
Abstract
Acute myocardial infarction (AMI) is a serious condition that occurs when part of the heart is subjected to ischemia episodes, following partial or complete occlusion of the epicardial coronary arteries. The resulting damage to heart muscle cells have a significant impact on patient’s health and quality of life. About that, recent research focused on the role of the sarcoplasmic reticulum (SR) and mitochondria in the physiopathology of AMI. Moreover, SR and mitochondria get in touch each other through multiple membrane contact sites giving rise to the subcellular region called mitochondria-associated membranes (MAMs). MAMs are essential for, but not limited to, bioenergetics and cell fate. Disruption of the architecture of these regions occurs during AMI although it is still unclear the cause-consequence connection and a complete overview of the pathological changes; for sure this concurs to further damage to heart muscle. The calcium ion (Ca2+) plays a pivotal role in the pathophysiology of AMI and its dynamic signaling between the SR and mitochondria holds significant importance. In this review, we tried to summarize and update the knowledge about the roles of these organelles in AMI from a Ca2+ signaling point of view. Accordingly, we also reported some possible cardioprotective targets which are directly or indirectly related at limiting the dysfunctions caused by the deregulation of the Ca2+ signaling.
Similar content being viewed by others

Introduction
Acute myocardial infarction (AMI) represents a consequence of ischemic heart diseases (IHD), mainly caused by the partial or the complete blockage of the epicardial coronary arteries leading to the death of myocardial cells. These cells are replaced by fibrotic tissue, resulting in a loss of cardiac contractility. AMI accounts for over 33% of mortality associated with IHD [1]. It can be classified into two types: ST-segment elevation myocardial infarction (STEMI) and non-STEMI (NSTEMI), based on the electrocardiographic presentation during diagnosis. The severity of ST-segment changes is influenced by the location of the myocardial region affected by acute ischemia [2, 3]. Patients with STEMI are at higher risk of mortality due to complete thrombotic occlusion of the vessel, transmural infarction, and the absence of collateral circulation [4]. On the other hand, patients with non-STEMI experience reduced blood flow in the affected coronary artery without complete occlusion, but they face higher long-term mortality risk due to the potential occurrence of multivessel coronary artery disease [5]. Throughout this review, we will focus mainly on STEMI cases because the total occlusion of the vessel greatly impacts on the biochemical pathways of interest, highlighting them. The ischemic episode is primarily caused by plaque rupture, leading to the formation of blood clots that reduce microcirculatory perfusion and limit blood flow to the heart. This imbalance between oxygen supply and demand accounts for about 70% of fatal events [6]. Then, reperfusion strategies, such as pharmacological approaches or percutaneous coronary intervention (PCI) and coronary artery bypass, have become the gold standard for treating AMI [6].
Early reperfusion has been demonstrated to limit ischemic cell death, but it can also result in cellular alterations and further tissue damage. The resulting damage can be categorized as reversible or irreversible. Reversible damage allows cardiac myocytes in the affected area to survive ischemia, and early reperfusion facilitates the recovery of cellular function. However, irreversible damage causes complete loss of resilience in myocytes, leading to cardiac tissue death. Irreversible damage is characterized by various changes in cardiomyocyte structure, including cell swelling, denaturation of intracellular proteins, and cell calcification [7]. During hypoxia, cardiomyocytes undergo anaerobic metabolism, leading to ATP production deficiencies, ion imbalances, and intracellular acidosis. Upon blood flow restoration, rapid reoxygenation of cardiac tissue restores ATP production and pH levels but also leads to the hyperproduction of reactive oxygen species (ROS) and intracellular calcium (Ca2+) overload, ultimately causing cardiomyocyte death and systolic dysfunction [8]. Arrhythmogenesis, whose onset is very common after ischemia and at reperfusion time, represents one of these dysfunctions. Here, mitochondria might have a central role through the control of the metabolism-excitation route, despite it is not yet fully clarified. Indeed, some mitochondrial-localized channels (i.e., inner membrane anion channel) when inhibited help in maintaining the mitochondrial membrane potential (ΨM) and action potentials during stress. Moreover, also Ca2+ and (mitochondrial ROS) appear to be determinant, by promoting mitochondrial dysfunctions dependent from a vicious cycle of Ca2+ overload and mitoROS production, consequent Ryanodine receptor 2 (RyR2) oxidation and Ca2+ leak from the sarcoplasmic reticulum (SR) [9].
As a messenger of cell death, but also stimulator of ATP synthesis and cardiac contraction [10], Ca2+ is sensed and captured by mitochondria from different intracellular stores (i.e., SR) and the extracellular milieu (Fig. 1). The events associated with ischemia reperfusion (I/R) have a strong impact on Ca2+ homeostasis, causing a variation in the expression and functionality of the proteins that bind and transport Ca2+; therefore, these changes in the intracellular Ca2+ fluxes can influence cell fate.

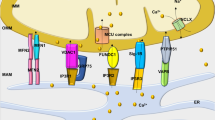
Representation of the Ca2+ signaling between mitochondria and sarcoplasmic reticulum in healthy condition and after IRI
This figure illustrates the dynamics of calcium (Ca2+) signaling between the sarcoplasmic reticulum (SR) and mitochondria associated membranes (MAMs) under normal physiological conditions and following ischemia-reperfusion injury (IRI). In healthy conditions, Ca2+ ions are released from the SR via channels such as the inositol 1,4,5-triphosphate receptor type 3 (IP3R3), inositol 1,4,5-triphosphate receptor type 1 (IP3R1), and ryanodine receptors (RyR2). These Ca2+ ions can then enter the mitochondria through the voltage-dependent anion-selective channel (VDAC) and the mitochondrial calcium uniporter (MCU). Following IRI, alterations in Ca2+ handling occur, affecting the balance between the SR and mitochondria. This dysregulation may involve changes in the activity of the Na+/ Ca2+ exchanger (NCX), plasma membrane Ca2+ ATPase, and sarcoplasmic reticulum Ca2+ ATPases (SERCA), as well as modulation of the phosphorylation status of phospholamban (PLB). Disruption of Ca2+ homeostasis during IRI can lead to mitochondrial dysfunction such as excessive reactive oxygen species (ROS) production, mPTP opening, impairment of ATP production and cell death
In this review, we will summarize the impairments in charge of mitochondria, SR and their contact sites named as mitochondria associated membranes (MAMs), that could be clinically relevant in cardiac I/R. We will highlight biochemical events regarding Ca2+ signaling dysregulation as contributor of cardiac damage, and whether they can be successfully targeted to reduce reperfusion injury. Despite advancements, patients with AMI continue to face substantial risks of mortality and morbidity. Novel treatments are imperative to shield the myocardium from the adverse impacts of AMI and reperfusion injury, aiming to minimize infarct size (IS), to sustain cardiac function, and to forestall heart failure onset [11, 12].
The importance of sarcoplasmic reticulum and mitochondria for Ca2+ homeostasis in acute myocardial infarction
The balance of intracellular Ca2+ at any level is crucial for normal cardiomyocyte function, from embryos to adulthood [13]. One of the main functions of the sarcoplasmic reticulum (SR) is to finely regulate the accumulation and the release of this ion close to the subsarcolemmal space, playing a key role in excitation-contraction coupling (ECC) in the heart. The importance of Ca2+ handling at SR and T tubules (TT) interface in ECC is widely reviewed elsewhere and it is already a reliable molecular target for cardioprotective drugs. What is almost unknown to date is the contribution of the dysregulation of Ca2+ signaling at the SR-mitochondria interface in ECC. Indeed, whether this pattern is essential for cardiac excitation-metabolism coupling, still few evidence connects mitochondria Ca2+ buffering to heart contraction. A seminal paper by Rizzuto’s group, in 2012 provided the first pioneer proof that mitochondria are able to taken up a significant fraction of Ca2+ released during systole and then released back into the cytosol during diastole [14]; this happens through the mitochondrial calcium uniporter (MCU) complex, a channel of the inner mitochondrial membrane (IMM). Moreover, the MCU-mediated Ca2+ uptake into mitochondria is of crucial importance for the direct ATP synthesis [15] and for the activation of dehydrogenases that feed electrons into the respiratory chain. Of note, MCU is essential for the control of “fight or flight” mechanisms, by regulating the Ca2+-mediated heart rate increase in cardiac pacemaker cells during stress [16]. Further investigations encompassing these noteworthy findings have been poor in the pathological field perhaps leaving the real role of the mitochondrion in ECC unknown. As mitochondria receive Ca2+ from SR, cytosol and from microdomains that are formed at MAMs, essential is the evaluation of what changes in correspondence of this route during AMI.
The proximity between mitochondria and SR facilitates reciprocal exchange between organelles, enabling tight integration between ATP generation by mitochondria and Ca2+ release mediated by Ca2+ channels in SR membrane, meeting the energy demands of the myocyte [17]. The activity of various mitochondrial enzymes in oxidation is influenced by the concentration of Ca2+, suggesting that an increase in Ca2+ influx into mitochondria promotes electron transport and ATP production. Indeed, it is reported that tricarboxylic acid (TCA) cycle is modulated by Ca2+ levels, in particular, the activation of ketoglutarate dehydrogenase (KGDH), isocitrate dehydrogenase (IDH) and pyruvate dehydrogenase (PDH) are directly dependent from matrix Ca2+ concentration [18, 19]. This leads to an increase in NADH, higher activity of the ETC and a positive loop for what concern mitochondrial metabolism. An additional dehydrogenase, named as FAD-glycerol phosphate dehydrogenase (GPDH) and localized in the IMM, mediates the transfer of reducing equivalents from NADH to the ETC by increasing ATP production [20, 21].
Overall, evidence of the Ca2+-dependent modulation of the oxidative phosphorylation derived by studies on skeletal muscle, which described an augmentation of conductance of the complexes I, III and IV of ETC [22]. Furthermore, experiments on isolated porcine heart mitochondria demonstrated that Ca2+ directly stimulates ATP synthase (complex V), enhancing respiratory chain activity and increasing ATP production [23].
However, intense ETC activity leads to increased production of mitoROS, which can overwhelm cellular antioxidant defenses, causing harmful damage, particularly during I/R when mitochondrial Ca2+ overload correlates with a significant increase of ROS and oxidative stress [24].
As anticipated above, RyR2 is a Ca2+ channel that can be found also in close proximity to MAMs to guarantee the correct release of Ca2+ towards mitochondria. Functional anomalies in RyR2 Ca2+ release have a noteworthy symptomatic impact in cardiac disease [9]; in a post-MI murine model, sustained Ca2+ leakage through RyR2 from SR leads to mitochondrial Ca2+ overload and a consequent mitoROS production, which cause oxidation of the receptor and an aberrant Ca2+ leak, creating a feedback loop [25]. RyR2 contain multiple cysteine residues, making them susceptible to oxidative changes. Their sensitivity to redox alterations means that the effects of ROS on RyR2 can vary based on concentration. Low levels of oxidants can boost RyR2 activity, while persistent high levels lead to irreversible inhibition, possibly affecting individual cysteine residues differently. Various oxidative modifications like S-glutathionylation and S-nitrosylation impact RyR2 function [23]. Additionally, oxidative stress can activate RyR2 by promoting disulfide bond formation between subunits, altering the channel’s structure and reducing SR Ca2+. Modulation of mitochondrial Ca2+ levels could be regulated also by RyR2 because, upon activation, RyR2 releases Ca2+ ions into the cytoplasm, which can subsequently enter the mitochondria through MCU [24]. This suggests that modulation of RyR2 function may have a protective effect against mitochondrial Ca2+ overload that occurs during IRI. Further evidence showed that RyR2 oxidation increases channel activity and worsens defective intracellular Ca2+ homeostasis [25].
In a time course manner after AMI, RyR2 expression was monitored with the recording of a transient decrease, followed by an increase only during the 4-week recovery, suggesting a role of this channel in the compensatory reaction of rat cardiac tissue to injury [26]. Of note, during I/R RyR2 is reported to be phosphorylated by Ca2+/calmodulin-dependent protein kinase II (CaMKII) at S2814; this site of phosphorylation is crucial for the cardiac damage probably linked to the increase in SR Ca2+ leak and consequent mitochondrial Ca2+ overload. Knock-in mice carrying a genetically inactivated site on RyR2 revealed a strongly reduced necrosis and apoptosis, thus a protection from I/R injury (IRI). By contrary, knock-in mice with the constitutively activated form of the aminoacidic site, presented a much more severe phenotype [27]. Accordingly, several studies demonstrated that ablation of CaMKIIδ, the main isoform in the myocardium, protects against I/R, suggesting a promising target for heart protection [28, 29].
Aberrations of other SR Ca2+ handling proteins are associated with AMI, such as SERCA2a. This pump is essential in controlling systole and diastole events as it modulates the cytosolic Ca2+ and thus the amount of Ca2+ bound to Troponin C. During I/R, there is an impairment of the normal influx of Ca2+ resulting in an overload of the amount of intracellular Ca2+ and a decrease in the activity of the SERCA2a isoform [30, 31]. Several studies over the years have demonstrated that increased SERCA2a expression can improve myocardial contractility and Ca2+ management following I/R myocardial injury [32, 33]. Of note, SERCA2a affinity for Ca2+ is carefully modulated by the interaction with phospholamban (PLB), whose expression levels and the phosphorylation status may limit or allow Ca2+ reuptake into the SR. This mechanism is regulated either by protein kinase A (PKA) or by CaMK, both proteins can phosphorylate PLB, releasing its inhibitory effects on SERCA2a [34]. A study by Shintani-Ishida demonstrated a significant PLB dephosphorylation associated to ischemic episodes in an in vivo model of AMI, that allows to cytosolic Ca2+ overload in early reperfusion, contributing to the formation of contraction bands [35].
The contractile dysfunction encountered following I/R and widely described until now can be further ascribed to the proteolytic modification of SR proteins like Junctophilins (JPH) 1 and 2 [36], calcineurin, protein kinase C (PKC), SERCA2a. This happens following the activation of Ca2+-dependent proteases, such as calpains, during Ca2+ overload that occurs in I/R [37]. Indeed, both JPH1 and JPH2 proteins are recognized as target of calpain-1 and 2, whose activity enhances during stress conditions [38]. Their expression, in particularly of JPH2, is downregulated during IRI [38].
Of note, in I/R, mitochondria are the first sensing organelles of the lower presence of oxygen; the ETC activity is inhibited and a consequent significant depletion in ATP production occurs. In this condition the cell favorizes a metabolic switch to anaerobic glycolysis. This leads to an intracellular Na+ and Ca2+ overload as a compensatory mechanism to buffer the excessive H+, mediated respectively by the activation of the Na+/H+ exchanger [39], inhibition of Na+/ Ca2+ antiporter (NCX) [40, 41]. Intracellular Ca2+ levels and membrane depolarization can also regulate the activity of large-conductance calcium-activated potassium ion channels (BKCa channels). These channels participate in a wide variety of fundamental physiological processes from vascular tone and cardiac rhythmicity [42]. BKCa channels have been detected in the IMM of adult cardiomyocytes where they increases K+conductance and improves mitochondria respiratory function by reducing the production of mitoROS and decreasing deleterious intra-mitochondrial Ca2+ accumulation occurring after I/R injury [43,44,45]. Increased production of mitoROS especially during cardiac reperfusion triggers the opening of another channel: the mitochondrial permeability transition pore (mPTP) [40, 41]. Opening of the mPTP is characterized by depolarization of the ΨMand from to exposure to high levels of mitoROS. During AMI, oxygen deprivation impairs oxidative phosphorylation, causing ion dysregulation. During reperfusion, the oxygen-starved heart undergoes oxidative damage due to the sudden abundance of nutrients and O2, triggering mPTP-dependent cell death [46, 47]. The intricate relationship between malfunctioning mitochondria and disrupted Ca2+ regulation underscores the intricacy of cardiomyocyte death during myocardial reperfusion injury. Key contributors to cell death in this context include hypercontracture and mitochondrial permeability transition (MPT), both exacerbated by incomplete repolarization of mitochondrial membranes. Hypercontracture arises from excessive contractile activity promoted by restored energy production in presence of elevated cytosolic Ca2+ levels. However, in tissues, the mechanical strain stemming from neighboring cell hypercontraction leads to mutual cellular breakdown and necrosis [48].
Whether the MCU-mediated Ca2+ influx is confirmed to be an important regulator of cardiac metabolism (i.e., ATP production) and in heart contractility in response to mechanisms of mild stress, its role in I/R remains controversial, at least from the analysis of data presented in the literature. In 2015, the groups of Elrod JW and Molkentin JD, independently showed how the inducible and conditional loss of cardiac MCU in a mouse model of I/R limits mitochondrial Ca2+ overload, the irreversible opening of mPTP and thus cell death; as consequence, the hearts resulted to be protected from bigger IS in reperfusion [24, 49]. By contrary, by taking advantage from a mouse model carrying a total deletion of MCU, Finkel’s group revealed the absence of IRI prevention. Hearts from WT and MCU−/− mice had the same levels of apoptosis, cell contracture and no differences in mPTP opening [50]. However, although MCU depletion limited mitochondrial Ca2+ uptake also in this animal model, the basal metabolism remained unchanged. It cannot be hidden that data reported until now are controversial. Different results might be explained by the mouse model studied: whether MCU is deleted in the whole body, the onset of some compensation mechanisms might interfere with the physio pathological readout; currently, the inducible and conditional loss of cardiac MCU can be the most reliable method to assess mitochondrial Ca2+ role during ischemia.
A very recent work by Ashok et al., demonstrated that mitochondrial NCX is the primary way of Ca2+ entry in mitochondria of neonatal mouse ventricular myocyte knock-out for MCU [51]. This work supports the hypothesis of the adaptation of the mouse model carrying a germline deletion of MCU, in which Ca2+ entry in mitochondria might be controlled in different ways.
More recently, without the use of transgenic preclinical models Guan L and colleagues reported a significant upregulation of MCU in IRI, confirming that the resulting mitochondrial Ca2+ overload led to dysregulation at multiple levels including the imbalance of mitochondrial quality control mechanisms like fusion, fission and mitophagy and the activation of calpains [52].
MCU is not the only one pore-forming protein of the complex. Also, MCUb plays crucial roles in Ca2+ channeling and it is considered as the dominant negative subunit of MCU [53]. To confirm in alternative ways the role of mitochondrial Ca2+ in I/R, in 2020 Molkentin JD and colleagues replicated the study carried out in 2015 to understand the behavior of this subunit under I/R conditions [54]. He described a significant upregulation of MCUb starting from 3 days after ischemia without detecting the protein at resting state or early at reperfusion time [54]. Although this could mean that MCUb adaptations can have protective roles during later stages of reperfusion, the exact ratio among MCU – MCUb – EMRE and its significance should be carefully evaluated under these experimental conditions.
Ca2+ dysregulation at MAMs in myocardial infarction
MAMs are specialized regions where mitochondria and the SR come close together. Recent advancements in biomedical techniques have allowed a better visualization of this compartment in living cells using fluorescence confocal microscopy. MAMs act as biochemically independent areas while serving as communication points between SR and mitochondria, maximizing their signaling interactions. As we previously highlighted the importance of SR and mitochondria in AMI, it’s worth noting that MAMs also play a crucial role in this context. Indeed, they constitute several microdomains with high Ca2+ concentrations, thus being essential for cardiomyocytes in regulating Ca2+ transfer to mitochondria and supporting energy production. Thanks to the use of electron tomography, it has been demonstrated that SR and mitochondria are connected by tethers with a distance of approximately 10–30 nm and it has been defined a fundamental reliance of cell function and viability on the preservation of appropriate spacing between the SR and mitochondria [55].
Evidence suggests that dysfunctions at MAMs may contribute to the pathogenesis of reperfusion injury following AMI, suggesting that a decrease in the tethering between the two organelles is associated with a disturbance of clearance and ER stress, and a decline of mitochondrial dynamics included mitochondrial Ca2+ uptake, which, in turn, contributes to a decrease in cell death in cardiac myocytes subjected to I/R [56].
From a historical point of view, the first protein complex identified at MAMs was the IP3Rs/GRP75/VDAC1 axis. The different isoforms of the ER membrane IP3R are linked to voltage-dependent anion channel 1 (VDAC1) localized at OMM by glucose-regulated protein 75 chaperone (GRP75). This complex plays a critical role in enabling Ca2+ transfer between the two organelles, consequently controlling either the apoptotic process or the energy production. Specifically, IP3R3-GRP75-VDAC1 complex is implicated in mitochondrial Ca2+ overload and subsequent cardiomyocyte death observed during the reperfusion phase after sustained ischemic insult. Furthermore, the mitochondrial matrix protein Cyclophilin D (CypD) is also known as a regulator of mPTP activity [46], can interact with IP3R1-GRP75-VDAC1 complex. This interaction acts to regulate Ca2+ exchange from SR to mitochondria (Fig. 1) [57]. Proofs highlighting the relevance of VDAC1 in this pathological context come from a study carried out on a rat model of MI and on human cardiac tissues obtained from post-MI patients. The findings from these investigations unveiled the upregulation of VDAC1 levels; when inhibited through the oligomerization inhibitor VBIT-4 it relieves the increased fibrosis in the atrial myocardium of rats subjected to MI [58]. Further investigations have focused on glycogen synthase kinase-3β (GSK3β), which in its active form can phosphorylate VDAC1 and increase mitochondrial Ca2+ uptake. Moreover it has been revealed that in perfused rat hearts treated with GSK inhibitors, the ischemic process is less harmful on heart function [59, 60].
FUN14 domain containing 1 (FUNDC1) is a highly conserved OMM protein which plays a crucial role at MAMs: it is activated under ischemic conditions to induce mitophagy; while after reperfusion it is phosphorylated and inactivated resulting in reduction of mitophagy and activation of apoptosis [61]. FUNDC1 interacts with IP3R2 to control ER Ca2+ release into mitochondria; indeed, when depleted it leads to disruption of MAMs and to mitochondrial dysfunction [62]. Cardiomyocyte-conditional deletion of FUNDC1 provokes heart failure in vivo, which is worsened by acute MI: at molecular level this is explained by the disruption of MAMs integrity, showed as a decrease of MAMs proteins expression and a dissociation between mitochondria and ER, and a consequent impairment in Ca2+ homeostasis [62]. Other evidence both in vitro and in vivo confirmed the significance of FUNDC1 in myocardial injury following ischemic events, its expression and the mitophagic control are repressed leading to mitochondrial impairments and cardiomyocyte apoptotic death [63].
A key protein is also represented by Mitofusin 2 (MFN2), usually involved in controlling mitochondrial fusion, it is localized at ER and OMM interface and through the formation of homo- or hetero-dimers with MFN1 or MFN2 promotes MAMs stabilization (Fig. 1). Mitofusins revealed to be essential in cardiovascular system as they control mitochondrial fusion and ensure the correct mitochondrial morphology necessary for cardiac respiration and contraction [64].
MFN2 exhibits contrasting roles in MI and myocardial IRI. Some evidence suggests that MFN1/MFN2 double-knockout mice died at the embryonic stage due to lethal cardiac damage and conditional deletion in adult mouse hearts leads to severe impairments in mitochondrial functionality and cardiac respiration. Despite that, hearts deficient in both MFN1 and MFN2 are protected against acute IRI, due to an increased resistance to mPTP opening in response to Ca2+ and a reduction in mitochondria–SR interaction, suggesting that hearts deleted of both proteins are resistant to AMI [65]. Notably, MFN2-deficient hearts showed a less severe phenotype compared to double MFNs knockout, suggesting a potential compensation by MFN1 in mitigating the loss of MFN2. It has been shown that the deletion of MFN2 protected mice hearts delaying mPTP opening after Ca2+ overload and the following cell death after IRI [66]. MFN2 strongly impacts also on platelet biology as it normally preserves mitochondrial and complex I activities [67]. Its absence determines reduced respiration and ROS generation, all determinants involved in oxygen consumption needed for thrombin-dependent procoagulant functions and mPTP opening, thus decreasing platelet response during I/R and limiting IS [67].
A very recent work by Yepuri et al. showed that DIAPH1, a Formin involved in generation of actin filaments, directly interacts with MFN2 and regulates mitochondria–SR tethering. Once defined the localization of DIAPH1 in mitochondria and at SR, it has been shown that its silencing leads to disruption of the tethering between these two organelles and the consequence is a protection from I/R both in cardiomyocytes and beating hearts [68]. This modulation of myocardial IRI has been previously demonstrated showing that DIAPH1 deletion regulates serum response factor (SRF) and early growth response 1 (EGR1) leading to modulation of Ca2+ transporters in cardiomyocytes such as SERCA2a expression [69].
However, there are also some controversies in the scientific literature about MFN2 knockout models. A cardiac MFN2 knock-out mouse exhibited a worsened response to IRI, due to an impairment of mitochondrial functionality and cellular homeostasis; in fact, the lack of this protein leads to increased accumulation of autophagosomes in response to I/R stress and to cardiac dysfunction [70]. Further research is needed to elucidate the role of MFN2 in the protection against IRI after MI, especially because recent discoveries bring to light different MFN2 variants essential to shape the ER or to tether it to mitochondria and it has to be clarified which of them is involved in pathobiology of IRI [71].
Another essential complex involved in proper functioning of MAMs is B cell receptor–associated protein 31 (BAP31)-Mitochondrial fission 1 protein (Fis1). Fis1 is an OMM protein, creating a complex involved in an early event during apoptosis induction, it transmits an apoptotic signal from the mitochondria to the ER by interacting with BAP31 and leading to its cleavage into the pro-apoptotic form p20BAP31 after the recruitment of procaspase-8. This apoptotic message triggers the release of Ca2+ from the ER to the mitochondria for apoptotic cascade activation [72]. During I/R, p20BAP31 form causes Bax and Bak translocation to the OMM and Cyt-C release from the mitochondria, these events lead to cardiomyocytes death and provide further evidence of the involvement of Ca2+ handling at MAMs in the pathophysiology of MI [73].
The regulation of MAM structures involves the tethering between SR-resident protein Vesicle-associated membrane protein-associated protein B (VAPB) and mitochondrial protein tyrosine phosphatase-interacting protein-51 (PTPIP51) (Fig. 1) [74]. This interaction is fundamental for the control of Ca2+ homeostasis and autophagic process [75].
The Sigma-1 receptor (Sig-1R) is a transmembrane protein of the SR, it mostly resides at MAMs where it interacts with binding immunoglobulin protein (BIP); in cases of Ca2+ depletion from the SR, Sig-1R dissociates from BIP and binds to IP3Rs leading to a prolonged Ca2+ influx into mitochondria [76].
In conclusion, the study of MAMs in AMI sheds light on the complex interplay between the SR and mitochondria and that Ca2+ takes essential roles in cardiac contraction and cell fate in response to ischemic events. Further studies into the mechanisms underlying MAMs function and dysfunction might potentially pave the way for the development of targeted interventions for future AMI therapies. Of note, the experimental work by Csordas G and colleagues, consisted in generate an engineered mitochondria-SR tether (linker) in mice which induced a strong mitochondrial and SR remodeling, enhancing the contacts, but also showed an increase resistance to injury in vivo and ex vivo I/R [77]. A specific transgene for cardiac muscle was introduced into mice, containing a monitorable protein. Through in vivo and in vitro clinical tests, the behavior of the transgene was identified, showing both expanding and contracting actions on mitochondrial-SR contacts, reducing mortality of cardiac muscle cells and damage from Ca2+ overload in I/R situations. This process balances the interaction between SR and mitochondria, enhancing resilience to stress conditions associated with abnormal Ca2+ regulation [77].
Cardioprotective strategies targeting Ca2+ signaling at MAMs
Over the years, a wide range of experimental strategies have been employed to improve patient outcomes in AMI clinics following IRI [78, 79]. The existing therapies are mainly aimed to prevent cardiac overload and fatigue after the ischemic episode; they also work to reduce the occurrence of secondary events by managing main risk factors, such as hypertension and atherosclerosis. However, many molecular mechanisms becoming impaired during I/R damage, often linked to Ca2+ homeostasis and mPTP opening are currently far from being considered as much as they should be (Table 1). Indeed, while our understanding of basic and preclinical research is expanding, translational studies directed at signaling between the SR and mitochondria are in the growing phase and require further attention. The current clinical picture poses also further challenges, as many cardiologists believe that the decline in mortality has stabilized and remains uncertain whether it can be reduced in either the short or the long term. Considering these issues, is it possible to study SR and mitochondrial environment and their contact sites as a therapeutic target in the pathophysiology of AMI?
Pharmacological regulation of mitochondrial Ca2+ intake
After its discovery, MCU targeting appeared the best reliable way for a direct therapeutic approach to treat the excessive Ca2+ intake into the mitochondrion during I/R. Several groups have developed molecules capable of reducing mitochondrial Ca2+ uptake, the first one was ruthenium red (RuRed), synthesized in 1892. RuRed is a compound that inhibits MCU without affecting Ca2+ efflux and mitochondrial respiration [80, 81]. The most important study investigating the use of RuRed in heart disease was by Grover et al. in which the treatment of rat hearts perfused with the compound in the micromolar range improved cardiac contractile function and oxygen reperfusion efficiency after ischemia [82]. Due to its low selectivity and permeability, RuRed has come under criticism over the years and stimulated the development of some additional derivatives like Ru360 and Ru265 with improved properties, like higher selectivity, cell permeability accompanied by low toxicity [83] (Table 1). MCU inhibition has been reached also by using KB-R7943, a compound originally developed as an inhibitor of the NCX [84], that has been shown to protect against myocardial IRI through the inhibition of MCU causing a reduced Ca2+ uptake within the mitochondrial matrix (Fig. 2) [84]. Studies carried out on rats have demonstrated that using KB-R7943 at concentration of 1µM had a protective effect in IRI through restoring left ventricular (LV) function, by reducing ventricular fibrillation and hypercontracture of cardiomyocytes [85].

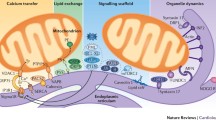
Representation of proteins involved in Ca2+ signaling and possible related cardioprotective strategies to prevent ischemia-reperfusion damage
This figure depicts the key proteins involved in calcium (Ca2+) signaling that are dysregulated during I/R and their potential cardioprotective strategies to mitigate I/R demange
Sarcoplasmic Reticulum (SR), Mitochondria associated membranes (MAM), Mitochondrial calcium uniporter (MCU), Voltage-dependent anion-selective channel (VDAC), Inositol 1,4,5-triphosphate receptor type 2 (IP3R2), Inositol 1,4,5-triphosphate receptor type 1 (IP3R1), Ryanodine receptors (RyR2), Plasma membrane Ca2+ ATPase, sarcoplasmic reticulum Ca2+ ATPases (SERCA), Phospholamban (PLB), Glucose-regulated protein 75 chaperone (GRP75), Mitofusins 1 and 2 (MFN1 and MFN2), Vesicle-associated membrane protein-associated protein B (VAPB), Protein tyrosine phosphatase-interacting protein-51 (PTPIP51), B cell receptor–associated protein 31 (BAP31), Mitochondrial fission 1 protein (Fis1)
DS16570511 and MCU-i4 synthesis recently updated the list of mitochondrial Ca2+ uptake inhibitors, giving to the target an appealing route and a good pharmacological profile to be translated in clinic [86, 87]. The use of DS1657051 in the 3–30 µM range blocked mitochondrial Ca2+ uptake with beneficial effects on increasing cardiac contractility and reducing IRI. An interesting property is reversible binding with the target after washout [86]. On the other hand, MCU-i4 binds a specific domain of MICU1, fundamental for the gating activity of MCU complex, but it affects mitochondrial depolarization (Fig. 2). Mainly at preclinical level, these treatments led to rapid beneficial effects like increased heart contractility and reduced IS [88] However, while these advancements are exciting, it’s crucial to approach them with a critical lens. Further research is necessary to fully elucidate their mechanisms of action, assess their safety profiles, and evaluate their efficacy in clinical trials. Their effects on mitochondrial depolarization and the necessity for translation to clinical settings warrant careful consideration.
Pharmacological regulation of SR Ca2+ intake and release
Chronic hypoxia leads to the deterioration of myocardial functions with impaired Ca2+ handling in charge of the SR, which may be mediated by oxidative stress. Studies have hypothesized that antioxidant administration would protect against cardiac IRI by improving SR Ca2+ management [89].
On the most known natural compounds and hormone acting at SR levels is Melatonin; the authors of this study [90] investigated its effects on intracellular Ca2+ handling as therapeutic strategy to counteract acute cardiac injury-related side effects. Taking advantage from the establishment of an in vivo mouse model of IRI, they described how Melatonin can impact on both SR and mitochondria subcellular compartments. Indeed, it is able to inhibit the IP3R phosphorylation and, at the same time also MCU expression, thus alleviating both cytoplasmic and mitochondrial Ca2+ overload which occurs after ischemia (Fig. 2). Melatonin appears to have additional properties without showing a peculiar molecular pathway; it has been shown to downregulate IP3R expression and to promote SERCA2a expression via the ERK1 pathway in cardiomyocytes preventing I/R damage. However, the lack of elucidation on its specific molecular pathway raises questions about its precise mode of action. Melatonin also inhibits apoptosis of cardiomyocytes and improves the organization of actin filaments in cardiomyocytes subjected to I/R [91]. As mentioned above, SERCA2a plays an important role in bringing Ca2+ from cytosol to SR lumen. Reduced SERCA2a activity and expression is one of the consequences that occur following IRI. Using viral vectors to promote its long-term overexpression in cardiac tissue was observed to markedly enhance cardiac function and reduce myocardial apoptosis. This improvement was ascribed to the augmentation of Ca2+ signaling, directly impacting the functionality of vascular endothelial cells and smooth muscle [92]. Studies conducted on rats have demonstrated that lentiviruses can integrate the gene of interest into the host genome with a long-term effect of genetic transduction [93]. Even in I/R models, overexpression of SERCA2a reduced the incidence of ventricular arrhythmia and improved hemodynamics [94, 95].
A recent study conducted in the field of cardiac function, showed that phosphorylation at serine 663 of SERCA2 emerges as a pivotal clinical and pathophysiological occurrence. Elevated phosphorylation at this site is evident in ischemic hearts of both patients and preclinical models. Inhibiting serine 663 phosphorylation, through a CRISPR/Cas9-mediated genome editing strategy allowed to generate a human cell line expressing a phosphoresistant SERCA2 mutant (SERCA2S663A), enhances SERCA2 activity, offering protection against cell death by mitigating cytosolic and mitochondrial Ca2+ overload [96]. The above-mentioned necrosis and apoptosis are only two of the plethora of cell death mechanisms by which cells die after stressors [97] and in which Ca2+ and inflammation are crucial factors. Parthanatos is one type of programmed necrotic cell death mainly described in neurodegenerative diseases and stroke and appropriately reviewed in [98]. Important molecular mechanisms behind Parthanatos relies on excessive intracellular ROS accumulation and an abnormal and dependent ER Ca2+ release directed to mitochondria; as consequence, mitochondrial membrane depolarizes and an irreversible energetic crisis occurs [99]. The authors did not investigate the proteins involved in this wrong ER-mitochondria Ca2+ communication, but found in Propofol a potent pharmacological agent able to counteract ROS production and ER-mediated Ca2+ release with beneficial outcomes against Parthanatos-mediated IRI [99].
From the literature, Propofol results to exert a protective action also in cardiac-related settings (i.e., open-heart surgery, cardioplegia, valve replacement, I/R). Almost all studies ascribe the effect to the increase in the antioxidant defense of the organism; no mention to specific Ca2+ signaling pathways being involved. It would be interesting to expand this knowledge in disease, to confirm the ER-mitochondria modulation of Ca2+ transfer and to unveil the responsible proteins.
Direct regulation of MAMs
Despite their importance, still few drugs have been reported to modulate Ca2+ signaling and architecture within MAMs. To the best of our knowledge, it has only been studied the involvement of PTPIP51 in regulating cardiac function controlling the mitochondria-SR junction. Studies performed on I/R mouse hearts, showed that the adenovirus-associated virus (AAV)-mediated knockdown of PTPIP51 significantly protected cardiac tissue after I/R due to a reduction of mitochondria-SR contacts and Ca2+ exchange [100] (Table 1).
The fact that only PTPIP51 has been studied overlooks potential alternative targets within MAMs, limiting our understanding of their modulation. Additionally, the reliance on a single study to support PTPIP51’s efficacy in I/R injury may present an oversimplified view of MAM involvement in cardiac function. Further exploration of other molecular targets within MAMs is warranted to fully understand their role in cardiac pathology and to develop more effective therapeutic strategies.
Recently, some reports are describing a change in expression of IP3Rs receptors in various cardiac diseases, as a maladaptive response in charge of the MAMs [101]. Indeed, in vivo knockdown of IP3R in murine hearts protected against MI and induced cardiac structural remodeling and fibrosis [102]. Studies conducted on hearts in which global ischemia is generated by the Langendorff system, have shown that the drug Resveratrol significantly improved LV pressure and coronary flow. The activity of lactate dehydrogenase and creatine phosphokinase were also decreased resulting in a reduction in IS. Resveratrol is also known to inhibit the upregulation of the expression of the anion channel VDAC1 triggered by IRI, confirming that VDAC1 plays an important role in resveratrol-mediated cardioprotection [103]. CypD seems to have a double role, being a modulator of the mPTP and triggering its opening following an increased accumulation of Ca2+ in the mitochondrial matrix [104]. Moreover, during I/R, CypD would synergistically act with the IP3R1-GRP75-VDAC1 complex and with an increase in mitochondrial Ca2+ load and induction of apoptosis. Inhibition or downregulation of CypD, prevented mitochondrial Ca2+ overload and cell death in an adult mouse cardiomyocyte I/R model [57]. In this context it should be reported also that CypD negatively regulates mitochondrial Ca2+ accumulation [105]. So, in contrast to what wrote above, CypD would actively participate in a pathological increase of mitochondrial Ca2+ which determines glucose rather than fatty acids oxidation with alterations in cellular bioenergetics and mPTP activity [105].
In cardioprotection mechanical strategies exist besides pharmacological treatments [106]. This is the case of the ischemic preconditioning (IPC) and the ischemic postconditioning (IPostC) in which brief episodes of intermittent I/R before or after the main ischemic event, respectively, protect the heart from the main features of reperfusion damage [106]. Although direct evidence about the impact of IPC and IPostC on SR-mitochondria tethering do not exist yet, it is known that these techniques concur to minimize ATP depletion, ROS generation and thus mitochondria-related dysregulations. Multiple studies involving the use of cultured cardiomyocytes and preclinical models of cardiac I/R confirmed that repeated interruption of blood reflow at reperfusion time (i.e., 3 cycles of 5 min hypoxia/reoxygenation) decreases mainly oxidative stress but also both cytosolic and mitochondrial Ca2+ overload [107] with the preservation of mitochondrial membrane potential [108] and a strong inhibition of mPTP opening [109]. The prevention of mitochondrial Ca2+ overload probably refers to the blockade of excessive Ca2+ release from SR, as investigated by Khalaf A. and co-workers by using a similar protocol of IPostC [110]. Instead, the increase in intracellular Ca2+ persistent can be avoided by the IPostC-mediated increase in PLB phosphorylation which guarantees a correct SR Ca2+ load [111]. To sum up, there is a deficiency in effective therapeutic approaches specifically targeting Ca2+ transfer at MAMs in myocardial reperfusion injury. However, among the strategies identified as protective in patients, most of their beneficial effects can be directly or indirectly attributed to the modulation of Ca2+-mediated damage. While there is growing interest in targeting MAMs to modulate Ca2+ signaling, the limited knowledge underlines the needs of further investigation to optimize the therapeutic potential both in preclinical and clinical settings.
Conclusions
The research on SR and mitochondria, and on their contacts, has highlighted the important role of these organelles in the pathophysiology of CVDs, especially in AMI. Although the molecular characterization of these pathways has reached the golden age, we must admit that the road to fully understand how they can be successfully targeted in therapy is still far. What we know is that the disruption of the architecture of these subcellular sites occurs during AMI and can lead to further damage to the heart muscle in preclinical models. Likewise, the use of Ca2+ handling/cycling correctors can help to protect the organelles from damage and to restore the normal function of the cardiac tissue. Despite that, we cannot understand the real impact on the clinical outcome of the patients affected, whether the translational studies and clinical trials do not give feedback to the scientific community.
References
Laforgia PL, Auguadro C, Bronzato S, Durante A. The reduction of Mortality in Acute myocardial infarction: from Bed Rest to Future directions. Int J Prev Med. 2022;13:56.
Ishida M, Kato S, Sakuma H. Cardiac MRI in ischemic heart disease. Circ J. 2009;73:1577–88.
Gregg RE, Babaeizadeh S. Detection of culprit coronary lesion location in pre-hospital 12-lead ECG. J Electrocardiol. 2014;47:890–4.
Manari A, Albiero R, De Servi S. High-risk non-ST-segment elevation myocardial infarction versus ST-segment elevation myocardial infarction: same behaviour and outcome? J Cardiovasc Med (Hagerstown). 2009;10(Suppl 1):S13–16.
Chan MY, Sun JL, Newby LK, Shaw LK, Lin M, Peterson ED, et al. Long-term mortality of patients undergoing cardiac catheterization for ST-elevation and non-ST-elevation myocardial infarction. Circulation. 2009;119:3110–7.
Reed GW, Rossi JE, Cannon CP. Acute myocardial infarction. Lancet. 2017;389:197–210.
Kingma JG. Myocardial infarction: an overview of STEMI and NSTEMI Physiopathology and Treatment. WJCD. 2018;08:498–517.
Morciano G, Giorgi C, Bonora M, Punzetti S, Pavasini R, Wieckowski MR, et al. Molecular identity of the mitochondrial permeability transition pore and its role in ischemia-reperfusion injury. J Mol Cell Cardiol. 2015;78:142–53.
Gambardella J, Sorriento D, Ciccarelli M, Del Giudice C, Fiordelisi A, Napolitano L, et al. Functional role of Mitochondria in Arrhythmogenesis. Adv Exp Med Biol. 2017;982:191–202.
Neubauer S. The failing heart–an engine out of fuel. N Engl J Med. 2007;356:1140–51.
Ramachandra CJA, Hernandez-Resendiz S, Crespo-Avilan GE, Lin Y-H, Hausenloy DJ. Mitochondria in acute myocardial infarction and cardioprotection. EBioMedicine. 2020;57:102884.
Zhang Y, Yao J, Zhang M, Wang Y, Shi X. Mitochondria-associated endoplasmic reticulum membranes (MAMs): possible therapeutic targets in heart failure. Front Cardiovasc Med. 2023;10:1083935.
Hernandez-Resendiz S, Prakash A, Loo SJ, Semenzato M, Chinda K, Crespo-Avilan GE, et al. Targeting mitochondrial shape: at the heart of cardioprotection. Basic Res Cardiol. 2023;118:49.
Drago I, De Stefani D, Rizzuto R, Pozzan T. Mitochondrial Ca2 + uptake contributes to buffering cytoplasmic Ca2 + peaks in cardiomyocytes. Proc Natl Acad Sci U S A. 2012;109:12986–91.
Jouaville LS, Pinton P, Bastianutto C, Rutter GA, Rizzuto R. Regulation of mitochondrial ATP synthesis by calcium: evidence for a long-term metabolic priming. Proc Natl Acad Sci U S A. 1999;96:13807–12.
Wu Y, Rasmussen TP, Koval OM, Joiner M-LA, Hall DD, Chen B, et al. The mitochondrial uniporter controls fight or flight heart rate increases. Nat Commun. 2015;6:6081.
Eisner V, Csordás G, Hajnóczky G. Interactions between sarco-endoplasmic reticulum and mitochondria in cardiac and skeletal muscle - pivotal roles in Ca2+ and reactive oxygen species signaling. J Cell Sci. 2013;126:2965–78.
Denton RM. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim Biophys Acta. 2009;1787:1309–16.
Rossi A, Pizzo P, Filadi R. Calcium, mitochondria and cell metabolism: a functional triangle in bioenergetics. Biochim Biophys Acta Mol Cell Res. 2019;1866:1068–78.
Cole ES, Lepp CA, Holohan PD, Fondy TP. Isolation and characterization of flavin-linked glycerol-3-phosphate dehydrogenase from rabbit skeletal muscle mitochondria and comparison with the enzyme from rabbit brain. J Biol Chem. 1978;253:7952–9.
Nesci S, Algieri C, Trombetti F, Fabbri M, Lenaz G. Two separate pathways underlie NADH and succinate oxidation in swine heart mitochondria: kinetic evidence on the mobile electron carriers. Biochim Biophys Acta Bioenerg. 2023;1864:148977.
Glancy B, Willis WT, Chess DJ, Balaban RS. Effect of calcium on the oxidative phosphorylation cascade in skeletal muscle mitochondria. Biochemistry. 2013;52:2793–809.
Territo PR, Mootha VK, French SA, Balaban RS. Ca(2+) activation of heart mitochondrial oxidative phosphorylation: role of the F(0)/F(1)-ATPase. Am J Physiol Cell Physiol. 2000;278:C423–435.
Kwong JQ, Lu X, Correll RN, Schwanekamp JA, Vagnozzi RJ, Sargent MA, et al. The mitochondrial calcium Uniporter selectively matches metabolic output to acute contractile stress in the heart. Cell Rep. 2015;12:15–22.
Santulli G, **e W, Reiken SR, Marks AR. Mitochondrial calcium overload is a key determinant in heart failure. Proc Natl Acad Sci U S A. 2015;112:11389–94.
Sallinen P, Mänttäri S, Leskinen H, Ilves M, Ruskoaho H, Saarela S. Time course of changes in the expression of DHPR, RyR(2), and SERCA2 after myocardial infarction in the rat left ventricle. Mol Cell Biochem. 2007;303:97–103.
Di Carlo MN, Said M, Ling H, Valverde CA, De Giusti VC, Sommese L, et al. CaMKII-dependent phosphorylation of cardiac ryanodine receptors regulates cell death in cardiac ischemia/reperfusion injury. J Mol Cell Cardiol. 2014;74:274–83.
Lebek S, Chemello F, Caravia XM, Tan W, Li H, Chen K, et al. Ablation of CaMKIIδ oxidation by CRISPR-Cas9 base editing as a therapy for cardiac disease. Science. 2023;379:179–85.
Yu J, Chen Y, Xu M, Sun L, Luo H, Bao X, et al. Ca2+/Calmodulin-Dependent protein kinase II regulation by inhibitor 1 of Protein Phosphatase 1 protects against myocardial ischemia-reperfusion Injury. J Cardiovasc Pharmacol Ther. 2019;24:460–73.
Talukder MAH, Kalyanasundaram A, Zuo L, Velayutham M, Nishijima Y, Periasamy M, et al. Is reduced SERCA2a expression detrimental or beneficial to postischemic cardiac function and injury? Evidence from heterozygous SERCA2a knockout mice. Am J Physiol Heart Circ Physiol. 2008;294:H1426–1434.
Wang L, Myles RC, Lee I-J, Bers DM, Ripplinger CM. Role of reduced Sarco-Endoplasmic Reticulum Ca2+-ATPase function on Sarcoplasmic Reticulum Ca2 + alternans in the intact rabbit heart. Front Physiol. 2021;12:656516.
Baker DL, Hashimoto K, Grupp IL, Ji Y, Reed T, Loukianov E, et al. Targeted overexpression of the sarcoplasmic reticulum Ca2+-ATPase increases cardiac contractility in transgenic mouse hearts. Circ Res. 1998;83:1205–14.
Talukder MAH, Kalyanasundaram A, Zhao X, Zuo L, Bhupathy P, Babu GJ, et al. Expression of SERCA isoform with faster Ca2 + transport properties improves postischemic cardiac function and Ca2 + handling and decreases myocardial infarction. Am J Physiol Heart Circ Physiol. 2007;293:H2418–2428.
Brittsan AG, Kranias EG. Phospholamban and cardiac contractile function. J Mol Cell Cardiol. 2000;32:2131–9.
Shintani-Ishida K, Yoshida K-I. Ischemia induces phospholamban dephosphorylation via activation of calcineurin, PKC-α, and protein phosphatase 1, thereby inducing calcium overload in reperfusion. Biochim Biophys Acta. 2011;1812:743–51.
Wang S, Zhou Y, Luo Y, Kan R, Chen J, Xuan H, et al. SERCA2a ameliorates cardiomyocyte T-tubule remodeling via the calpain/JPH2 pathway to improve cardiac function in myocardial ischemia/reperfusion mice. Sci Rep. 2021;11:2037.
Singh RB, Chohan PK, Dhalla NS, Netticadan T. The sarcoplasmic reticulum proteins are targets for calpain action in the ischemic-reperfused heart. J Mol Cell Cardiol. 2004;37:101–10.
Guo A, Hall D, Zhang C, Peng T, Miller JD, Kutschke W, et al. Molecular determinants of calpain-dependent cleavage of Junctophilin-2 protein in Cardiomyocytes. J Biol Chem. 2015;290:17946–55.
Pike MM, Luo CS, Clark MD, Kirk KA, Kitakaze M, Madden MC, et al. NMR measurements of na + and cellular energy in ischemic rat heart: role of na(+)-H + exchange. Am J Physiol. 1993;265:H2017–2026.
Morciano G, Naumova N, Koprowski P, Valente S, Sard?o VA, Potes Y, Rimessi A, Wieckowski MR, Oliveira PJ. The mitochondrial permeability transition pore: an evolving concept critical for cell life and death. Biol Rev Camb Philos Soc. 2021 Dec;96(6):2489–2521. https://doi.org/10.1111/brv.12764. Epub 2021 Jun 21. PMID: 34155777.
Bonora M, Giorgi C, Pinton P. Molecular mechanisms and consequences of mitochondrial permeability transition. Nat Rev Mol Cell Biol. 2022 Apr;23(4):266–285. https://doi.org/10.1038/s41580-021-00433-y. Epub 2021 Dec 8. PMID: 34880425.
Lai MH, Wu Y, Gao Z, Anderson ME, Dalziel JE, Meredith AL. BK channels regulate sinoatrial node firing rate and cardiac pacing in vivo. Am J Physiol Heart Circ Physiol. 2014;307:H1327–1338.
Singh H, Lu R, Bopassa JC, Meredith AL, Stefani E, Toro L. MitoBK(ca) is encoded by the Kcnma1 gene, and a splicing sequence defines its mitochondrial location. Proc Natl Acad Sci U S A. 2013;110:10836–41.
Goswami SK, Ponnalagu D, Hussain AT, Shah K, Karekar P, Gururaja Rao S, et al. Expression and activation of BKCa channels in mice protects against Ischemia-Reperfusion Injury of isolated hearts by modulating mitochondrial function. Front Cardiovasc Med. 2018;5:194.
Szteyn K, Singh H. BKCa channels as targets for Cardioprotection. Antioxid (Basel). 2020;9:760.
Robichaux DJ, Harata M, Murphy E, Karch J. Mitochondrial permeability transition pore-dependent necrosis. J Mol Cell Cardiol. 2023;174:47–55.
Bauer TM, Murphy E. Role of mitochondrial calcium and the permeability transition pore in regulating cell death. Circ Res. 2020;126:280–93.
Garcia-Dorado D, Ruiz-Meana M, Inserte J, Rodriguez-Sinovas A, Piper HM. Calcium-mediated cell death during myocardial reperfusion. Cardiovasc Res. 2012;94:168–80.
Luongo TS, Lambert JP, Yuan A, Zhang X, Gross P, Song J, et al. The mitochondrial calcium Uniporter matches energetic supply with Cardiac workload during stress and modulates permeability transition. Cell Rep. 2015;12:23–34.
Pan X, Liu J, Nguyen T, Liu C, Sun J, Teng Y, et al. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat Cell Biol. 2013;15:1464–72.
Ashok D, Papanicolaou K, Sidor A, Wang M, Solhjoo S, Liu T, et al. Mitochondrial membrane potential instability on reperfusion after ischemia does not depend on mitochondrial Ca2 + uptake. J Biol Chem. 2023;299:104708.
Guan L, Che Z, Meng X, Yu Y, Li M, Yu Z, et al. MCU Up-regulation contributes to myocardial ischemia-reperfusion Injury through calpain/OPA-1-mediated mitochondrial fusion/mitophagy inhibition. J Cell Mol Med. 2019;23:7830–43.
Giorgi C, Marchi S, Pinton P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat Rev Mol Cell Biol. 2018 Nov;19(11):713–730. https://doi.org/10.1038/s41580-018-0052-8. Erratum in: Nat Rev Mol Cell Biol. 2018 Sep 24; PMID: 30143745.
Huo J, Lu S, Kwong JQ, Bround MJ, Grimes KM, Sargent MA, et al. MCUb induction protects the Heart from Postischemic Remodeling. Circ Res. 2020;127:379–90.
Csordás G, Renken C, Várnai P, Walter L, Weaver D, Buttle KF, et al. Structural and functional features and significance of the physical linkage between ER and mitochondria. J Cell Biol. 2006;174:915–21.
Gong Y, Lin J, Ma Z, Yu M, Wang M, Lai D, et al. Mitochondria-associated membrane-modulated Ca2 + transfer: a potential treatment target in cardiac ischemia reperfusion injury and heart failure. Life Sci. 2021;278:119511.
Paillard M, Tubbs E, Thiebaut P-A, Gomez L, Fauconnier J, Da Silva CC, et al. Depressing mitochondria-reticulum interactions protects cardiomyocytes from lethal hypoxia-reoxygenation injury. Circulation. 2013;128:1555–65.
Klapper-Goldstein H, Verma A, Elyagon S, Gillis R, Murninkas M, Pittala S, et al. VDAC1 in the diseased myocardium and the effect of VDAC1-interacting compound on atrial fibrosis induced by hyperaldosteronism. Sci Rep. 2020;10:22101.
Das S, Wong R, Rajapakse N, Murphy E, Steenbergen C. Glycogen synthase kinase 3 inhibition slows mitochondrial adenine nucleotide transport and regulates voltage-dependent anion channel phosphorylation. Circ Res. 2008;103:983–91.
Gomez L, Thiebaut P-A, Paillard M, Ducreux S, Abrial M, Da Crola C, et al. The SR/ER-mitochondria calcium crosstalk is regulated by GSK3β during reperfusion injury. Cell Death Differ. 2016;23:313–22.
Zhou H, Zhu P, Guo J, Hu N, Wang S, Li D, et al. Ripk3 induces mitochondrial apoptosis via inhibition of FUNDC1 mitophagy in cardiac IR injury. Redox Biol. 2017;13:498–507.
Wu S, Lu Q, Wang Q, Ding Y, Ma Z, Mao X, et al. Binding of FUN14 domain containing 1 with Inositol 1,4,5-Trisphosphate receptor in Mitochondria-Associated endoplasmic reticulum membranes maintains mitochondrial dynamics and function in hearts in vivo. Circulation. 2017;136:2248–66.
Yu W, Xu M, Zhang T, Zhang Q, Zou C. Mst1 promotes cardiac ischemia-reperfusion injury by inhibiting the ERK-CREB pathway and repressing FUNDC1-mediated mitophagy. J Physiol Sci. 2019;69:113–27.
Chen Y, Liu Y, Dorn GW. Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ Res. 2011;109:1327–31. https://doi.org/10.1161/CIRCRESAHA.111.258723
Hall AR, Burke N, Dongworth RK, Kalkhoran SB, Dyson A, Vicencio JM, et al. Hearts deficient in both Mfn1 and Mfn2 are protected against acute myocardial infarction. Cell Death Dis. 2016;7:e2238.
Papanicolaou KN, Khairallah RJ, Ngoh GA, Chikando A, Luptak I, O’Shea KM, et al. Mitofusin-2 maintains mitochondrial structure and contributes to stress-induced permeability transition in cardiac myocytes. Mol Cell Biol. 2011;31:1309–28.
Jacob S, Kosaka Y, Bhatlekar S, Denorme F, Benzon H, Moody A, et al. Mitofusin-2 regulates platelet mitochondria and function. Circ Res. 2024;134:143–61.
Yepuri G, Ramirez LM, Theophall GG, Reverdatto SV, Quadri N, Hasan SN, et al. DIAPH1-MFN2 interaction regulates mitochondria-SR/ER contact and modulates ischemic/hypoxic stress. Nat Commun. 2023;14:6900.
O’Shea KM, Ananthakrishnan R, Li Q, Quadri N, Thiagarajan D, Sreejit G, et al. The Formin, DIAPH1, is a key modulator of myocardial Ischemia/Reperfusion Injury. EBioMedicine. 2017;26:165–74.
Zhao T, Huang X, Han L, Wang X, Cheng H, Zhao Y, et al. Central role of mitofusin 2 in autophagosome-lysosome fusion in cardiomyocytes. J Biol Chem. 2012;287:23615–25.
Naón D, Hernández-Alvarez MI, Shinjo S, Wieczor M, Ivanova S, de Martins O, et al. Splice variants of mitofusin 2 shape the endoplasmic reticulum and tether it to mitochondria. Science. 2023;380:eadh9351.
Iwasawa R, Mahul-Mellier A-L, Datler C, Pazarentzos E, Grimm S. Fis1 and Bap31 bridge the mitochondria-ER interface to establish a platform for apoptosis induction. EMBO J. 2011;30:556–68.
Lu F, Tian Z, Zhang W, Zhao Y, Li H, Ren H, et al. Calcium-sensing receptors regulate cardiomyocyte Ca2 + signaling via the sarcoplasmic reticulum-mitochondrion interface during hypoxia/reoxygenation. J Biomed Sci. 2010;17:50.
Stoica R, De Vos KJ, Paillusson S, Mueller S, Sancho RM, Lau K-F, et al. ER-mitochondria associations are regulated by the VAPB-PTPIP51 interaction and are disrupted by ALS/FTD-associated TDP-43. Nat Commun. 2014;5:3996.
Gomez-Suaga P, Paillusson S, Stoica R, Noble W, Hanger DP, Miller CCJ. The ER-Mitochondria Tethering Complex VAPB-PTPIP51 regulates Autophagy. Curr Biol. 2017;27:371–85.
Hayashi T, Su T-P. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate ca(2+) signaling and cell survival. Cell. 2007;131:596–610.
Nichtová Z, Fernandez-Sanz C, De La Fuente S, Yuan Y, Hurst S, Lanvermann S, et al. Enhanced Mitochondria-SR Tethering triggers Adaptive Cardiac muscle remodeling. Circ Res. 2023;132:e171–87.
Campo G, Pavasini R, Morciano G, Lincoff AM, Gibson CM, Kitakaze M, et al. Clinical benefit of drugs targeting mitochondrial function as an adjunct to reperfusion in ST-segment elevation myocardial infarction: a meta-analysis of randomized clinical trials. Int J Cardiol. 2017;244:59–66.
Campo G, Pavasini R, Morciano G, Lincoff MA, Gibson C, Kitakaze M. Data on administration of cyclosporine, nicorandil, metoprolol on reperfusion related outcomes in ST-segment elevation myocardial infarction treated with percutaneous coronary intervention. Data Brief. 2017;14:197–205.
Moore CL. Specific inhibition of mitochondrial ca + + transport by ruthenium red. Biochem Biophys Res Commun. 1971;42:298–305.
Rossi CS, Vasington FD, Carafoli E. The effect of ruthenium red on the uptake and release of ca 2 + by mitochondria. Biochem Biophys Res Commun. 1973;50:846–52.
Grover GJ, Dzwonczyk S, Sleph PG. Ruthenium red improves postischemic contractile function in isolated rat hearts. J Cardiovasc Pharmacol. 1990;16:783–9.
Woods JJ, Nemani N, Shanmughapriya S, Kumar A, Zhang M, Nathan SR, et al. A selective and cell-permeable mitochondrial calcium uniporter (MCU) inhibitor preserves mitochondrial bioenergetics after Hypoxia/Reoxygenation Injury. ACS Cent Sci. 2019;5:153–66.
Santo-Domingo J, Vay L, Hernández-Sanmiguel E, Lobatón CD, Moreno A, Montero M, et al. The plasma membrane Na+/Ca2 + exchange inhibitor KB-R7943 is also a potent inhibitor of the mitochondrial Ca2 + uniporter. Br J Pharmacol. 2007;151:647–54.
Ren Y, Deng L, Cai Y, Lv Y, Jia D. The protective effect of Na+/Ca2 + exchange blocker kb-r7943 on myocardial ischemia-reperfusion injury in hypercholesterolemic rat. Cell Biochem Biophys. 2014;70:1017–22.
Kon N, Murakoshi M, Isobe A, Kagechika K, Miyoshi N, Nagayama T. DS16570511 is a small-molecule inhibitor of the mitochondrial calcium uniporter. Cell Death Discov. 2017;3:17045.
Di Marco G, Vallese F, Jourde B, Bergsdorf C, Sturlese M, De Mario A, et al. A high-throughput screening identifies MICU1 targeting compounds. Cell Rep. 2020;30:2321–e23316.
Rasmussen TP, Wu Y, Joiner MA, Koval OM, Wilson NR, Luczak ED, et al. Inhibition of MCU forces extramitochondrial adaptations governing physiological and pathological stress responses in heart. Proc Natl Acad Sci U S A. 2015;112:9129–34.
Yeung HM, Hung MW, Fung ML. Melatonin ameliorates calcium homeostasis in myocardial and ischemia-reperfusion injury in chronically hypoxic rats. J Pineal Res. 2008;45:373–82.
Wang J, Toan S, Li R, Zhou H. Melatonin fine-tunes intracellular calcium signals and eliminates myocardial damage through the IP3R/MCU pathways in cardiorenal syndrome type 3. Biochem Pharmacol. 2020;174:113832.
Hu S, Zhu P, Zhou H, Zhang Y, Chen Y. Melatonin-Induced Protective effects on cardiomyocytes against Reperfusion Injury partly through Modulation of IP3R and SERCA2a Via activation of ERK1. Arq Bras Cardiol. 2018;110:44–51.
Lipskaia L, Hadri L, Lopez JJ, Hajjar RJ, Bobe R. Benefit of SERCA2a gene transfer to vascular endothelial and smooth muscle cells: a new aspect in therapy of cardiovascular diseases. Curr Vasc Pharmacol. 2013;11:465–79.
Niwano K, Arai M, Koitabashi N, Watanabe A, Ikeda Y, Miyoshi H, et al. Lentiviral vector-mediated SERCA2 gene transfer protects against heart failure and left ventricular remodeling after myocardial infarction in rats. Mol Ther. 2008;16:1026–32.
Sakata S, Lebeche D, Sakata N, Sakata Y, Chemaly ER, Liang LF, et al. Restoration of mechanical and energetic function in failing aortic-banded rat hearts by gene transfer of calcium cycling proteins. J Mol Cell Cardiol. 2007;42:852–61.
Lipskaia L, Chemaly ER, Hadri L, Lompre A-M, Hajjar RJ. Sarcoplasmic reticulum ca(2+) ATPase as a therapeutic target for heart failure. Expert Opin Biol Ther. 2010;10:29–41.
Gonnot F, Boulogne L, Brun C, Dia M, Gouriou Y, Bidaux G, et al. SERCA2 phosphorylation at serine 663 is a key regulator of Ca2 + homeostasis in heart diseases. Nat Commun. 2023;14:3346.
Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25:486–541.
Tang D, Kang R, Berghe TV, Vandenabeele P, Kroemer G. The molecular machinery of regulated cell death. Cell Res. 2019;29:347–64.
Zhong H, Song R, Pang Q, Liu Y, Zhuang J, Chen Y, et al. Propofol inhibits parthanatos via ROS-ER-calcium-mitochondria signal pathway in vivo and vitro. Cell Death Dis. 2018;9:932.
Qiao X, Jia S, Ye J, Fang X, Zhang C, Cao Y, et al. PTPIP51 regulates mouse cardiac ischemia/reperfusion through mediating the mitochondria-SR junction. Sci Rep. 2017;7:45379.
Nakayama H, Bodi I, Maillet M, DeSantiago J, Domeier TL, Mikoshiba K, et al. The IP3 receptor regulates cardiac hypertrophy in response to select stimuli. Circ Res. 2010;107:659–66.
Garcia MI, Karlstaedt A, Chen JJ, Amione-Guerra J, Youker KA, Taegtmeyer H, et al. Functionally redundant control of cardiac hypertrophic signaling by inositol 1,4,5-trisphosphate receptors. J Mol Cell Cardiol. 2017;112:95–103.
Liao Z, Liu D, Tang L, Yin D, Yin S, Lai S, et al. Long-term oral resveratrol intake provides nutritional preconditioning against myocardial ischemia/reperfusion injury: involvement of VDAC1 downregulation. Mol Nutr Food Res. 2015;59:454–64.
Di Lisa F, Canton M, Menabò R, Kaludercic N, Bernardi P. Mitochondria and cardioprotection. Heart Fail Rev. 2007;12:249–60.
Elrod JW, Wong R, Mishra S, Vagnozzi RJ, Sakthievel B, Goonasekera SA, et al. Cyclophilin D controls mitochondrial pore-dependent ca(2+) exchange, metabolic flexibility, and propensity for heart failure in mice. J Clin Invest. 2010;120:3680–7.
Hausenloy DJ, Barrabes JA, Bøtker HE, Davidson SM, Di Lisa F, Downey J, et al. Ischaemic conditioning and targeting reperfusion injury: a 30 year voyage of discovery. Basic Res Cardiol. 2016;111:70.
Sun H-Y, Wang N-P, Kerendi F, Halkos M, Kin H, Guyton RA, et al. Hypoxic postconditioning reduces cardiomyocyte loss by inhibiting ROS generation and intracellular Ca2 + overload. Am J Physiol Heart Circ Physiol. 2005;288:H1900–1908.
Quarrie R, Lee DS, Steinbaugh G, Cramer B, Erdahl W, Pfeiffer DR, et al. Ischemic preconditioning preserves mitochondrial membrane potential and limits reactive oxygen species production. J Surg Res. 2012;178:8–17.
Argaud L, Gateau-Roesch O, Raisky O, Loufouat J, Robert D, Ovize M. Postconditioning inhibits mitochondrial permeability transition. Circulation. 2005;111:194–7.
Khalaf A, Babiker F. Discrepancy in calcium release from the sarcoplasmic reticulum and intracellular acidic stores for the protection of the heart against ischemia/reperfusion injury. J Physiol Biochem. 2016;72:495–508.
Hu L, Wang J, Zhu H, Wu X, Zhou L, Song Y, et al. Ischemic postconditioning protects the heart against ischemia-reperfusion injury via neuronal nitric oxide synthase in the sarcoplasmic reticulum and mitochondria. Cell Death Dis. 2016;7:e2222.
Funding
The Signal Transduction Laboratory is supported by the Italian Association for Cancer Research grants IG-23670 (to P.P.) and IG-19803 (to C.G.), A-ROSE (Associazione Ricerca Oncologica Sperimentale Estense); Italian Ministry of Health grants GR-2018-12367114 and GR-2019-12369862 (to G.M.); European Research Council grant 853057-InflaPML (to C.G.); local funds from the University of Ferrara (to P.P. and C.G.).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interest
The authors report no relationships that could be construed as a conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Agyapong, E.D., Pedriali, G., Ramaccini, D. et al. Calcium signaling from sarcoplasmic reticulum and mitochondria contact sites in acute myocardial infarction. J Transl Med 22, 552 (2024). https://doi.org/10.1186/s12967-024-05240-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-024-05240-5