
Abstract
Background
Adenoviruses are common pathogens infecting animals and humans. They are classified based on serology, or genome sequence information. These methods have limitations due to lengthy procedures or lack of infectivity data. Adenoviruses are easy to produce and amenable to genetic and biochemical modifications, which makes them a powerful tool for biological studies, and clinical gene-delivery and vaccine applications. Antibodies directed against adenoviral proteins are important diagnostic tools for virus identification in vivo and in vitro, and are used to elucidate infection mechanisms, often in combination with genomic sequencing and type specific information from hyper-variable regions of structural proteins.
Results
Here we describe a novel and readily useable method for cloning, expressing and purifying small fragments of hyper-variable regions 1-6 of the adenoviral hexon protein. We used these polypeptides as antigens for generating polyclonal rabbit antibodies against human adenovirus 3 (HAdV-B3), mouse adenovirus 1 (MAdV-1) and MAdV-2 hexon. In Western immunoblots with lysates from cells infected from thirteen human and three mouse viruses, these antibodies bound to homologous full-length hexon protein and revealed variable levels of cross-reactivity to heterologous hexons. Results from immuno-fluorescence and electron microscopy studies indicated that HAdV-B3 and MAdV-2 hexon antibodies recognized native forms of hexon.
Conclusions
The procedure described here can in principle be applied to any adenovirus for which genome sequence information is available. It provides a basis for generating novel type-specific tools in diagnostics and research, and extends beyond the commonly used anti-viral antibodies raised against purified viruses or subviral components.
Similar content being viewed by others


Background
Adenoviruses (AdV) are ubiquitous pathogens, and affect vertebrates, including humans, livestock and wild animals. They undergo genetic recombination and periodically emerge in the human population, often in geographically distinct patterns [1]. Human adenoviruses (HAdV) are comprised of ~60 types classified into seven species, A to G based on serology and DNA genome sequence [2, 3]. The genetic information of adenoviruses is encoded in a double-stranded linear DNA molecule. It is imported into the cell nucleus upon stepwise disassembly of incoming virions [4,5,6,7]. Adenovirus vectors are widely used in clinical gene therapy, mostly in cancer treatment and vaccination boost regimes [8], in part due to strong innate immunity reactions due to viral danger signals [9, 10]. Since the viral genome remains episomal throughout the infection cycle [11], this gives rise to a robust, but transient viral gene expression. In addition, AdV vectors are widely used in clinical oncolytic applications due to the ease of genetic engineering and biochemical modifications [12,13,14,15].
Most humans have been exposed to adenoviruses, and raise neutralizing antibodies and protective cytotoxic T cell responses [16,17,18]. In immune-competent individuals, AdVs cause only mild or no symptoms, with no observable long-term effects on health [19]. Persistent infections can occur [20] which may be caused by interferon based anti-viral defense mechanisms [21]. Adenoviruses can be live threatening to immune-compromised individuals [21,22,23]. The high seroprevalence and immunogenicity of adenoviruses represents a considerable drawback when using these viruses as vectors for gene therapy, as pre-existing immunity leads to virus immune-complexes which can lead to inflammatory reactions [13] and reduced treatment efficacy.
The AdV capsid is icosahedral, non-enveloped and consists of three major structural proteins fiber, penton base and hexon [24]. The facets of the capsid are composed of 240 copies of the homotrimeric hexon protein, while 12 copies of the fiber protein are attached to pentameric penton protein bases on each of the vertices. Each hexon trimer has a pseudo-hexagonal base, which allows for close packaging within the facet, and three tower domains that are exposed on the exterior surface of the virion. All three major capsid proteins are immunogenic [18, 25,26,27,28]. Occurrence of neutralizing antibodies against fiber protein in naturally infected individuals has been reported [18, 25, 29, 30].
Importantly, the bulk of functionally significant neutralizing antibodies are directed against the hexon protein [31,32,33]. The serotype-specific residues recognized by these antibodies are located in seven hyper-variable regions (HVRs 1-7) protruding from the outer side of the viral capsid as deduced by comprehensive alignment of hexon sequences [34,35,36,37]. Experiments with hexon chimeric viruses, in which the complete hexon, single or combinations of HVRs were replaced by those of another serotype, revealed escape of humoral immune detection [31, 32, 38,39,40,41,42]. Purified trimeric native hexon inhibited neutralization, but monomeric heat-denatured hexon did not, suggesting that neutralizing epitopes are complex and conformational but not linear [27, 34]. In contrast to these findings, other groups reported generation of neutralizing antibodies using small HVR peptides only [28, 43]. Non-neutralizing and cross-reactive hexon antibodies with common species- and genus-specific reactivity have been suggested to recognize sites that are accessible on the purified hexon but partially masked in the intact virion and outside of the HVRs [27, 44]. Such antibodies were characterized using complement fixation, immunodiffusion, immunoprecipitation and immunoblot assays [45,46,47].
Mouse adenoviruses (MAdV) represent an attractive model for studying viral and host factors involved in acute and persistent infections, testing oncolytic vectors in syngenic tumor models, and the development of anti-viral drugs [48,49,50,51,52,53]. Like HAdVs they belong to the genus of Mastadenoviruses. Reagents such as antibodies against MAdV are scarce. Here we devised and tested the use of minimal hexon fragments containing the HVRs 1-6 for production of specific hexon antibodies. Recombinant proteins generated in E. coli included hexon fragments derived from MAdV-1, −2, and HAdV-B3, −C5. Rabbit antibodies raised against MAdV-1, −2, and HAdV-3 fragments were tested in immunoblot assays, immunofluorescence, neutralization assays and EM for binding to denatured and native hexon. All three hexon antibodies recognized full-length hexon in Western immunoblots of lysates from infected cells, and two of three antibodies recognized native virus protein in immunofluorescence and transmission electron microscopy, but not in neutralization assays.
Methods
Cells and viruses
All cell lines including the human lung carcinoma cell line A549, the human cervical carcinoma cell line HeLa Ohio, the mouse colon carcinoma CMT93 and 3T6 fibroblast cells were grown in DMEM plus 8% FCS [15, 54, 55]. The cell lines were routinely screened for the absence of mycoplasma contamination. Human HAdV-C5 wt300 was obtained from T. Shenk [56]. All other human prototype viruses were kindly provided by the late T. Adrian (Medizinische Hochschule Hannover, Germany) and were verified by DNA restriction analysis [57] and in part by hexon sequence analysis (R. Hendrickx et al., manuscript in preparation). All human wild type viruses were amplified in A549 cells and viral titers were determined by plaque assay using 911 cells as described previously [54]. Recombinant E1/E3-deleted HAd-B3 and HAdV-C5 vectors expressing firefly luciferase were described previously [58]. MAdV-1, −2 and −3 were kind gifts of K. Spindler (University of Michigan, Ann Arbor, USA), S. Compton (Yale University School of Medicine, USA) and D. Krüger (Charité Campus Mitte, Berlin Germany), respectively, and were amplified in CMT93 or 3T6 cells and titered by qPCR (R. Hendrickx et al., manuscript in preparation). The replication-competent HAdV-B3-pIX-FS2A-eGFP contains an eGFP open reading frame (ORF) genetically fused to the downstream end of the pIX gene using an autocleavage FS2A sequence ([51] and L. Studer manuscript in preparation). Similarly, the replication-competent MAdV-1-E1A-FS2A-GL contains a Gaussia luciferase (GL) ORF genetically fused to the downstream end of the E1A gene (R. Hendrickx, manuscript in preparation). The MAdV2-∆E1A-eGFP contains an E1A exon 1 deletion which was replaced by an eGFP cassette (R. Hendrickx, manuscript in preparation).
Construction of adenoviral hexon fragment expression vectors
For production of the four different hexon fragments the expression vector pGEX-6P-1 vector (GE Healthcare) was used. DNA fragments containing the hexon HVRs 1-6 were PCR-amplified using viral genomic DNA as template and inserted into the EcoRI and SalI cloning sites of pGEX-6P-1. The oligonucleotide sequences (Microsynth AG, Switzerland) used for cloning are listed in STab 1. Hexon HVRs 1-6 protein sequences, and relative molecular sizes of the GST-hexon and final hexon fragments are summarized in Additional file 1: Figure S1 and Additional file 2: Table S1.
Purification of adenoviral hexon protein fragments, production of rabbit polyclonal antibodies
Five grams of cell paste corresponding to a 4-l culture of E.coli BL21-CodonPlus(DE3)-RIPL cells, induced at 30 °C for 4 h with 0.3 mM IPTG, were resuspended in 25 ml of buffer T + 300 mM KCl (25 mM Tris-HCl, 10% glycerol, protease inhibitors (aprotinin, leupeptin, pepstatin A, PMSF), 0.5 mM EDTA, 1 mM dithiothreitol, 0.01% Nonidet-P40, pH 7.5). The cells were then disrupted by sonication and the lysate was clarified using ultracentrifugation (45 min, 4 °C, 100,000 × g).
The lysate was incubated with 1 ml of Glutathione-Sepharose beads (GE Healthcare) equilibrated with buffer T + 300 mM KCl for 1 h at 4 °C. The beads were then spun down and washed twice with 10 ml of T + 300 mM KCl and once with T + 50 mM KCl, followed by repeated protein elution with 1.5 ml of T + 100 mM KCl containing 30 mM glutathione. Fractions were analyzed by SDS-PAGE, pooled and treated with 100 units of PreScission Protease (GE Healthcare) for 22 h at 4 °C.
The solution was diluted with buffer 1xT until conductivity reached T + 50 mM KCl and was loaded on a 1 ml Mono Q column (GE Healthcare) equilibrated in T + 50 mM KCl. The protein was eluted using an 8 ml gradient of 50 – 500 mM KCl in buffer T.
Peak fractions were pooled, and loaded on a 5 ml Q5 Bio-Scale column (Bio-Rad) equilibrated in T + 50 mM KCl following adjustment of conductivity. The protein was eluted using a 100 ml gradient of 50 – 600 mM KCl in buffer 1xT. The elution times varied between different fragments but under these conditions, all of the fragments bound to the column, with HAdV-B3 eluting between 150 and 210 mM KCl, HAdV-C5 eluting between 270 and 350 mM KCl, MAdV-1 eluting between 90 and 130 mM KCl and MAdV-2 eluting between 60 and 110 mM KCl. Peak fractions were analyzed by SDS-PAGE, pooled and briefly incubated with a small amount of Glutathione-Sepharose beads to bind residual GST. The proteins were then concentrated using a 15 ml Vivaspin Turbo 15 concentration column with 10 kDa cut-off (Sartorius Stedim Biotech). All purification steps were performed at 4 °C and the final purified proteins were stored in buffer x at −80 °C until further use.
Purified hexon HVRs 1-6 fragments were used to immunize rabbits. Antibody development was carried out at BioGenes GmbH (Berlin, Germany). The animals were intramuscularly immunized using BioGenes’ adjuvant. The adjuvant was mixed 3:1 with the antigen. Final bleeds were done following four immunizations. The obtained sera were mixed with Thimerosal at 0.02% as preservative.
Neutralization assay
The in vitro neutralization assay was performed as described in [59]. In brief, 2.5 × 104 cells (3T6 for mouse viruses, A549 for human viruses) were seeded in 100 μl DMEM-FCS medium in 96 well plates and grown overnight at 37 °C. Serial five-fold dilutions or fixed dilutions of the hexon neutralizing antibody were prepared in 50 μl DMEM-FCS medium and mixed with 25 μl of either medium or increasing amounts of the hexon protein fragment. The mixes were incubated for 30 min at 37 °C. Subsequently, 25 μl of the reporter virus were added to the solutions and the mixes were incubated for another 30 min. Positive controls omitting the antibody and negative controls omitting the virus were included. The virus concentration was adjusted to a multiplicity of infection (MOI) of 5 in the case of the human viruses, whereas for MAdV-1-E1A-FS2A-GL 103 genome equivalent virus particles (geq vp) per cell were used. The solutions were subsequently added to the cells and incubated for 24 h at 37 °C. For measuring the luminescence signal, the medium was aspirated and replaced with 40 μl of SteadyGLO lysis / substrate (Promega). The plates were incubated for 10 min at room temperature with orbital shaking. Then, 20 μl from each well was transferred to a Greiner LumiTrac plate and analyzed using a Tecan Plate reader with luminescence unit. When using MAdV-1-E1A-FS2A-GL, the renilla luciferase assay system (Promega) was used containing the coelenterazin substrate. All tests were performed in triplicates and repeated twice. The dog HAdV-C5 neutralizing serum was kindly provided by Anja Ehrhardt [60, 61]. The hyperimmune rabbit anti-HAdV-B3 was a kind gift from A. Kajon (Lovelace Respiratory Program, Albuquerque, USA).
PAGE and western blot
For analysis of viral proteins derived from infected cells, cells were lysed in NETN (10 mM Tris pH 8.0, 200 mM NaCl, 1 mM EDTA, 0.5% NP40) complemented with protease inhibitors (Mini-Complete, Roche). Analyses of cleared cell lysates and hexon fragments from bacteria were performed by polyacrylamide gel electrophoresis [62] followed by Coomassie Blue or PageBlue™ (Thermo Scientific) staining, or by Western blotting of electrotransfered protein to Immobilon-P membranes as described previously [63]. Membranes were saturated in TBS-T containing 5% dry milk and incubated with primary antibodies including the rabbit anti-hexon fragment antibodies produced here, the rabbit anti-pIIIa antibodies (kindly provided by P. Hearing, School of Medicine, Stony Brook, USA) [64] (both 1:1000), and the mouse monoclonal anti-actin antibody (5A7 biorbyt UK), anti-GAPDH antibody (MA5-15738, Thermo Scientific), and anti-tubulin antibody (DM1A, Sigma) (all at 1 μg/ml). Incubation with secondary antibodies included HRP-conjugated donkey anti-rabbit IgG, sheep anti-mouse IgG (GE Healthcare, both 1:4000), and sheep anti-dog IgG (GE Healthcare) for 1 h. The immunoreactivity was determined using the Luminata Crescendo Western HRP substrate (Millipore) and scored using the ImageQuant LAS 4000 imager (GE Healthcare).
Immunofluorescence imaging
1 × 104 human A549 or mouse 3T6 cells were seeded in 50 μl of DMEM-FCS medium and were incubated at 37 °C for 3 h. The cells were then infected with virus using an MOI of 1 for HAdV-B3-pIX-FS2A-eGFP, or alternatively, 100 geq vp per cell for MAdV-1-ΔE1A-eGFP and MAdV-2-ΔE1A-eGFP. The replication-competent human virus gave rise to a strong GFP signals and cells were incubated for 1 day, whereas the mouse cells were incubated for 5 days, due to the weaker GFP signals produced by these viruses. The cells were fixed using 3% PFA for 15 min at room temperature. The cells were washed three times with PBS-N3 and quenched for 10 min at room temperature with PBS-N3 + 25 mM NH4Cl. The cells were washed again three times and blocked for 1 h at room temperature in PBS-N3 + 0.5% BSA. The cells were then incubated with primary antibodies diluted 1:500 in blocking buffer for one hour at 4 °C, washed three times and incubated with secondary antibodies for 1 h at room temperature. The cells were washed and 50 μl of PBS-N3 + DAPI was added to the wells at least 30 min prior to imaging. The cells were then imaged at a 20× magnification using a ImageXpress Micro XLS system.
Immunogold-labelled electron microscopy imaging
Staining and analyses by transmission microscopy (TEM) was performed as described earlier [65]. In brief, 10 μl of concentrated CsCl-purified adenovirus solutions were pipetted onto glow discharged TEM mesh grids. Virus was left to adsorb for 1-2 min, excess buffer was dried using a filter paper and grids were washed four times for 1 min in 10 μl of PBS with 10% goat serum. Grids were then incubated in 10 μl of primary antibody diluted 1:50 in PBS with 1% goat serum for 30 min at room temperature. This was followed by additional washing four times for 1 min in PBS with 10% goat serum and then incubation with 10 μl of 1:50 diluted secondary colloidal 10 nm gold-conjugated antibodies for 30 min at room temperature. Grids were washed twice in PBS with 0.1% BSA, three times in PBS and twice with deionized water for 1 min each. Virus was counterstained with 3% uranyl acetate for 20-40 s and the excess staining solution was drained with filter paper. Grids were allowed to air dry and were then imaged using a TEM FEI Tecnai G2 electron microscope. The 9C12 antibody included as primary antibody was developed by Laurence Fayadat and Wiebe Olijve, and was obtained from Developmental Studies Hybridoma Bank developed under the auspices of the National Institute of Child Health and Human Development and maintained by the University of Iowa, Department of Biology, Iowa City, IA.
Results
Design and production of adenoviral hexon fragments
In the past, either purified virus or purified virus proteins were used for immunization to generate hexon-specific antibodies [44, 66]. Since in our hands, purified MAdVs yields were lower compared to HAdVs and purification of MAdV hexon protein from lysates of infected cells was not efficient (not shown), we decided to produce recombinant hexon fragments containing HVRs to induce specific antibodies. To this end, hexon protein sequences from recently described MAdV-2 [48] and −3 [49] were aligned to MAdV-1 and other known hexon sequences (Fig. 1a and Additional file 1: Figure S1). All seven HVRs characteristic for the sequence diversity across different adenoviral sequences [34,35,36,37] coincided with the highest variable stretches in the MAdV sequences.

Alignment of partial adenovirus hexon sequences and hexon protein trimer structure. a Sequence alignment of 19 adenovirus hexon sequences containing HVRs 1-6. Protein sequences were obtained from GenBank and the alignment was performed using the MUSCLE algorithm. The color intensity indicates how highly an amino acid in a particular position is conserved amongst species. Highly conserved epitopes are highlighted in dark blue while variable epitopes are white or light blue. The HVRs 1-6 are highlighted by red bars and the cloned hexon fragments are highlighted by a blue bar. Hexon N- and C-terminal ends were omitted (for alignment of full-length hexon sequences see Additional file 1: Figure S1). b Graphical representation of the HAdV-C5 hexon protein trimer structure (PDB ID: 3TG7), side and top view. The HVRs 1-6 are highlighted in red and HVR 7 is highlighted in green
Based on the previously published crystal structure of the trimeric form of the hexon protein (PDB ID: 3TG7) we then mapped these regions to the structural model of the HAdV-C5 hexon (Fig. 1b). We next designed adenoviral hexon fragments of about 20 kDa mass containing the HVRs 1-6, plus small parts of conserved regions at each end of the fragment to maximize the chances of preserving the secondary structure (Fig. 1a, Additional file 1: Figure S1 and Additional file 2: Table S1). In order to exclude sequences possibly containing common species- or genus-specific reactivity, and due to molecular size constraints, the HVR 7 was not included in the designed fragment as there is a highly extended region between the HVR 6 and 7.
Four HVRs 1-6 hexon fragments were generated including those of HAdV-B3, HAdV-C5, MAdV-1 and MAdV-2. The HAdV-C5 fragment was included as control to check for blocking capacities of such fragments (see below). The HAdV-B3 fragment and production of HAdV-B3 hexon antibodies was included to characterize HAdV-B3-derived vectors (L. Studer manuscript in preparation and [67]). All fragments were expressed in E.coli BL21-CodonPlus(DE3)-RIPL cells as N-terminal GST-fusion proteins containing a PreScission protease cleavage site, that allowed efficient enzymatic removal of the tag.
A three-step purification procedure was employed to obtain highly pure hexon fragments (Fig. 2a). The first step was affinity purification using Glutathione-Sepharose beads. The GST-tagged protein bound to the affinity beads and could be separated from the majority of the contaminants. The GST-fusion hexon fragments as well as the final hexon fragments had a slightly slower mobility in SDS-PAGE than expected (Additional file 3: Figure S2A, Fig. 2b-e), which could be due to their relatively high negative charge (pI ~4-5) [68]. The eluted protein was treated with PreScission protease to cleave the GST-tag.

Purification of recombinant adenoviral hexon fragments. a Schematic representation of the purification process. Transformed E.coli BL21-CodonPlus(DE3)-RIPL cells were lysed to release cellular contents. The recombinant GST-fusion proteins were pulled down using Glutathione-Sepharose beads. After elution, the GST tag was cleaved and the hexon fragments were further purified using Mono Q and Q5 ion exchange chromatography. The resulting protein fractions were concentrated. b-e SDS-PAGE analyses of 1, 2 and 4 μl of purified HAdV-B3, HAdV-C5, MAdV-1 and MAdV-2 hexon fragments. Arrows indicate the bands of the purified proteins
The second purification step consisted of an ion exchange separation on a Mono Q column which allowed to separate the cleaved hexon fragment from the GST tag. Apart from the HAdV-5 hexon fragment, the untagged hexon protein fragments were found in the flow through fraction, while the majority of the GST bound to the column (Additional file 2: Figure S2B). For the third purification step the Mono Q peak fractions were pooled and loaded on a second ion exchange Q5 column. All of the four fragments bound to the column and, after applying a KCl gradient, the hexon fragments were eluted revealing variable elution conditions (Additional file 3: Figure S2C). The purified protein fractions were briefly incubated with a small amount of Glutathione-Sepharose beads to bind residual GST and concentrate the hexon fragments. SDS-PAGE analysis revealed highly pure ≥90% hexon protein preparations (Fig. 2b-e). Apart from the MAdV-2 fragment, all other hexon fragments migrated as a double band, either after Q5 elution or after concentration. Mass-spectrometry analysis confirmed the identity of both bands, although the cause of the differential migration remained unclear. Possible reasons for heterogeneity are degradation, oxidation or post-translational modifications. It is difficult to ascertain the effect of this second band on immunisation. We believe that even if the other band were a slightly shortened product, most of the HVR epitopes should still be present and yield specific antibodies after immunisation. In the animal, professional antigen presenting cells process complex antigens into small peptide fragments and present these fragments on their surface which gives rise to the expansion of T-cells and eventually B-cells, giving rise to a systemic humoral immune response, including the production of immunoglobulins, which are of particular interest in this study.
In summary, our three-step purification procedure starting from a 4-l bacterial culture gave rise to highly pure hexon fragments with overall protein yields ranging from 600 to 850 μg. This material was further used for functional analysis and immunization of rabbits to raise polyclonal antibodies.
The HAdV-C5 HVRs 1-6 hexon fragment blocks the neutralizing activity of a polyclonal antibody
We tested whether the hexon fragment was able to bind HAdV-C5 neutralizing anti-hexon antibodies. If this were the case, it could improve the production of native hexon-recognizing antibodies. We evaluated the blocking activity of one of the generated fragments, the HAdV-C5 hexon fragment in a neutralization assay using a dog anti-HAdV-C5 neutralizing serum [60, 61]. As expected, the dog anti-HAdV-C5 serum stained various viral proteins including a protein corresponding in size to hexon (Additional file 4: Figure S3). We first tested the neutralization capacity of the dog serum against HAdV-C5-CMVLuc. When HAdV-C5-CMVLuc was pre-incubated with five-fold dilutions of the dog sera, the dog sera potently blocked expression mediated by HAdV-C5-CMVLuc in human A549 cells (Fig. 3a). The same serum did not block HAdV-B3-CMVLuc-mediated expression (Fig. 3b), confirming the neutralization specificity of the dog serum. A reduced luciferase activity effect of the dog serum at the 1:50 dilution against HAdV-B3-CMVLuc was obtained, but it was considered to be unspecific due to cytopathic effects observed on the cells, which resulted in a lower luciferase signal due to the reduced cell count. A possible reason for the cell cytopathic effect includes the presence of Thimerosal added as preservative to the sera, amounting at the 1:50 dilution to concentrations that had been reported to be toxic for human and mouse cells [69].

HAdV-C5 HVRs 1-6 hexon fragment efficiently blocks the neutralizing activity of an anti HAdV-C5 antibody. a, b Titration of the HAdV-C5 neutralizing serum. Five-fold serial dilutions of the neutralizing serum were pre-incubated with either HAdV-C5-CMVLuc or HAdV-B3-CMVLuc for 30 min before adding to human A549 cells. Luciferase activity was determined as relative light unit signal (RLU) in lysed cells 24 h post infection. c-f Blocking of HAdV-C5 neutralizing activity by the HAdV-C5 HVRs 1-6 hexon fragment. Two neutralizing antibody dilutions, 1:250 and 1:1250, which gave rise to <0.075% and ~5% residual HAdV-C5 activity, respectively, were used to repeat the neutralization assays in the presence of increasing amounts of hexon fragment. Blocking of neutralization could be seen in the homologous system using HAdV-C5-CMVLuc, but not in the heterologous system for HAdV-B3-CMVLuc-mediated expression
To test if the HAdV-C5 HVRs 1-6 hexon fragment acted as an antibody decoy, we repeated the neutralization assays in the presence of the hexon fragment. We used two neutralizing antibody dilutions, 1:250 and 1:1250, which previously had shown to gave rise to <0.075% and ~ 5% residual HAdV-C5 infection, respectively (Fig. 3a). Addition of increasing amounts of the hexon fragment increased the luciferase signal of HAdV-C5-CMVLuc, but not of HAdV-B3-CMVLuc-mediated expression (Fig. 3c-f). At the highest concentration of the HAdV-C5 HVRs 1-6 hexon fragment, blocking of the neutralizing activity was almost complete for both serum dilutions, suggesting that the dog serum did contain mainly hexon directed neutralizing antibodies. In summary, these data indicated that the HAdV-C5 HVRs 1-6 hexon fragment existed in a quasi native structure that sequestered and blocked virus-neutralizing antibodies.
Hexon fragment antibodies reveal cross-reactivity and bind to full-length hexon protein in immunoblot assays
The purified HAdV-B3, MAdV-1 and MAdV-2 HVRs 1-6 hexon fragments were used to produce hyperimmune sera in rabbits. The resulting sera, hereafter referred to as HAdV-B3- / MAdV-1- / MAdV-2-hexon antibody (Ab), were tested in several assays to examine their properties.
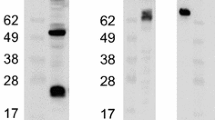
To test the specificity of the generated antibodies, we performed immunoblot assays using lysates from cells infected with thirteen human and three mouse viruses. Human HeLa cells were infected with HAdVs representing four species and included HAdV-B3, HAdV-B7, HAdV-B16 and HAdV-B21 of the B1 subspecies, HAdV-B11, HAdV-B14, HAdV-B34 and HAdV-B35 of the B2 subspecies, HAdV-D8, HAdV-D30 and HAdV-D37 of the D species, and HAdV-C5 and HAdV-E4 from the C and E species, respectively. Mouse CMT93 cells were infected with all three known MAdVs. In addition to the hexon antibodies, all blots were also immunostained using a HAdV cross-reactive anti-pIIIa antibody [64] to check for efficient infection, and an actin antibody to check for sample loading. pIIIa is a conserved structural protein contributing to capsid structure and stability. In our experiments, it serves as loading control and a marker for production of late transcribed genes, including hexon.
The HAdV-B3 hexon Ab strongly bound to the full-length HAdV-B3 hexon, and also cross-reacted strongly with most of the other hexons of the B1 and B2 subspecies viruses, and with the hexons of the D serotypes included here (Fig. 4a). Of note, weak signals of HAdV-B14 and B34 of B2 subspecies were paralleled by relative weak pIIIa signals, whereas pIIIa signals of all D serotypes were rather robust. Hexons from HAdV-C5 and HAdV-E4, as well as HAdV-B16 revealed weak binding and cross-reactivity.

Antibodies raised against hexon fragments reveal variable cross-reactivity against hexon proteins in Western immunoblotting. Lysates from HeLa cells infected with thirteen HAdVs representing the four species B, C, D and E and CMT93 lysates with all three different MAdVs were analyzed. Uninfected HeLa or CMT93 cells were included as negative controls, respectively. The blots were stained with the HAdV-B3 hexon Ab (a), the MAdV-1 hexon Ab (b), or the MAdV-2 hexon Ab (c). In addition, all blots were immunostained using a (HAdV) cross-reactive anti-pIIIa antibody to check for efficient infection, and an actin antibody for loading control. It was noticed that the actin signal in mouse cells infected with MAdVs strongly faded in the course of infection. d A second set of CMT93 cells infected with all three MAdVs at about five-fold higher virus input were harvested 48, 72 and 96 h post infection. The blot was immunostained using the MAdV-1 and MAdV-2 hexon Ab and GAPDH loading control antibody. The same lysates were stained for protein content using PageBlue. Marker proteins of 118, 85, 48, 34, 26 and 20 kDa were included in the first lane
When testing the MAdV-1 hexon Ab in a first experiment with mouse CMT93 lysates harvested 72 h post infection, it bound strongly to the MAdV-1 full-length hexon, and weakly to the MAdV-3 hexon, which are highly related in sequence [48] (Fig. 4b). In addition, the MAdV-1 hexon Ab exhibited low affinity towards the HAdV-B11 and HAdV-B35 hexon. The MAdV-2 hexon Ab displayed low cross-reactivity to MAdV-1 hexon, but strongly bound to the MAdV-2 full length hexon (Fig. 4c). It was also noticed that the cross-reactive anti-pIIIa antibody did not efficiently recognize pIIIa from MAdVs which could be explained by the low sequence similarity to human adenoviruses in certain segments of the protein [20]. Further, in contrast to cell lysates obtained from HAdV infections, the actin signal in mouse cells infected with MAdVs faded in the course of infection.
We repeated the infection experiment using an about 5-fold higher virus input followed by harvesting cell lysates 48, 72 and 96 h post infection. In addition to immunoblotting analyses, the cell lysates were also analyzed for protein levels by PageBlue staining (Fig. 4d). Surprisingly, all MAdV-infected lysates revealed a robust reduction in cell protein contents at all three time points analyzed, when compared to uninfected cells. When immunostaining for GAPDH, weak signals were seen for MAdV-1 and -3 only at 48 h post infection Staining for actin and tubulin showed similar results (not shown), in agreement with the reduced levels of cellular proteins. In this experiment with higher virus input, the MAdV-1 hexon Ab bound to similar extents to MAdV-1 and MAdV-3 full-length hexon proteins. The MAdV-2 hexon Ab displayed low cross-reactivity to MAdV-1 as well as to MAdV-3 hexon protein.
In summary, all three sera produced against hexon HVRs 1-6 fragments revealed binding to homologous full-length hexon protein demonstrating variable degrees of cross-reactivity to heterologous hexons when tested for binding in immunoblot assays representing mostly non-native forms of hexons.
HAdV-B3- and MAdV-2- but not MAdV-1-hexon fragment antibodies display hexon signals in IF analysis of infected cells and in immuno-EM of intact virions
Immunofluorescence (IF) microscopy is widely used to study the infectious life cycle of viruses. To investigate whether the hexon fragment antibodies were suitable for use in IF, human and mouse cells were infected with eGFP expressing reporter viruses followed by analysis for eGPF and hexon signals. When human A549 cells were infected with replication-competent HAdV-B3-pIX-FS2A-eGFP we were able to observe a strong hexon signal that overlapped with the GFP signals (Fig. 5a). Of note, uninfected cells revealed a weak background signal when using the HAdV-B3 hexon Ab. Similarly, when mouse 3T6 cells were infected with recombinant MAdV-2-∆E1A-eGFP, hexon and GFP signals coincided to a large degree, whereas uninfected cells revealed a very low background staining with the MAdV-2 hexon Ab (Fig. 5b). In contrast, when 3T6 cells infected with MAdV-1-∆E1A-eGFP virus were stained with the MAdV-1 hexon Ab, a high background was observed with signals that did not overlap with the GFP signals (Fig. 5c).

Use of hexon fragment antibodies in immunofluorescence analysis of GFP-reporter virus infected cells. a Human A549 cells were infected with replication-competent HAdV-3-pIX-FS2A-eGFP at an MOI of 1 and cells were fixed and stained one day post infection with the HAdV-B3 hexon Ab. Co-localization of a HAdV-B3 hexon specific signal with the GFP signal was observed in the overlays. b Mouse 3T6 cells were infected with 100 geq vp of MAdV-2-ΔE1A-eGFP and cells were fixed and stained five days post infection with the MAdV-2 hexon Ab. Co-localization of the MAdV-2 hexon signal with GFP was observed in channel overlays. c Mouse 3T6 cells were infected with 100 geq vp of MAdV-1-ΔE1A-eGFP and cells were fixed and stained five days post infection with the MAdV-1 hexon Ab. No co-localization of GFP and hexon signal was observed. DAPI staining is highlighted in blue, the GFP signal is highlighted in green and the hexon antibody signal is highlighted in red
These findings were corroborated when the same antibodies were used for staining of CsCl-purified viruses by high-resolution immunogold EM. For this, wild type HAdV-B3, HAdV-C5, MAdV-1 and -2 were first incubated with the three hexon-fragment antibodies or with the mouse monoclonal 9C12 antibody specifically binding to HAdV-C5 [59], followed by incubation with gold-labeled secondary antibodies (Fig. 6). When staining HAdV-C5 with the 9C12 antibody, we observed multiple gold grains on the HAdV-5 capsid surface. This staining was specific, since none of the other virus capsids were bound by the 9C12 antibody. Gold grains were also seen when HAdV-B3 was incubated with the HAdV-B3 hexon Ab, or MAdV-2 was incubated with the MAdV-2 hexon Ab, albeit at lower numbers than for 9C12. In contrast, in the case of MAdV-1 we were not able to detect immune-gold particles on the intact viral capsids. In summary, our IF and EM results suggest that HAdV-B3 and MAdV-2 hexon Abs recognized in addition to non-native also native forms of hexon, whereas the MAdV-1 hexon HVRs 1-6 antibodies recognized only non-native hexon forms.

Transmission electron microscope analysis of immunogold-labeled viral capsids. Purified HAdV-B3, HAdV-C5, MAdV-1 and MAdV-2 were first incubated with the indicated rabbit hexon antibodies for HAdV-B3, MAdV-1, MAdV-2, or the monoclonal mouse 9C12 antibody specific for HAdV-C5, respectively. Following staining with colloidal 10 nm gold-labeled anti-rabbit or anti-mouse antibodies, uranyl-acetate staining was performed. TEM images were acquired at 135 000× magnification
HAdV-B3- and MAdV-1-hexon Abs exhibit no neutralizing activity on virus infection
Since two of our hexon Abs recognized native hexon forms, we tested if they also had neutralizing activity. To test this we repeated the neutralizing assays using HAdV-B3-CMV-Luc and MAdV-1-E1A-FS2A-GL for infection of human and mouse cells, respectively. When compared to the pre-immune sera, neither the HAdV-B3 hexon Ab (Fig. 7a) nor the MAdV-1 hexon Ab (Fig. 7b) revealed any neutralizing activity. As seen previously with the dog anti-HAdV-C5 serum, a small cytopathic effect induced reduction of luciferase expression at the 1:50 dilution of both sera. When including as positive control a hyperimmune rabbit anti-HAdV-B3 serum, HAdV-B3-CMV-Luc-mediated luciferase expression was strongly blocked (Fig. 7c). The neutralizing activity of the MAdV-2 hexon Ab could not be tested here, due to the lack of a MAdV-2 luciferase reporter virus.

HAdV-B3 and MAdV-1 hexon Abs do not reveal neutralizing activity. a Five-fold serial dilutions of preimmune serum or HAdV-B3 hexon Ab were pre-incubated with HAdV-B3-CMVLuc for 30 min before adding to human A549 cells. b Five-fold serial dilutions of preimmune serum or MAdV-1 hexon Ab were pre-incubated with MAdV-1-E1A-FS2A-GL for 30 min before adding to mouse CMT93 cells. For both viruses, luciferase activity was measured as RLU in lysed cells 24 h post infection. c Comparison of the neutralizing activities of the HAdV-3 hexon Ab with a neutralizing rabbit anti HAdV-B3 serum. Dilutions and experimental set-up were as described above
Discussion
Standard procedures to generate adenovirus hexon-specific antibodies include preparations of purified virions, or viral proteins for the purpose of immunization [44, 66]. However, not all adenoviruses can be amplified efficiently in cell culture. For instance the two HAdVs belonging to species F (HAdV-F40 and −41) are notoriously difficult to grow to high titers [70]. When trying to isolate MAdV-1 and-2 hexon protein from infected cell cultures using standard protocols [71] we failed to obtain purified proteins. Since hexon is a large protein of 108 kDa that requires a viral chaperone activity [72], production in E.coli was found to be inefficient (unpublished data).
We decided to generate a small hexon subfragment consisting of about one fifth of the full-length size, but still containing six of the seven HVRs (Fig. 1a, Additional file 1: Figure S1 and Additional file 2: Table S1). HVR 7 was omitted since inclusion of this region increases the fragment size by about two-fold. In addition, neutralization profile analysis of hexon chimeric HAdV-C5 containing either HVRs 1-7 or only HVRs 1-6 of HAdV-C2 as well as epitope map** of chimpanzee Ad68 hexon using a panel of neutralizing antibodies had suggested that most reactivity was contained in the HVRs 1-6 sequence [27, 38]. Similar data were also reported with HAdV-B3 and -B7 [73, 59, 75].
Following efficient expression in bacteria, our three-step purification procedure of the hexon HVRs 1-6 fragments using standard biochemical methods resulted in >90% pure protein preparations based on SDS-PAGE analysis, with sufficient quantities for immunization and production of polyclonal antibodies in rabbits (Fig. 2). It was somewhat surprising to find that the HAdV-C5 fragment was almost completely blocking a dog-anti HAdV-C5 neutralizing activity although the fragment consisted only of HVRs 1-6 (Fig. 3). Hexon fragment proteins consisting of HVRs 1-7 have been used for similar blocking experiments [42], as well as a HVR 5-derived small peptide of HAdV-C5 [28]. However, blocking activity by peptides may be rather exceptional, since it has been estimated that only a small fraction of <10% of the antibodies directed against native antigens bind to linear epitopes [76]. In the case of HAdV-D48 hexon, neutralizing antibodies did not recognize linear peptides [34]. Whether all of the four hexon fragments are capable of blocking neutralizing antibodies remains to be determined once the recombinant reporter viruses are available.
Hexon antigenic determinants and antibodies raised against hexon will not only be influenced by the nature of antigen used for immunization, e.g., purified virus, or hexon or partial fragment, but also by the immunization procedure which can include use of alum precipitation, adjuvants, and terminal boost with virus only. Thus, generation of mouse monoclonal antibodies resulted in cross-reactive genus-, species- or type-specific antibodies [44, 66]. Type-specific and neutralizing epitopes have been suggested to be conformation dependent [27, 34], whereas non-neutralizing and cross-reactive hexon antibodies with common species- and genus-specific reactivity have been suggested to recognize sites that are accessible on the purified hexon but partially masked in the intact virion and outside of the HVRs [27, 44, 45].
A variable degree of cross-reactivity binding to heterologous hexons was observed when testing our three hexon Abs for binding in immunoblot assays, representing binding to mostly non-native forms of hexons. In the case of the HAdV-B3 hexon Ab, extensive cross-reactivity with most of the other tested hexons of the B1 and B2 subspecies viruses, and with the hexons of the D types were noticed (Fig. 4a). The observed cross-reactivity between species B and D is confirmed by phylogenetic tree analyses data of conserved and variable hexon regions, revealing that the B and D species viruses are more closely related than the B and C species viruses [77]. The reduced cross-reactivity to hexon of HAdV-B16 is in line with the previously noted clustering of the HAdV-B16 hexon sequence to the HAdV-E4 sequence, suggested to have resulted from a recombination between sequences of these viruses [77, 78]. Since the MAdVs clade is rather distant from the other Mastadenovirus clades [48], it is not surprising to find lower levels of cross-reactivity of the MAdV-1 hexon antibodies and almost no cross-reactivity with the MAdV-2 hexon antibodies (Fig. 4b, c). The MAdV-1 hexon directed antibodies bound stronger to the MAdV-3 hexon than to the MAdV-2 hexon, reflecting the closer ancestor relationship of MAdV-1 and -3 compared to MAdV-2 [48].
When used for detection of hexon proteins in IF of infected cells and for EM of purified virus, the HAdV-B3 and MAdV-2 hexon antibodies, but not the MAdV-1 antibody bound native hexon proteins (Figs. 5 and 6). However, when used for virus neutralizing assays, neither the HAdV-B3 nor the MAdV-1 antibodies revealed any blocking effect (Fig. 7), suggesting that the MAdV-1 antibody exclusively detects nonnative and denatured epitopes, whereas the HAdV-B3 antibody recognizes native hexon epitopes to some degree, but not those epitopes contained on trimeric hexons leading to neutralization.
Our results with the HAdV-B3 hexon antibody is in contrast to a study by Yuan et al. which generated HAdV-B3 neutralizing antibodies immunizing with various short HVR peptides bound to KLH carrier protein [43]. However, the Yuan et al. study did not include other virus serotypes to confirm the specificity. Previous studies have shown that not all hexon binding antibodies exert neutralizing activity, but those that do bind with high affinity and function by rapid, single-hit kinetics, acting at a post-entry and TRIM21-dependent step [18, 27, 79, 80]. Non-neutralizing antibodies were found to bind with lower avidity to native hexon than neutralizing antibodies [27], which could explain why two of our three hexon antibodies still bound to intact virus in EM analysis, despite their lack of neutralization.
Conclusions
The procedure described here can be applied to generate monoclonal or polyclonal antibodies against adenoviruses that are difficult to grow in cell culture. Such antibodies can greatly speed up the process of optimizing the cultivation and purification conditions of the virus, and enhance studies of the infection cycle and serve as diagnostic tools in tissue analyses. Purified hexon fragments can be used in a variety of biochemical assays apart from immunizing animals, for instance blocking neutralization assays, pull-down assays or competition assays. The method described here is cost effective and yields milligram quantities of recombinant soluble hexon protein. It extends traditional methods, and improves adenoviral research and diagnostics.
Abbreviations
- Ab:
-
Antibody
- AdV:
-
Adenoviruses
- geq vp:
-
Genome equivalent virus particles
- GL:
-
Gaussia luciferase
- HAdV:
-
Human adenoviruses
- HAdV-B3:
-
Human adenovirus 3
- HVRs:
-
Hyper-variable regions
- IF:
-
Immunofluorescence
- MAdV:
-
Mouse adenoviruses
- MOI:
-
Multiplicity of infection
- ORF:
-
Open reading frame
- RLU:
-
Relative light unit signal
- TEM:
-
Transmission microscopy
References
Lynch JP 3rd, Fishbein M, Echavarria M. Adenovirus. Semin Respir Crit Care Med. 2011;32:494–511.
Aoki K, Benko M, Davison AJ, Echavarria M, Erdman DD, Harrach B, Kajon AE, Schnurr D, Wadell G, Members of the Adenovirus Research C. Toward an integrated human adenovirus designation system that utilizes molecular and serological data and serves both clinical and fundamental virology. J Virol. 2011;85:5703–4.
Seto D, Chodosh J, Brister JR, Jones MS, Members of the Adenovirus Research C. Using the whole-genome sequence to characterize and name human adenoviruses. J Virol. 2011;85:5701–2.
Greber UF, Arnberg N, Wadell G, Benko M, Kremer EJ. Adenoviruses - from pathogens to therapeutics: a report on the 10th International Adenovirus Meeting. Cell Microbiol. 2013;15:16–23.
Greber UF. Virus and host mechanics support membrane penetration and cell entry. J Virol. 2016;90:3802–5.
Suomalainen M, Greber UF. Uncoating of non-enveloped viruses. Curr Opin Virol. 2013;3:27–33.
Wolfrum N, Greber UF. Adenovirus signalling in entry. Cell Microbiol. 2013;15:53–62.
Lopez-Gordo E, Podgorski II, Downes N, Alemany R. Circumventing antivector immunity: potential use of nonhuman adenoviral vectors. Hum Gene Ther. 2014;25:285–300.
Flatt JW, Greber UF. Misdelivery at the nuclear pore complex-stop** a virus dead in its tracks. Cell. 2015;4:277–96.
Hendrickx R, Stichling N, Koelen J, Kuryk L, Lipiec A, Greber UF. Innate immunity to adenovirus. Hum Gene Ther. 2014;25:265–84.
Wang IH, Suomalainen M, Andriasyan V, Kilcher S, Mercer J, Neef A, Luedtke NW, Greber UF. Tracking viral genomes in host cells at single-molecule resolution. Cell Host Microbe. 2013;14:468–80.
Kasala D, Choi JW, Kim SW, Yun CO. Utilizing adenovirus vectors for gene delivery in cancer. Expert Opin Drug Deliv. 2014;11:379–92.
Khare R, Chen CY, Weaver EA, Barry MA. Advances and future challenges in adenoviral vector pharmacology and targeting. Curr Gene Ther. 2011;11:241–58.
Luisoni S, Greber UF. Biology of adenovirus cell entry – receptors, pathways, mechanisms. In: Curiel DT, editor. Adenoviral vectors for gene therapy. London: Academic Press, Elsevier; 2016. p. 27–58.
Nagel H, Maag S, Tassis A, Nestle FO, Greber UF, Hemmi S. The alphavbeta5 integrin of hematopoietic and nonhematopoietic cells is a transduction receptor of RGD-4C fiber-modified adenoviruses. Gene Ther. 2003;10:1643–53.
Flomenberg P, Piaskowski V, Truitt RL, Casper JT. Characterization of human proliferative T cell responses to adenovirus. J Infect Dis. 1995;171:1090–6.
Flomenberg P, Piaskowski V, Truitt RL, Casper JT. Human adenovirus-specific CD8+ T-cell responses are not inhibited by E3-19K in the presence of gamma interferon. J Virol. 1996;70:6314–22.
Wohlfart C. Neutralization of adenoviruses: kinetics, stoichiometry, and mechanisms. J Virol. 1988;62:2321–8.
Wold WS, Ison MG. Adenoviruses. In: Knipe DM, Howley PM, editors. Fields virology, vol. 2. Philadelphia, New York: Wolters Kluwer, Lippincott Williams & Wilkins; 2013. p. 1732–67.
Garnett CT, Talekar G, Mahr JA, Huang W, Zhang Y, Ornelles DA, Gooding LR. Latent species C adenoviruses in human tonsil tissues. J Virol. 2009;83:2417–28.
Zheng Y, Stamminger T, Hearing P. E2F/Rb family proteins mediate interferon induced repression of adenovirus immediate early transcription to promote persistent viral infection. PLoS Pathog. 2016;12:e1005415.
Kosulin K, Geiger E, Vecsei A, Huber WD, Rauch M, Brenner E, Wrba F, Hammer K, Innerhofer A, Potschger U, et al. Persistence and reactivation of human adenoviruses in the gastrointestinal tract. Clin Microbiol Infect. 2016;22:381 e381–8.
Lion T. Adenovirus infections in immunocompetent and immunocompromised patients. Clin Microbiol Rev. 2014;27:441–62.
Nemerow GR, Stewart PL, Reddy VS. Structure of human adenovirus. Curr Opin Virol. 2012;2:115–21.
Gahery-Segard H, Farace F, Godfrin D, Gaston J, Lengagne R, Tursz T, Boulanger P, Guillet JG. Immune response to recombinant capsid proteins of adenovirus in humans: antifiber and anti-penton base antibodies have a synergistic effect on neutralizing activity. J Virol. 1998;72:2388–97.
Hong SS, Magnusson MK, Henning P, Lindholm L, Boulanger PA. Adenovirus strip**: a versatile method to generate adenovirus vectors with new cell target specificity. Mol Ther. 2003;7:692–9.
Pichla-Gollon SL, Drinker M, Zhou X, Xue F, Rux JJ, Gao GP, Wilson JM, Ertl HC, Burnett RM, Bergelson JM. Structure-based identification of a major neutralizing site in an adenovirus hexon. J Virol. 2007;81:1680–9.
Toogood CI, Crompton J, Hay RT. Antipeptide antisera define neutralizing epitopes on the adenovirus hexon. J Gen Virol. 1992;73(Pt 6):1429–35.
Bradley RR, Lynch DM, Iampietro MJ, Borducchi EN, Barouch DH. Adenovirus serotype 5 neutralizing antibodies target both hexon and fiber following vaccination and natural infection. J Virol. 2012;86:625–9.
Cheng C, Gall JG, Nason M, King CR, Koup RA, Roederer M, McElrath MJ, Morgan CA, Churchyard G, Baden LR, et al. Differential specificity and immunogenicity of adenovirus type 5 neutralizing antibodies elicited by natural infection or immunization. J Virol. 2010;84:630–8.
Roy S, Clawson DS, Calcedo R, Lebherz C, Sanmiguel J, Wu D, Wilson JM. Use of chimeric adenoviral vectors to assess capsid neutralization determinants. Virology. 2005;333:207–14.
Roberts DM, Nanda A, Havenga MJ, Abbink P, Lynch DM, Ewald BA, Liu J, Thorner AR, Swanson PE, Gorgone DA, et al. Hexon-chimaeric adenovirus serotype 5 vectors circumvent pre-existing anti-vector immunity. Nature. 2006;441:239–43.
Sumida SM, Truitt DM, Lemckert AA, Vogels R, Custers JH, Addo MM, Lockman S, Peter T, Peyerl FW, Kishko MG, et al. Neutralizing antibodies to adenovirus serotype 5 vaccine vectors are directed primarily against the adenovirus hexon protein. J Immunol. 2005;174:7179–85.
Crawford-Miksza L, Schnurr DP. Analysis of 15 adenovirus hexon proteins reveals the location and structure of seven hypervariable regions containing serotype-specific residues. J Virol. 1996;70:1836–44.
Rux JJ, Burnett RM. Type-specific epitope locations revealed by X-ray crystallographic study of adenovirus type 5 hexon. Mol Ther. 2000;1:18–30.
Rux JJ, Kuser PR, Burnett RM. Structural and phylogenetic analysis of adenovirus hexons by use of high-resolution x-ray crystallographic, molecular modeling, and sequence-based methods. J Virol. 2003;77:9553–66.
Takeuchi S, Itoh N, Uchio E, Aoki K, Ohno S. Seroty** of adenoviruses on conjunctival scra**s by PCR and sequence analysis. J Clin Microbiol. 1999;37:1839–45.
Gall JG, Crystal RG, Falck-Pedersen E. Construction and characterization of hexon-chimeric adenoviruses: specification of adenovirus serotype. J Virol. 1998;72:10260–4.
Tian X, Su X, Li H, Li X, Zhou Z, Liu W, Zhou R. Construction and characterization of human adenovirus serotype 3 packaged by serotype 7 hexon. Virus Res. 2011;160:214–20.
Wu H, Dmitriev I, Kashentseva E, Seki T, Wang M, Curiel DT. Construction and characterization of adenovirus serotype 5 packaged by serotype 3 hexon. J Virol. 2002;76:12775–82.
Youil R, Toner TJ, Su Q, Chen M, Tang A, Bett AJ, Casimiro D. Hexon gene switch strategy for the generation of chimeric recombinant adenovirus. Hum Gene Ther. 2002;13:311–20.
Yu B, Wang C, Dong J, Zhang M, Zhang H, Wu J, Wu Y, Kong W, Yu X. Chimeric hexon HVRs protein reflects partial function of adenovirus. Biochem Biophys Res Commun. 2012;421:170–6.
Yuan X, Qu Z, Wu X, Wang Y, Liu L, Wei F, Gao H, Shang L, Zhang H, Cui H, et al. Molecular modeling and epitopes map** of human adenovirus type 3 hexon protein. Vaccine. 2009;27:5103–10.
Adam E, Lengyel A, Takacs M, Erdei J, Fachet J, Nasz I. Grou** of monoclonal antibodies to adenovirus hexons by their cross-reactivity. Arch Virol. 1986;87:61–71.
Bauer U, Flunker G, Bruss K, Kallwellis K, Liebermann H, Luettich T, Motz M, Seidel W. Detection of antibodies against adenovirus protein IX, fiber, and hexon in human sera by immunoblot assay. J Clin Microbiol. 2005;43:4426–33.
Willcox N, Mautner V. Antigenic determinants of adenovirus capsids. I. Measurement of antibody cross-reactivity. J Immunol. 1976;116:19–24.
Willcox N, Mautner V. Antigenic determinants of adenovirus capsids. II. Homogeneity of hexons, and accessibility of their determinants, in the virion. J Immunol. 1976;116:25–9.
Hemmi S, Vidovszky MZ, Ruminska J, Ramelli S, Decurtins W, Greber UF, Harrach B. Genomic and phylogenetic analyses of murine adenovirus 2. Virus Res. 2011;160:128–35.
Klempa B, Kruger DH, Auste B, Stanko M, Krawczyk A, Nickel KF, Uberla K, Stang A. A novel cardiotropic murine adenovirus representing a distinct species of mastadenoviruses. J Virol. 2009;83:5749–59.
Lenaerts L, Kelchtermans H, Geboes L, Matthys P, Verbeken E, De Clercq E, Naesens L. Recovery of humoral immunity is critical for successful antiviral therapy in disseminated mouse adenovirus type 1 infection. Antimicrob Agents Chemother. 2008;52:1462–71.
Robinson M, Li B, Ge Y, Ko D, Yendluri S, Harding T, VanRoey M, Spindler KR, Jooss K. Novel immunocompetent murine tumor model for evaluation of conditionally replication-competent (oncolytic) murine adenoviral vectors. J Virol. 2009;83:3450–62.
Smith K, Spindler K. Murine adenoviruses. New York: John Wiley and Sons Ltd; 1999.
Weinberg JB, Stempfle GS, Wilkinson JE, Younger JG, Spindler KR. Acute respiratory infection with mouse adenovirus type 1. Virology. 2005;340:245–54.
Hemmi S, Geertsen R, Mezzacasa A, Peter I, Dummer R. The presence of human coxsackievirus and adenovirus receptor is associated with efficient adenovirus-mediated transgene expression in human melanoma cell cultures. Hum Gene Ther. 1998;9:2363–73.
Peter I, Graf C, Dummer R, Schaffner W, Greber UF, Hemmi S. A novel attenuated replication-competent adenovirus for melanoma therapy. Gene Ther. 2003;10:530–9.
Hearing P, Shenk T. The adenovirus type 5 E1A transcriptional control region contains a duplicated enhancer element. Cell. 1983;33:695–703.
Adrian T, Wadell G, Hierholzer JC, Wigand R. DNA restriction analysis of adenovirus prototypes 1 to 41. Arch Virol. 1986;91:277–90.
Fleischli C, Sirena D, Lesage G, Havenga MJ, Cattaneo R, Greber UF, Hemmi S. Species B adenovirus serotypes 3, 7, 11 and 35 share similar binding sites on the membrane cofactor protein CD46 receptor. J Gen Virol. 2007;88:2925–34.
Varghese R, Mikyas Y, Stewart PL, Ralston R. Postentry neutralization of adenovirus type 5 by an antihexon antibody. J Virol. 2004;78:12320–32.
Hausl MA, Zhang W, Muther N, Rauschhuber C, Franck HG, Merricks EP, Nichols TC, Kay MA, Ehrhardt A. Hyperactive slee** beauty transposase enables persistent phenotypic correction in mice and a canine model for hemophilia B. Mol Ther. 2010;18:1896–906.
Yakimovich A, Gumpert H, Burckhardt CJ, Lutschg VA, Jurgeit A, Sbalzarini IF, Greber UF. Cell-free transmission of human adenovirus by passive mass transfer in cell culture simulated in a computer model. J Virol. 2012;86:10123–37.
Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–5.
Ebbinghaus C, Al-Jaibaji A, Operschall E, Schoffel A, Peter I, Greber UF, Hemmi S. Functional and selective targeting of adenovirus to high-affinity Fcgamma receptor I-positive cells by using a bispecific hybrid adapter. J Virol. 2001;75:480–9.
Ma HC, Hearing P. Adenovirus structural protein IIIa is involved in the serotype specificity of viral DNA packaging. J Virol. 2011;85:7849–55.
Meier O, Gastaldelli M, Boucke K, Hemmi S, Greber UF. Early steps of clathrin-mediated endocytosis involved in phagosomal escape of Fcgamma receptor-targeted adenovirus. J Virol. 2005;79:2604–13.
Russell WC, Patel G, Precious B, Sharp I, Gardner PS. Monoclonal antibodies against adenovirus type 5: preparation and preliminary characterization. J Gen Virol. 1981;56:393–408.
Hemminki O, Bauerschmitz G, Hemmi S, Lavilla-Alonso S, Diaconu I, Guse K, Koski A, Desmond RA, Lappalainen M, Kanerva A, et al. Oncolytic adenovirus based on serotype 3. Cancer Gene Ther. 2011;18:288–96.
Guan Y, Zhu Q, Huang D, Zhao S, Jan Lo L, Peng J. An equation to estimate the difference between theoretically predicted and SDS PAGE-displayed molecular weights for an acidic peptide. Sci Rep. 2015;5:13370.
Park EK, Mak SK, Kultz D, Hammock BD. Evaluation of cytotoxicity attributed to thimerosal on murine and human kidney cells. J Toxicol Environ Health A. 2007;70:2092–5.
Sherwood V, Burgert HG, Chen YH, Sanghera S, Katafigiotis S, Randall RE, Connerton I, Mellits KH. Improved growth of enteric adenovirus type 40 in a modified cell line that can no longer respond to interferon stimulation. J Gen Virol. 2007;88:71–6.
Boulanger PA, Puvion F. Large-scale preparation of soluble adenovirus hexon, penton and fiber antigens in highly purified form. Eur J Biochem. 1973;39:37–42.
Szolajska E, Chroboczek J. Faithful chaperones. Cell Mol Life Sci. 2011;68:3307–22.
Liu M, Tian X, Li X, Zhou Z, Li C, Zhou R. Generation of neutralizing monoclonal antibodies against a conformational epitope of human adenovirus type 7 (HAdv-7) incorporated in capsid encoded in a HAdv-3-based vector. PLoS One. 2014;9:e103058.
Qiu H, Li X, Tian X, Zhou Z, **ng K, Li H, Tang N, Liu W, Bai P, Zhou R. Serotype-specific neutralizing antibody epitopes of human adenovirus type 3 (HAdV-3) and HAdV-7 reside in multiple hexon hypervariable regions. J Virol. 2012;86:7964–75.
Myers ND, Skorohodova KV, Gounder AP, Smith JG. Directed evolution of mutator adenoviruses resistant to antibody neutralization. J Virol. 2013;87:6047–50.
Chiron Mimotopes Pty. L. Determination of antibody binding parameters using biotinylated peptides. Pinnacles. 1993;3:7–11.
Ebner K, Pinsker W, Lion T. Comparative sequence analysis of the hexon gene in the entire spectrum of human adenovirus serotypes: phylogenetic, taxonomic, and clinical implications. J Virol. 2005;79:12635–42.
Madisch I, Harste G, Pommer H, Heim A. Phylogenetic analysis of the main neutralization and hemagglutination determinants of all human adenovirus prototypes as a basis for molecular classification and taxonomy. J Virol. 2005;79:15265–76.
McEwan WA, Hauler F, Williams CR, Bidgood SR, Mallery DL, Crowther RA, James LC. Regulation of virus neutralization and the persistent fraction by TRIM21. J Virol. 2012;86:8482–91.
Smith JG, Cassany A, Gerace L, Ralston R, Nemerow GR. Neutralizing antibody blocks adenovirus infection by arresting microtubule-dependent cytoplasmic transport. J Virol. 2008;82:6492–500.
Acknowledgements
We thank Maarit Suomalainen for careful reading of the manuscript and Anja Ehrhardt (Ludwig Maximilians University of Munich, Germany) and A. Kajon (Lovelace Respiratory Program, Albuquerque, USA) for their generous gifts of neutralizing anti-HAdV-C5 and anti-HAdV-B3 antisera, respectively and P. Hearing (School of Medicine, Stony Brook, USA) for the rabbit anti-pIIIa serum. We also thank the Functional Genomics Centre Zurich for performing mass-spectrometry analysis and the Centre for Microscopy and Image Analysis, UZH for providing assistance with electron microscopy analysis.
Funding
This work was financially supported by an Initial Training Network grant “ADVance” from the European Union supporting RH, UFG and SH (to UFG and SH and other principal investigators of ADVance, coordinated by Dr. A. Baker, University of Glasgow, United Kingdom), by SNF grant number 31003A_146286 (SH) and by the University of Zürich (UFG, SH). MB was supported by the Novartis foundation for medical-biological research (grant 16C222). The funders of this work had no impact on the design or the interpretation of the study.
Availability of data and materials
The datasets used and/or analyzed during the current study and newly generated reagents are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
MP, UG and SH designed the study and wrote the manuscript. MP developed the methodology, purified proteins, carried out Western blot assays, blocking assays and prepared EM samples. RH performed the IF studies and neutralization assays in Fig. 7. MB performed Western blot assays in Fig. 4. JF prepared EM samples and acquired EM images. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1: Figure S1.
Protein sequence alignment of 19 full-length adenoviral hexon proteins. Protein sequences were obtained from GenBank and the alignment was performed using the MUSCLE algorithm. The HVRs1-6 are highlighted by red bars, HVR7 by a green bar and the cloned hexon fragment is highlighted by a blue bar. (PDF 795 kb)
Additional file 2: Table S1.
Hexon fragment data and oligonucleotides used for generation of expression constructs. (DOC 45 kb)
Additional file 3: Figure S2.
Adenoviral HVRs 1-6 hexon fragment purification. SDS-PAGE analyses of HVRs1-6 hexon fragments of HAdV-B3 HAdV-C5, MAdV-1 and -2 were performed after GSH affinity purification (A), Mono Q ion-exchange purification (B), and Q5 ion-exchange purification (C). The Flow fraction represents proteins that did not bind to the chromatography matrix, the Wash fraction represents proteins washed out during the washing step of the purification, and the Beads fraction represents proteins that were eluted from the matrix by SDS boiling. Elution from the Q5 ion-exchange column was performed applying a slow KCl gradient of 50-600 mM. The hexon fragments were eluted at different KCl concentrations, including 150-210 mM KCl for the HAdV-B3 fragment, 270-350 mM KCl for the HAdV-C5 fragment, 90-130 mM KCl for the MAdV-1 ragment and 60-110 mM KCl for the MAdV-2 fragment. (PDF 2150 kb)
Additional file 4: Figure S3.
Immunostaining of hexon by the neutralizing dog anti-HAdV-C5 serum. Lysates from uninfected and infected Hela cells were analyzed by Western immunoblot using the polyclonal dog anti-HAdV-C5 serum. Several viral proteins including a protein corresponding in relative size to hexon (108 kDa) were detected. (PDF 346 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Pacesa, M., Hendrickx, R., Bieri, M. et al. Small-size recombinant adenoviral hexon protein fragments for the production of virus-type specific antibodies. Virol J 14, 158 (2017). https://doi.org/10.1186/s12985-017-0822-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12985-017-0822-5