein WL, Luo Y. Heat shock treatment reduces beta amyloid toxicity in vivo by diminishing oligomers. Neurobiol Aging. 2010;31(6):1055–8.
**ao Q, Gil S-C, Yan P, Wang Y, Han S, Gonzales E, Perez R, Cirrito JR, Lee J-M. Role of phosphatidylinositol clathrin assembly lymphoid-myeloid leukemia (PICALM) in intracellular amyloid precursor protein (APP) processing and amyloid plaque pathogenesis. J Biol Chem. 2012;287(25):21279–89.
**ao X, Liu H, Liu X, Zhang W, Zhang S, Jiao B. APP, PSEN1, and PSEN2 Variants in Alzheimer’s Disease: Systematic Re-evaluation According to ACMG Guidelines. Front Aging Neurosci. 2021;13:695808.
Xu W, Tan L, Yu J-T. The role of PICALM in alzheimer’s disease. Mol Neurobiol. 2015;52(1):399–413.
Yan X, Hu Y, Wang B, Wang S, Zhang X. Metabolic dysregulation contributes to the progression of alzheimer’s disease. Front Neurosci. 2020;14:530219.
Yerbury JJ, Ooi L, Dillin A, Saunders DN, Hatters DM, et al. Walking the tightrope: proteostasis and neurodegenerative disease. J Neurochem. 2016;137(4):489–505.
Yu R, Nielsen J. Big data in yeast systems biology. FEMS Yeast Res. 2019;19(7):foz070.
Yu Y, Niccoli T, Ren Z, Woodling NS, Aleyakpo B, Szabadkai G, Partridge L. PICALM rescues glutamatergic neurotransmission, behavioural function and survival in a Drosophila model of Aβ42 toxicity. Hum Mol Genet. 2020;29(14):2420–34.
Zambrano CA, Egaña JT, Núñez MT, Maccioni RB, González-Billault C. Oxidative stress promotes tau dephosphorylation in neuronal cells: the roles of cdk5 and PP1. Free Radic Biol Med. 2004;36(11):1393–402.
Zarouchlioti C, Parfitt DA, Li W, Gittings LM, Cheetham ME. DNAJ Proteins in neurodegeneration: essential and protective factors. Philos. Trans R Soc Lond B Biol Sci. 2018;373(1738):20160534.
Zeng Q, Siu W, Li L, ** Y, Liang S, et al. Autophagy in Alzheimer’s disease and promising modulatory effects of herbal medicine. Exp Gerontol. 2019;119:100–10.
Zetterberg M, Sjölander A, von Otter M, Palmér MS, Landgren S, et al. Ubiquitin carboxy-terminal hydrolase L1 (UCHL1) S18Y polymorphism in Alzheimer’s disease. Mol Neurodegener. 2010;5:11.
Zhang Y, Thompson R, Zhang H, Xu H. APP processing in Alzheimer’s disease. Mol Brain. 2011;4:3.
Zhao R, Hu W, Tsai J, Li W, Gan W-B. Microglia limit the expansion of β-amyloid plaques in a mouse model of Alzheimer’s disease. Mol Neurodegener. 2017;12(1):47.
Zhu X, Lee H, Raina AK, Perry G, Smith MA. The role of mitogen-activated protein kinase pathways in Alzheimer’s disease. Neurosignals. 2002;11(5):270–81.
Zolkiewski M, Zhang T, Nagy M. Aggregate reactivation mediated by the Hsp100 chaperones. Arch Biochem Biophys. 2012;520(1):1–6.
Author information
Authors and Affiliations
Contributions
YJ and LTM wrote the paper. YJ and LTM generated figures and analyzed data.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Table S1. Genetic modifiers of AD identified in S. cerevisiae models. Table S2. Genetic modifiers of AD identified in C. elegans models. Table S3. Genetic modifiers of AD identified in Drosophila models. Table S4. Human orthologs of genetic modifiers identified in Aβ models. Human orthologs of S. cerevisiae genes were retrieved from the Saccharomyces Genome Database (SGD) YeastMine online tool. Orthologs of C. elegans genes were queried from the OrthoList2 online tool. Orthologs of Drosophila genes were obtained from FlyBase. AD genes identified from GWAS studies are highlighted in red. Table S5. Human orthologs of genetic modifiers identified in Tau models. Orthologs of S. cerevisiae genes were retrieved from the SGD YeastMine online tool. Orthologs of C. elegans genes were queried from the OrthoList2 online tool. Orthologs of Drosophila genes were obtained from FlyBase. AD genes identified from GWAS studies are highlighted in red. Table S6. Functional classes enrichment of Aβ modifiers identified in S. cerevisiae models analyzed using FunSpec. Gene list was queried to multiple yeast databases including GO Molecular Function, GO Biological Process, GO Cellular Component, MIPS Functional Classification, MIPS Phenotypes, MIPS Subcellular Localization, MIPS Protein Complexes using FunSpec. Table S7. Cellular pathways and processes linked to Tau modifiers. GO analysis of Tau modifiers identified in S. cerevisiae models was performed using FunSpec. Tau modifiers identified in C.elegansmodels were analyzed using the WormCat online tool. Tau modifiers identified in Drosophila models were grouped using the GLAD database. Table S8. Functional annotation and enrichment of Aβ modifiers identified in C. elegans models analyzed using WormCat. Gene list were input to WormCat using default settings for analyzation. Promoting genes indicated that deletion/reduction suppresses AD phenotypes and/or overexpression enhances AD phenotypes. Preventing genes indicated that deletion/reduction enhances AD phenotypes and/or overexpression suppresses AD phenotypes. Table S9. Functional annotation and enrichment of Tau modifiers identified in C. elegans models analyzed using WormCat. Gene lists were analyzed using WormCat. Promoting genes indicated that deletion/reduction suppresses AD phenotypes and/or overexpression enhances AD phenotypes. Preventing genes indicated that deletion/reduction enhances AD phenotypes and/or overexpression suppresses AD phenotypes. Table S10. Functional classes enrichment of Aβ modifiers identified in S. cerevisiae models analyzed using GO Slim Mapper. Gene list was queried to SGD Yeast GO-Slim database. Table S11. Functional annotation and enrichment of Aβ modifiers identified in C. elegans models analyzed using WormCat. Gene list were input to WormCat using default settings for analyzation. Promoting genes indicated that deletion/reduction suppresses AD phenotypes and/or overexpression enhances AD phenotypes. Preventing genes indicated that deletion/reduction enhances AD phenotypes and/or overexpression suppresses AD phenotypes. Table S12. Functional annotation and enrichment of Aβ modifiers identified in Drosophila models analyzed using GLAD. Gene list was analyzed using Find Group Membership function in the GLAD online tool [5, 6, 12, 19, 26, 28, 31, 33, 38, 39, 44, 47, 48, 52, 56, 57, 61, 68, 71, 73,74,75, 77, 79, 85, 90, 95,96,97,98, 100, 102, 111, 113, 115, 121, 131, 137, 145, 146, 152, 153, 155, 157, 161, 163, 164, 166, 170, 174, 175, 177,178,179, 181, 186, 188, 190, 192, 197, 199, 200, 202, 203, 211, 217,218,219, 223, 228, 230, 231, 237, 239, 241, 244, 246, 247, 253, 259, 262, 265, 272, 273, 280, 290, 292, 293, 295, 296, 298, 303, 306, 307, 310, 315, 316, 318, 319, 323, 326, 332,333,334, 336, 337].
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Jiang, Y., MacNeil, L.T. Simple model systems reveal conserved mechanisms of Alzheimer’s disease and related tauopathies. Mol Neurodegeneration 18, 82 (2023). https://doi.org/10.1186/s13024-023-00664-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13024-023-00664-x