
Abstract
Background
N6-methyladenosine (m6A) is the most prevalent epigenetic modification in eukaryotic messenger RNAs and plays a critical role in cell fate transition. However, it remains to be elucidated how m6A marks functionally impact the transcriptional cascades that orchestrate stem cell differentiation. The present study focuses on the biological function and mechanism of m6A methylation in dental pulp stem cell (DPSC) differentiation.
Methods
m6A RNA immunoprecipitation sequencing was utilized to assess the m6A-mRNA landscape during DPSC differentiation. Ectopic transplantation of DPSCs in immunodeficient mice was conducted to verify the in vitro findings. RNA sequencing and m6A RNA immunoprecipitation sequencing were combined to identify the candidate targets. RNA immunoprecipitation and RNA/protein stability of Noggin (NOG) were evaluated. The alteration in poly(A) tail was measured by 3′-RACE and poly(A) tail length assays.
Results
We characterized a dynamic m6A-mRNA landscape during DPSC mineralization with increasing enrichment in the 3′ untranslated region (UTR). Methyltransferase-like 3 (METTL3) was identified as the key m6A player, and METTL3 knockdown disrupted functional DPSC differentiation. Moreover, METTL3 overexpression enhanced DPSC mineralization. Increasing m6A deposition in the 3′ UTR restricted NOG expression, which is required for DPSC mineralization. This stage-specific m6A methylation and destabilization of NOG was suppressed by METTL3 knockdown only in differentiated DPSCs. Furthermore, METTL3 promotes the degradation of m6A-tagged NOG by shortening the poly(A) tail length in the differentiated stage.
Conclusions
Our results address an essential role of dynamic m6A signaling in the temporal control of DPSC differentiation and provide new insight into epitranscriptomic mechanisms in stem cell-based therapy.
Similar content being viewed by others


Introduction
Mesenchymal stem cells (MSCs) residing in various tissues can still undergo self-renewal and differentiate into specific cell types to maintain tissue homeostasis and fulfill regenerative needs [1]. Multiple adult stem cell types with diverse biological properties have been reported in many tissues and organs. Beyond their similarity in cellular and molecular functions, MSCs exhibit distinct features related to their original phenotypes. Dental pulp stem cells (DPSCs) have superior pluripotency capacity and high mineralization potential, which are essential for hard tissue formation [2]. DPSCs can differentiate into odontoblasts and secrete mineralized matrix known as tertiary dentin bridge formation to seal the vital pulp chamber and prevent pulpal infection from potential insult [3]. The differentiation of DPSCs is critical for tertiary dentin formation and dental repair in vital pulp therapy [3, 4]. Meanwhile, accumulating evidence also supports that DPSCs are capable of differentiating into osteoblasts and forming lamellar bone, which is a promising stem cell source in bone engineering [5]. The initiate signaling reprograms cellular differentiation and extracellular matrix secretion will greatly benefit therapeutic approaches in engineering DPSC-based vital pulp procedures. Epigenetic modulations are capable of temporally controlling the transcription programs in a heritable manner and subsequently guiding quiescent stem cells to undergo preferential trajectories toward differentiation [6]. The key epigenetic regulators could reversibly modulate endogenous stem cell activities and promote functional mineralized tissue formation, which present therapeutic opportunities in regenerative strategies.
N6-methyladenosine (m6A) is the most prevalent posttranscriptional modification of messenger RNA (mRNA) and regulates almost every step of RNA metabolism in mammals [7]. The methyltransferase complex formed by methyltransferase-like 3 (METTL3), METTL14, and Wilms tumor 1-associated protein (WTAP) is responsible for the m6A modification of mRNA, which catalyzes adenosines with methyl groups from metabolite substrates [8]. Large-scale transcripts are dynamically and timely tagged by m6A marks to orchestrate the different stages of stem cells. RNA m6A modification has emerged as a critical epitranscriptomic mechanism that regulates embryonic development, cell reprogramming and differentiation [9, 10]. The disruption of m6A modification in the stem cell program displays diverse effects across cell types and specific fate stages. Reduction of m6A deposition improved pluripotency and blocked regeneration of embryonic stem cells [11], while it limited self-renewal and triggered cell differentiation in epiblast stem cells and induced pluripotent stem cells [12, 13]. It is critical to identify the regulatory mechanism of dynamic m6A marks in the DPSC fate transition. Our previous study characterized the m6A-tagged landscape in immature DPSCs, which is related to cell senescence and apoptosis [14]. Meanwhile, how m6A methylation participates in DPSC differentiation remains unclear. Clarifying the RNA epigenetic mechanism during DPSC differentiation and manipulating the key modulators in therapeutic applications would advance vital pulp therapy.
In this study, we revealed a dynamic and unique m6A-mRNA landscape with m6A RNA immunoprecipitation-sequencing (m6A RIP-seq), which provides an entry point to uncover the potential function of m6A methylation in DPSC differentiation. METTL3 was identified as a key molecule that mediates m6A modification in DPSC mineralization. Remarkably, dynamic m6A methylation of noggin (NOG) confers its stabilization by shortening the poly(A) tail in a stage-specific manner. Our results provide evidence for the critical role of m6A modification in DPSC differentiation and shed light on the epitranscriptomic mechanism in the temporal control of cell fate transition.
Materials and methods
DPSC isolation and culture
Dental pulp tissues were collected according to the appropriate guidelines after written informed consent was obtained by a protocol approved by the Human Research Committee of Stomatological Hospital, Southern Medical University (ethical code 2019(16)). Primary DPSCs were harvested and cultured as previously described [15]. Briefly, dental pulp tissues were removed from extracted third molars and then digested with 3 mg/mL collagenase I (Gibco-Invitrogen, Carlsbad, CA, USA). DPSCs were collected and cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum and 1% penicillin and streptomycin (all from Gibco-Invitrogen) at 37 °C with 5% CO2. The medium was changed every 2 days, and DPSCs at passages 3–5 were used for subsequent experiments.
DPSCs (at a density of 2 × 105 cells/mL) were seeded in 6-well or 24-well plates (Corning Life Sciences, NY, US) and cultured until the cells reached 80–90% confluence. The culture medium was changed to osteo/odontogenic medium (OM) containing 10 mmol/L β-glycerophosphate, 50 μg/mL ascorbic acid and 0.1 μmol/L dexamethasone (all from Sigma‒Aldrich, St. Louis, MO, USA). The induction medium was changed every other day from day 0 to day 21 (the day of induction was defined as day 0). After 7 days of induction, the cells were fixed and stained for alkaline phosphatase (ALP) (Beyotime Biotechnology, Shanghai, China) (n = 5). Alizarin red staining (ARS) (Beyotime) was used to stain the accumulated mineralized matrix on day 14 (n = 5). For adipogenic differentiation, DPSCs were induced by the Adipogenesis Differentiation Kit (HUXXC-90031, Cyagen Biosciences, Guangzhou, China). Cells were cultured with adipogenic inducing solution A for 3 days, and the medium was replaced with solution B for 1 day and then replaced with solution A. This cycle was repeated four times and then subjected to oil red O staining (Cyagen Biosciences) (n = 5).
Total m6A and methylated metabolite measurements
DPSCs were subjected to osteo/odontogenic induction for 0, 7, and 14 days, and RNA was extracted using TRIzol solution (Takara Biotechnology, Shiga, Japan). Total m6A was measured with an m6A RNA methylation quantification kit (P-9019-96, Epigentek, Farmingdale, NY, US) according to the manufacturer’s protocol.
Liquid chromatography with tandem mass spectrometry (LC‒MS/MS) analysis was utilized to assess the metabolite compounds related to m6A methylation, including S-adenosylmethionine (SAM) and S-adenosylhomocysteine (SAH). Cell samples were subjected to methanol and homogenized before analysis with an ultrahigh-performance liquid chromatography (UHPLC) column (1290 Infinity LC, Agilent Technologies). Next, during MS/MS acquisition, a m/z range of 25–1000 Da was used, and the ion accumulation time was screened. Metabolite compounds were identified with a database of available standards and subjected to multivariate data analysis. Metabolites with a variable importance in projection (VIP) value > 1 were further subjected to statistical analysis.
m6A RIP-seq and m6A RIP-qPCR
m6A RIP-seq was used to characterize m6A modification during DPSC mineralization (osteo/odontogenic medium-induced DPSCs, OM-DPSCs). Total RNA was isolated from DPSCs induced for 0, 7, and 14 days with TRIzol solution. The extracted RNA enriched and purified with oligo(dT)-attached magnetic beads and an m6A RIP kit (17-10499, Millipore, Burlington, MA, US) according to the kit’s protocol. The purified m6A-RIP RNA fragments were then fragmented into small pieces with fragmentation buffer for sequencing. The RNA fragments were incubated with magnetic beads conjugated with an m6A-specific antibody in buffer. The IP RNA and input RNA were reverse transcribed into cDNA and subjected to deep sequencing on an Illumina NovaSeq™ 6000 platform for m6A RIP-seq [14].
RNA sequencing and profile analysis
Total RNA was extracted from DPSCs, and poly(A) mRNA was purified with poly(T)-conjugated magnetic beads. Then, the mRNA was fragmented into small pieces and converted into double-stranded cDNA. Paired‐end runs with a read length of approximately 300 base pairs (bp) were used for RNA‐sequencing with the Illumina deep sequence platform by LC-BIO Technologies Co., Ltd. Significantly differentially expressed genes (DEGs) were identified in this study as genes with a fold change in expression ≥ 2.0 and corresponding p value < 0.05. The DEGs were subjected to Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses with the R packages GOseq and DAVID.
Lentiviral vector construction and infection
Two independent METTL3 shRNA sequences (shMETTL3-1 and shMETTL3-2) within lentiviral vectors were used. shMETTL3-1 lentivirus encoding METTL3-shRNA and shCTR-1 lentivirus encoding scrambled control (shCTR) sequences were constructed by GeneChem Company (Shanghai, China); shMETTL3-2 and shCTR-2 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, US). cDNA encoding the full-length METTL3 gene was amplified and used to construct a lentiviral vector for METTL3 overexpression (LV-METTL3) by GeneChem Company. For lentivirus transfection, DPSCs were transfected with lentivirus at a multiplicity of infection (MOI) of 50 and cultured for 72 h before subsequent experiments. DPSCs with METTL3 knockdown were treated with 2 µg/mL human Noggin peptide (ab16380, Abcam, Cambridge, UK) to neutralize noggin protein during DPSC differentiation.
Quantitative polymerase chain reaction (qPCR)
The cells were digested and total RNA was extracted from DPSCs with a Total RNA Isolation Kit (Foregene Biotechnology, Chengdu, China). The RNA was then reverse transcribed with RT Master Mix (Takara) according to the manufacturer’s instructions to obtain complementary DNA (cDNA). Real-time qPCR was performed with TB Green qPCR Mix (Takara) according to the manufacturer’s protocol. Relative target gene expression was analyzed with a standard curve and normalized to Glyceraldehyde-3-Phosphate Dehydrogenase (GAPDH) expression. The primer sequences used in qPCR are summarized in Additional file 1: Table S1.
Western blot analysis
DPSCs were lysed and assayed with a BCA protein assay kit (Beyotime). Samples containing 15–30 μg of protein were subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis, and the proteins were then transferred to polyvinylidene fluoride membranes (Millipore). The membranes containing the transferred proteins were blocked with 5% bovine serum albumin and reacted with the primary antibody overnight at 4℃. The membranes were then labeled with corresponding secondary antibody of horseradish peroxidase (HRP)-conjugated anti-mouse IgG or anti-rabbit IgG for 1 h at room temperature before visualization with SuperSignal enhanced chemiluminescence substrate (Thermo Fisher Scientific, Waltham, MA, US). Primary and secondary antibodies against the following proteins were used in this study: METTL3 (96391, 1:1000); METTL14 (51104, 1:1000); WTAP (56501, 1:1000); p-Smad1/5 (9516, 1:1000) were purchased from Cell Signaling Technology (CST, Danvers, MA, USA). NOG (sc-293439, 1:1000); RUNX Family Transcription Factor 2 (RUNX2, sc-390351, 1:1000); Dentin Sialophosphoprotein (DSPP, sc-73632, 1:1000); Smad1/2/3 (sc-7960, 1:1000); p-Smad3 (sc-517575, 1:1000) were obtained from Santa Cruz Biotechnology. GAPDH (60004-1-Ig, 1:3000); goat anti-mouse IgG (SA00001-1, 1:3000) and goat anti-rabbit (SA00001-2, 1:3000) were purchased from ProteinTech Group (ProteinTech, Wuhan, China).
Animal model construction
The animal experiments were conducted in compliance with ARRIVE guidelines and the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All procedures followed protocols approved by the Ethics Committees of Stomatological Hospital, Southern Medical University (ethical code 2019(16)). For ectopic transplantation studies, porous beta-tricalcium phosphate/hydroxyapatite (β-TCP/HA) discs (diameter: 4 mm, thickness: 2 mm) were obtained from Biological Materials Manufacturing Core, Sichuan University. Approximately 1 × 106 transfected DPSCs were seeded on β-TCP/HA discs and cultured in a 24-well plate with odontogenic medium for 24 h [17, 18]. The composites of DPSCs and the β-TCP/HA scaffold were transplanted into the subcutaneous dorsal pockets of 6-week-old BALB/c immunodeficient nude mice (n = 10) [18, 19]. Two subcutaneous pockets were made on the right and left side of the dorsal surface, each allowing for one composite. The shCTR and shMETTL3 groups were carefully transplanted into the left and right subcutaneous regions, respectively (n = 5), as were the LV-METTL3 and LV-CTR groups (n = 5). After 4 weeks, the harvested implants were fixed with paraformaldehyde, followed by decalcification with Ethylenediaminetetraacetic acid (EDTA) for 2 weeks. The formation of new mineralized tissue was evaluated by Masson-trichrome staining.
Immunofluorescence staining
The composites of DPSCs and the β-TCP/HA scaffold were fixed, dehydrated, embedded in paraffin and then cut at 5 µm. The tissue slides were subjected to immunofluorescence with a standard protocol. The slides were incubated with primary antibody overnight at 4℃. After washing, samples were interacted with the corresponding secondary HRP-conjugated antibody (1:1000, ProteinTech) for 1 h and then labeled with Cy3 Tyramine (11065, AAT Bioquest, Inc. Sunnyvale CA, US) or AF 488 Tyramide reagent (11070 AAT Bioquest). Primary antibodies against the following proteins were used in this study: METTL3 (15073-1-AP, 1:200, ProteinTech); NOG (sc-293439, 1:200, Santa Cruz); p-Smad3 (sc-517575, 1:100, Santa Cruz) and Smad1/2/3 (sc-7960, 1:100, Santa Cruz).
Immunofluorescence staining of the induced DPSCs was performed as described in a previous study [15]. Fixed cells were blocked and incubated with the primary antibodies anti-METTL3 and anti-NOG overnight and then the corresponding fluorescent secondary antibody of Cy3–conjugated anti-rabbit IgG (SA00009-2, 1:500, ProteinTech) or fluorescein (FITC)–conjugated anti-mouse (SA00003-1, 1:500, ProteinTech). Fluorescent images showing subcellular expression were obtained with a confocal microscope (LSM 900; Zeiss, Oberkohen, Germany).
m6A site-specific mutant plasmid construction
To investigate the biological effect of the m6A methylated sites in the 3′ untranslated region (UTR) of NOG, full-length NOG cDNA was constructed and cloned into the NheI- and BamHI-digested Flag-PCDNA3.1(+) vector by Genecreate Biotechnology Co., Ltd. (Wuhan, China). The vector used to express NOG with the wild-type m6A motif was referred to as NOG-WT. Two different mutants (NOG-MUT1 and NOG-MUT2) were generated by introducing single-nucleotide mutations (A to T) in the four m6A motifs of the 3′ UTR after map** the conserved m6A motif sequence in m6A RIP-seq. To examine mRNA expression, 293T cells were transfected with NOG-WT vector or a mutant NOG vector and subjected to qPCR [20].
RNA and protein stability analysis
RNA transcription in DPSCs was inhibited by treatment with 5 µg/mL actinomycin D (ActD, 7240-37-1, Sigma‒Aldrich) as described in a previous study to analyze mRNA decay rates [21]. mRNA was isolated after 0, 4, and 8 h and subjected to qPCR. The half-life of NOG mRNA has been reported.
Protein translation in DPSCs was inhibited by treatment with 100 µg/mL cycloheximide (CHX, A8244, APExBIO Technology, Houston, TX, US) to analyze protein stability. Total protein was isolated after 0, 4, and 8 h and subjected to western blot analysis. The protein expression level of NOG was used to analyze its stability.
3′-Rapid amplification of cDNA ends (3′-RACE)
3′-RACE to obtain the 3′ UTR sequences of NOG from shMETTL3 or shCTR DPSCs after osteogenic induction was performed with a 3′-RACE kit (6106, Takara) according to the protocol. The 3′-RACE products after two rounds of amplification were purified and subcloned into a vector, and the amplified fragments were further identified by sequencing [22]. The NOG-specific primers used for 3′-RACE analysis in this study were as follows: 5′-CATGGTGTGCAAGCCGTCCAAGTC-3′, 5′-TCACGGTGCTGCGGTGGCGCTGTC-3′.
Poly(A) tail assay
A poly(A) tail assay was performed with a Poly(A) Tail-Length Assay Kit (764551KT, Thermo Fisher) according to the protocol. Briefly, poly(A) polymerase was used to add G and I to the 3′ ends of the RNA, and the newly tailed RNA was converted to cDNA by reverse transcription. Then, NOG-specific forward and reverse primers and universal reverse primers were used to generate a product consisting of NOG with a poly(A) tail. The PCR products from METTL3 knockdown DPSCs in differentiated/undifferentiated stages were separated on agarose gels [23]. The specific primers for NOG were F: 5′-TAACCTGCTATTTATATTCCAGTGCCCTTC-3′ and R: 5′-TGAACTCTATAGCTTCTTCGAGGTCCAA-3′.
Quantification and statistical analysis
The experiments in this study were carried out biologically repeated at least three times, and the data are presented as the mean ± standard deviation. Statistical differences were evaluated by one‑way analysis of variance (ANOVA) and corresponding post hoc tests for multiple comparisons. Unpaired two-tailed Student’s t test was used to compare two groups. A p value < 0.05 was considered to be statistically significant and was analyzed by GraphPad Prism 7.0 (La Jolla, CA, US).
Results
The dynamic m6A epitranscriptomic landscape in DPSC differentiation
We first examined the total m6A content and epitranscriptomic profile of m6A-tagged mRNA. The total m6A level in the RNA pool was upregulated upon osteo/odontogenic induction for 7 and 14 days, as quantified by a colorimetric method (Fig. 1A). m6A RIP-seq analysis showed that the majority of m6A peaks were enriched in the DNA coding sequence (CDS), 3′ UTR and stop codon, with small subsets located in the 5′ UTR and start codon (Fig. 1B, C). Notably, the density of m6A peaks in the 3′ UTR increased over time, and the representative motif sequence of m6A sites also underwent slight modification (Fig. 1B–D). Next, conjoint analysis of m6A RIP and RNA-seq was used to identify the differentially m6A-methylated and expressed molecules and some representative genes were marked (Fig. 1E). These m6A-tagged genes were found to be mainly enriched in biological processes related to signal transduction, transcriptional regulation and cell differentiation (Fig. 1F). The signaling pathways as transforming growth factor-β (TGF-β) and Rap1 were identified in KEGG pathway analysis (Additional file 1: Fig. S1A).

The dynamic m6A epitranscriptomic landscape in DPSC differentiation. A DPSCs were induced with osteo/odontogenic medium for 0, 7, and 14 days, and then the total m6A level in the RNA polls was quantified by the colorimetric method. B The m6A methylation landscape in the genome-wide transcriptome was evaluated by m6A RIP-seq. Distribution of m6A peaks in the 3′ UTR, CDS region and 5′ UTR. C Pie chart analysis of the m6A peak fraction in transcript segments. D Specific and conserved motif sequences of high-confidence m6A peaks identified by the HOMER database. E Epitranscriptome profile of m6A-tagged mRNAs in DPSCs after odontogenic differentiation. F Gene Ontology enrichment analysis of differentially m6A methylated and expressed transcripts. G The concentrations of SAM, SAH and potential of m6A establishment were qualified by LC‒MS/MS. Metabolites with a VIP value > 1 were further subjected to statistical analysis (n = 6). H The mRNA expression of METTL3, METTL14, and WTAP during DPSC mineralization, as evaluated by qPCR (n = 3). I The protein expression of METTL3, METTL14, and WTAP in DPSCs was evaluated by western blotting. SAM: S-adenosylmethionine; SAH: S-adenosylhomocysteine. Significance was determined via ANOVA or Student’s t test. The quantitative data are presented as the mean ± SD *p < 0.05. **p < 0.01. ***p < 0.001
The establishment of m6A marks relies on the riboswitch from the methyl donor SAM to SAH conducted by the methyltransferase complex. We next evaluated the methylation potential and expression pattern of m6A-related molecules during DPSC mineralization. The concentrations of the methyl metabolite (SAH) were increased after induction, while the methyl donor (SAM) and methylated potential (SAM/SAH ratio) showed no significant variation (Fig. 1G). RNA-seq analysis showed that the relative gene expression of METTL3 was markedly increased on days 7 and 14 of induction among all the m6A-related molecules (Additional file 1: Fig. S1B). Regarding methyltransferases, the mRNA and protein levels of METTL3 also increased after odontogenic induction and peaked on day 14, while METTL14 exerted low protein expression levels, and WTAP showed no obvious trend (Fig. 1H, I). Taken together, these data suggest that METTL3 is the key player in m6A modification during DPSC mineralization.
METTL3 is required for functional DPSC differentiation
To identify the potential impact of METTL3 deletion on DPSC differentiation, two shRNA lentiviral vectors were used to knockdown METTL3 expression and both sufficiently suppressed the mRNA and protein expression by more than half (Fig. 2A, B). ALP, RUNX2 and DSPP were also downregulated at the mRNA level (Fig. 2C). The protein expression of RUNX2 was significantly reduced after 3 and 14 days of induction, and DSPP was also suppressed on days 3 and 7 (Fig. 2D). METTL3 knockdown led to weaker ALP activity and less mineralized nodule formation after 7 and 14 days of odontogenic induction (Fig. 2G, H). These results indicated that METTL3 inhibition impaired the odontogenic differentiation of DPSCs. Meanwhile, the expression of METTL3 was suppressed during adipogenic induction (Fig. 2E). METTL3 inhibition enhanced the mRNA expression of Lipoprotein Lipase (LPL) and Peroxisome Proliferator Activated Receptor (PPAR) and promoted lipid droplet formation (Fig. 2F, I). Overall, METTL3 inhibition compromised odontogenesis but promoted adipogenesis, implying a critical role of m6A methylation and METTL3 in controlling DPSC differentiation.

METTL3 is required for functional DPSC differentiation. A Two independent shRNA lentiviruses were used to inhibit METTL3 expression, as confirmed by qPCR. B Western blotting detected the protein expression of METTL3 in DPSCs after METTL3 shRNA treatment (n = 3). C qPCR analysis of the mRNA expression of ALP, RUNX2 and DSPP in differentiated DPSCs (n = 3). D The protein expression levels of METTL3, RUNX2 and DSPP in METTL3 knockdown DPSCs under odontogenic induction were evaluated by western blotting. E The mRNA expression level of METTL3 in DPSCs after adipogenic induction for 0, 3, 7, and 14 days. F The mRNA expression of PPAR and LPL after adipogenic induction as detected by qPCR (n = 3). G The odontogenic differentiation of shMETTL3-1 and shCTR-1 lentivirus-transduced DPSCs was evaluated by ALP and ARS staining (n = 5). H The differentiation capacity of shMETTL3-2- and shCTR-2-transduced DPSCs after induction. I Representative images of Oil Red O staining in METTL3 knockdown DPSCs. OM-DPSCs: DPSCs induced by odontogenic medium. Significance was determined via ANOVA or Student’s t test; the data are presented as the mean ± SD (n ≥ 3). *p < 0.05. **p < 0.01. ***p < 0.001
METTL3 overexpression enhances the mineralization of DPSCs
Considering the negative effect of METTL3 inhibition, we hypothesized that METTL3 overexpression would benefit DPSC mineralization. The elevated mRNA and protein levels of METTL3 upon overexpression lentivirus transfection were confirmed (Fig. 3A, B). METTL3 overexpression induced ALP activity and calcium nodule formation (Fig. 3C). The favorable effect of METTL3 overexpression was also evidenced by the increased mRNA expression of ALP, RUNX2, and DSPP and protein expression of RUNX2 and DSPP (Fig. 3D, E). These results support the promoting effect of METTL3 overexpression on DPSC odontogenic differentiation and matrix mineralization.

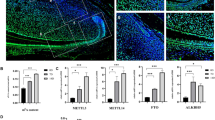
METTL3 overexpression enhances the mineralization of DPSCs. A Lentivirus was constructed to overexpress METTL3, as confirmed by qPCR. B Western blotting detected the protein expression of METTL3 in DPSCs after METTL3 overexpression. C The odontogenic differentiation of DPSCs after METTL3 overexpression was evaluated by ALP staining and ARS staining (n = 5). D qPCR analysis of the mRNA expression levels of ALP, RUNX2 and DSPP in METTL3-overexpressing OM-DPSCs. E The protein expression levels of METTL3, RUNX2 and DSPP were assayed by western blotting (n = 3). F Masson trichrome staining of the composites of β-TCP/HA scaffolds and DPSCs treated with METTL3 shRNA after subcutaneous transplantation in nude mice for 4 weeks (n = 5). G Subcutaneous transplantation of DPSCs transduced with METTL3 overexpression and control lentivirus (n = 5). H Collagen fibers and newly formed mineralized tissue were stained blue and evaluated by ImageJ. Significance was determined via two-tailed Student’s t test; the data are presented as the mean ± SD (n ≥ 3). *p < 0.05. **p < 0.01. ***p < 0.001
To evaluate the role of METTL3 in DPSC mineralization in vivo, we conducted subcutaneous transplantation of β-TCP/HA scaffolds with DPSCs in BALBc nude mice. DPSCs were subjected to METTL3 knockdown or overexpression prior to the incubation with β-TCP/HA scaffolds. Hematoxylin and eosin staining was performed to evaluate cellularity alteration (Additional file 1: Fig. S2) and Masson-trichrome staining assess extracellular matrix production. Immature mineralized tissue and collagen fiber (blue staining) formation were reduced by approximately two-thirds in the METTL3 knockdown DPSC groups compared to the control groups (Fig. 3F, H). Moreover, METTL3 overexpression enhanced collagen tissue formation by more than twofold after 4 weeks (Fig. 3G, H), indicating the therapeutic potential of METTL3 in DPSC differentiation and mineralized tissue formation.
Dynamic m6A modification of NOG orchestrates the differentiation stage
To dissect the potential targets of METTL3-mediated m6A modification in DPSC differentiation, transcriptome sequencing was used to screen the gene expression profile after METTL3 deletion in OM-DPSCs (Additional file 1: Fig. S3). GO analysis showed that these DEGs were mainly related to mRNA 3′ UTR binding, signal transduction and cell differentiation (Fig. 4A, B), which corresponds to the elevated m6A accumulation in the 3′ UTR during DPSC mineralization. Signaling pathways regulating the pluripotency of stem cells, TGF-β and Wnt signaling pathways were also enriched in the KEGG pathway analysis (Fig. 4C). In the m6A-RIP and transcriptome sequencing of DPSC mineralization, 279 genes with significant differences in both m6A peaks and expression were identified (Figs. 1E, 4D). Next, we filtered the potential m6A target genes of METTL3 by taking the intersection of these 279 genes and the shMETTL3-related DEGs in both the early and late stages of differentiation (Fig. 4E). Among the six candidate targets, NOG is a well-known inhibitor of bone morphogenetic protein (BMP) signaling and the downstream Smad pathway.

NOG is a target gene of METTL3-mediated m6A modification. A The top terms identified by Gene Ontology enrichment analysis of overlap** DEGs in METTL3-knockdown OM-DPSCs after induction for both 7 and 14 days. B Scatter plots of the top items in Gene Ontology enrichment. C The enrichment analysis of KEGG pathways. D The differentially m6A methylated and expressed genes in the m6A-mRNA profile after induction for 7 and 14 days by m6A RIP & RNA sequencing. E Venn diagram showing overlap** genes between DEGs in transcriptome sequencing after METTL3 knockdown and m6A-methylated transcripts during DPSC mineralization. DEGs: differentially expressed genes; D(m6A)MGs: differentially m6A methylated genes
The gene and protein expression levels of NOG were significantly reduced by half in DPSCs upon odontogenic induction, which was consistent with the transcriptome sequencing data (Fig. 5A, B). Immunofluorescence staining showed that the cytoplasmic expression of NOG was significantly suppressed after induction (Additional file 1: Fig. S4A). Moreover, the colocalization coefficient of METTL3 and NOG was reduced in differentiated DPSCs (Additional file 1: Fig. S4B). Visualization of m6A RIP-seq data showed that high-confidence and specific m6A peaks in the 3′ UTR of NOG mRNA markedly increased after induction (Fig. 5C). The two ascending m6A peaks in the 3′ UTR of NOG appeared to be the center of four m6A sites after map** the conserved motif sequence. To explore the potential impact of these m6A marks on gene expression, NOG expression vectors containing wild-type m6A sites (WT) and two A-to-T m6A mutations (MUT1 and MUT2) were constructed (Fig. 5D). Both m6A mutations resulted in increased mRNA expression of NOG compared with WT expression in 293 T cells (Fig. 5E). These data demonstrated that m6A tag accumulation in the 3′ UTR of NOG resulted in diminished gene expression, which is required for DPSC differentiation.

Dynamic m6A modification of NOG orchestrates the differentiation stage. A The relative gene expression and mRNA expression of NOG during DPSC differentiation. B The protein expression level of NOG detected by western blotting. C Visual data of high-confidence m6A peaks enriched in NOG mRNA detected by m6A RIP-seq. D Details of vectors containing fragments of the NOG 3′ UTR with the wild-type m6A motif or two independent m6A mutants (A-to-T mutation). The numbers (450, 463 and 571, 583) represent the positions of the m6A sites relative to the 3′ UTR. E qPCR assay of the mRNA expression of NOG in 293T cells transduced with the wild-type or m6A-mutant vector (n = 3). F m6A RIP-qPCR demonstrated the m6A deposition alteration in NOG mRNA after METTL3 inhibition. G Immunofluorescence staining showed the METTL3 and NOG expression in the composites of β-TCP/HA scaffolds with METTL3-knockdown DPSCs after subcutaneous transplantation in nude mice for 4 weeks. H Subcutaneous transplantation of DPSCs transduced with METTL3 overexpression and control lentivirus. I The expression level of METTL3 and NOG evaluated by imageJ. Significance was determined via ANOVA or Student’s t test; the data are presented as the mean ± SD (n ≥ 3). *p < 0.05. **p < 0.01. ***p < 0.001
Next, we investigated the potential effect of METTL3 depletion on m6A-tagged NOG. The m6A methylation level of NOG decreased sharply after METTL3 knockdown in OM-DPSCs (Fig. 5F), indicating that the elevated m6A enrichment in NOG was mediated by METTL3. In the ectopic mineralization models, the composites of β-TCP/HA scaffolds with METTL3-knockdown DPSCs showed a remarkable upregulation in NOG expression (Fig. 5G, I), while the NOG secretion were significantly suppressed in METTL3-overexpression transfected composites after subcutaneous transplantation (Fig. 5H, I).
Stage-specific NOG expression is regulated by METTL3
NOG antagonizes BMP signaling and selectively inhibits BMP-related Smad pathway activity, which is essential for cell differentiation. Notably, METTL3 knockdown enhanced the relative expression of NOG in the differentiated state but not in the undifferentiated state, and qPCR confirmed the inhibitory effect of METTL3 on NOG mRNA expression in only OM-DPSCs (Fig. 6A). Additionally, METTL3 deletion significantly upregulated the protein expression of NOG in OM-DPSCs, and METTL3 overexpression exerted the opposite effect (Fig. 6B). Meanwhile, no stable trend was observed for NOG expression changes in undifferentiated DPSCs. These data suggested that METTL3 regulated m6A-tagged NOG expression to orchestrate the differentiation process. Furthermore, we evaluated the downstream signal transduction of NOG by assessing phosphorylation of Smad1/3/5 [24]. METTL3 deletion suppressed the phosphorylation of Smad3 and Smad1/5, while METTL3 overexpression enhanced the activation of the Smad pathway in differentiated DPSCs (Fig. 6C). Consistently, the phosphorylation level of Smad3 were dramatically inhibited in METTL3-knockdown DPSCs with β-TCP/HA scaffolds, while significantly enhanced in METTL3-overexpression composites in the ectopic mineralization models (Additional file 1: Fig. S5).

Stage-specific NOG expression is regulated by METTL3. A The relative gene expression and mRNA expression of NOG in differentiated and undifferentiated DPSCs. B The protein expression level of NOG after METTL3 alteration determined by western blotting (n = 3). C Activation of the downstream Smad signaling pathway was evaluated by measuring the protein expression of p-Smad3, p-Smad1/5 and Smad in OM-DPSCs. D ALP activity and calcium mineralization formation in METTL3-knockdown DPSCs treated with NOG blocking antibody (n = 3). E The RUNX2 and DSPP mRNA expression levels assayed by qPCR. Significance was determined via ANOVA or Student’s t test; the data are presented as the mean ± SD (n ≥ 3). *p < 0.05. **p < 0.01. ***p < 0.001
The impaired ALP activity and calcium mineralization formation induced by METTL3 inhibition were significantly upregulated after neutralizing NOG protein with a blocking antibody (Fig. 6D), as was the mRNA expression of RUNX2 and DSPP (Fig. 6E). These data demonstrated that neutralizing excess NOG secretion can partially rescue insufficient odontogenic differentiation of METTL3-deleted DPSCs.
METTL3 programs NOG destabilization via poly(A) tail shortening
Clustered m6A hallmarks in the 3′ UTR around the stop codon mainly influence the stability and localization of RNA. RNA stability assays with actinomycin D to suppress transcription showed that METTL3 knockdown increased the half-life of NOG mRNA in differentiated DPSCs (Fig. 7A). Furthermore, in the undifferentiated state, the mRNA decay of NOG was not significantly decelerated (Fig. 7A). Then, we blocked protein translation and synthesis by cycloheximide. The protein degradation of NOG in differentiated DPSCs was also suppressed after METTL3 inhibition (Fig. 7B). Consistent with previous data, METTL3 specifically restricted the stabilization and promoted the degradation of m6A-tagged NOG in differentiated DPSCs.

METTL3 programs NOG destabilization via poly(A) tail shortening. A The remaining NOG mRNA in differentiated and undifferentiated DPSCs treated with actinomycin D was quantified by qPCR. B Protein translation was blocked by cycloheximide, and the remaining expression level of NOG was detected by western blotting (n = 3). C 3′-RACE characterization of the polyadenylation site and 3′ UTR information of NOG. D Diagram of 3′-RACE analysis of NOG mRNA. E The poly(A) tail length of the NOG transcript in METTL3 knockdown DPSCs was assayed by poly(A) tail length measurement. F Schematic of the m6A modification machinery in DPSC mineralization. Significance was determined via ANOVA or Student’s t test; the data are represented as the mean ± SD (n ≥ 3). *p < 0.05. **p < 0.01. ***p < 0.001
The poly(A) tail determines the mRNA stability and translation initiation of most eukaryotic mRNAs. Transcriptome sequencing suggested the presence of only one transcript of NOG without alternative splicing, which is in line with the transcript reported in the National Center for Biotechnology Information (NCBI) database (reference sequence: NC_000017.11). 3′-RACE analysis was conducted to obtain the 3′ UTR sequence information of NOG after METTL3 depletion. The 3′-RACE results showed no obvious alterations in the polyadenylation site or 3′ UTR length, while the number of A residues appeared to be increased in OM-DPSCs (Fig. 7C, D). Traditional RACE might result in PCR bias in the poly(A) tail due to the use of universal adaptor primers and nested amplification [22]. The poly (A) tail length was further quantified by PCR with NOG-specific primers after random oligomer addition of guanosine and inosine residues. The poly(A) tail was dozen bps longer after METTL3 knockdown in differentiated DPSCs, but this difference was not detected in undifferentiated DPSCs (Fig. 7E). These data support that METTL3 exerts temporal control over NOG stabilization to orchestrate DPSC differentiation (Fig. 7F).
Discussion
Emerging evidence has proven that m6A RNA methylation is a critical epitranscriptomic mechanism that permits additional specificity and plasticity to the transcriptome [25]. Here, we revealed the dynamic and unique m6A mRNA landscape in DPSC mineralization, and elevated m6A marks in the 3′ UTR of certain transcripts are required for transcriptional prepatterning. METTL3 was identified as the essential m6A modulator in regulating DPSC differentiation. Furthermore, increasing m6A hallmarks in the 3′ UTR restricted the gene expression of NOG during DPSC mineralization. METTL3 mediated the m6A modification of NOG and promoted its degradation via poly(A) tail shortening in a stage-specific manner. The present study addressed a critical role of m6A modification in the temporal control of DPSC differentiation and provided new insight into the transcriptional coordination of stem cell regulation.
RNA m6A deposition is redundant in the consensus motif RRm6ACH ([G/A/U][G > A]m6AC[U > A > C]), which is enriched in the CDS and 3′ UTR of RNA transcripts [8, 26]. During embryonic cortical neurogenesis, m6A-methylated transcripts are enriched in biological processes, such as neural stem cells, the cell cycle, and differentiation, which are essential to control the transcriptome composition of different stages [25]. We characterized the dynamic and unique m6A landscape in DPSC mineralization, and the m6A-mRNA profile was mainly related to transcriptional regulation and cell differentiation. Moreover, the increasing total m6A content and m6A distribution in the 3′ UTR might result from a pronounced elevation of METTL3 expression. METTL3 was reported to participate in tooth root development by modulating translational efficiency [27]. The inhibition of DPSC proliferation and osteogenesis by METTL3 knockdown was associated with an impaired glycolytic pathway [28]. METTL3 is also involved in bone mesenchymal stem cell (BMSC) differentiation and function [30, 31]. Ectopic mineralization models were used in this study and subcutaneous transplantation in immunocompromised mice supported that METTL3 is a positive regulator of DPSC differentiation and mineralized tissue formation. More evidences from orthotopic models are needed to support the therapeutic application in vital pulp procedures and DPSC-based therapy.
Various transcripts and signals are tagged in a timely manner by m6A modification, which in turn controls proper development and differentiation. METTL3-mediated m6A modification regulates the expression of some osteogenic markers and other related genes involved in bone metabolism [32]. Parathyroid hormone (PTH)/Pth1r, TGF-β/SMAD, WNT and other signaling pathways are modulated by m6A marks, which are essential in the cellular differentiation and cancer development [16, 33, 34]. Here, NOG and downstream Smad pathway were identified as the target of METTL3-mediated m6A modification during DPSC differentiation. NOG is a key player in ectoderm development, and its disruption can lead to organogenesis defects such as craniofacial defects and hypoplastic teeth [35, 36]. Noggin is capable of binding and inactivating members of the TGF-β superfamily proteins as BMPs, subsequently blocking BMP-induced Smad pathway activation [37]. BMSC osteogenesis and DPSC odontogenesis are regulated by NOG via the downstream Smad1/5 signaling pathway [38, 39]. We found that the m6A peaks in the 3′ UTR of NOG mRNA increased during DPSC mineralization, which restricted its gene expression. METTL3 inhibited m6A-tagged NOG expression and promoted its degradation in differentiated DPSCs. Consistent with our data, m6A modification modulates RNA degradation and gene expression in neural stem cells, which is a critical epigenetic mechanism in the temporal control of neurogenesis [25]. m6A signaling clustered in the 3′ UTR is mainly responsible for cytoplasmic events related to RNA stability and translation [40,41,42], and METTL3 can independently read and modulate m6A marks in the 3′ UTR of certain transcripts [40]. Taken together, these findings suggest that m6A modification dynamically modulates the stability of specific transcripts, which is required for the transcriptional prepatterning of DPSC mineralization.
Poly(A) tails are 150–250 adenosine nucleotides acquired by the end of the 3′ UTR in the nucleus that subsequently undergo deadenylation in the cytoplasm. The length of a poly(A) tail changes throughout the lifetime of mRNA and has essential effects on its stability, degradation and translation [43]. In the global transcriptome, transcripts with a longer poly(A) tail possess a longer average mRNA half-life [23]. The deadenylation of shorter poly(A) tails can cause RNA decay or translational defects [44]. A recent study also noted the correlation between m6A marks and poly(A) tail regulation. The transcriptional dynamics of certain genes are related to differences in poly(A) tail length via m6A modification and deadenylase complexes [45]. m6A signaling is capable of controlling RNA structural switching and RNA‒protein interactions [46]. METTL3 and WATP can modulate RNA stabilization in an m6A-HuR-dependent manner [47, 48]. The m6A reader YTH N6-methyladenosine RNA binding protein (YTHDF) 2 is reported to directly interact with the CCR4-NOT complex. YTHDF3 can recruit the poly(A) specific ribonuclease subunit (PAN) 2-PAN3 complex, contributing to its deadenylation and degradation [49, 50]. Nonadenosine residues, such as G modifications, are also related to high quality and delayed degradation of the poly(A) tail [51, 52]. In our study, the temporal control of NOG stabilization by METTL3 relied on poly(A) tail shortening in the differentiation stage. Further studies are needed to identify the specific mechanism of how m6A marks lead to shortened poly(A) tails in DPSC differentiation.
The osteo/odontogenic differentiation of DPSCs and tertiary dentin formation are of particular interest in relation to dental repair. Identifying the key signaling in DPSC differentiation and mineralized matrix formation, and recapitulating these processes in clinical strategies could preserve pulp vitality. In the present study, we demonstrated that dynamic m6A RNA methylation is essential for heightened transcriptional coordination during DPSC differentiation. METTL3-mediated m6A marks tag the 3′ UTR of NOG and inhibit its stabilization via poly(A) tail regulation in a stage-specific manner. The present study identifies a critical role of METTL3-mediated m6A methylation in the temporal control of cell fate transition and sheds light on the epitranscriptomic machinery of m6A-dependent poly(A) tail regulation in transcriptional dynamics.
Availability of data and materials
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
References
Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science (New York, NY). 2007;318(5858):1917–20. https://doi.org/10.1126/science.1151526.
Sui B, Chen C, Kou X, Li B, Xuan K, Shi S, et al. Pulp stem cell-mediated functional pulp regeneration. J Dent Res. 2019;98(1):27–35. https://doi.org/10.1177/0022034518808754.
Albuquerque MT, Valera MC, Nakashima M, Nor JE, Bottino MC. Tissue-engineering-based strategies for regenerative endodontics. J Dent Res. 2014;93(12):1222–31. https://doi.org/10.1177/0022034514549809.
AAE position statement on vital pulp therapy. J Endod. 2021;47(9):1340–4. https://doi.org/10.1016/j.joen.2021.07.015.
Campos JM, Sousa AC, Caseiro AR, Pedrosa SS, Pinto PO, Branquinho MV, et al. Dental pulp stem cells and Bonelike(®) for bone regeneration in ovine model. Regen Biomater. 2019;6(1):49–59. https://doi.org/10.1093/rb/rby025.
Farahzadi R, Fathi E, Vietor I. Mesenchymal stem cells could be considered as a candidate for further studies in cell-based therapy of Alzheimer’s disease via targeting the signaling pathways. ACS Chem Neurosci. 2020;11(10):1424–35. https://doi.org/10.1021/acschemneuro.0c00052.
Narayan P, Rottman FM. An in vitro system for accurate methylation of internal adenosine residues in messenger RNA. Science (New York, NY). 1988;242(4882):1159–62. https://doi.org/10.1126/science.3187541.
Wang X, Feng J, Xue Y, Guan Z, Zhang D, Liu Z, et al. Structural basis of N(6)-adenosine methylation by the METTL3–METTL14 complex. Nature. 2016;534(7608):575–8. https://doi.org/10.1038/nature18298.
Wang Y, Li Y, Toth JI, Petroski MD, Zhang Z, Zhao JC. N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat Cell Biol. 2014;16(2):191–8. https://doi.org/10.1038/ncb2902.
Xu K, Yang Y, Feng GH, Sun BF, Chen JQ, Li YF, et al. Mettl3-mediated m(6)A regulates spermatogonial differentiation and meiosis initiation. Cell Res. 2017;27(9):1100–14. https://doi.org/10.1038/cr.2017.100.
Batista PJ, Molinie B, Wang J, Qu K, Zhang J, Li L, et al. m(6)A RNA modification controls cell fate transition in mammalian embryonic stem cells. Cell Stem Cell. 2014;15(6):707–19. https://doi.org/10.1016/j.stem.2014.09.019.
Geula S, Moshitch-Moshkovitz S, Dominissini D, Mansour AA, Kol N, Salmon-Divon M, et al. Stem cells. m6A mRNA methylation facilitates resolution of naive pluripotency toward differentiation. Science (New York, NY). 2015;347(6225):1002–6. https://doi.org/10.1126/science.1261417.
Wu R, Liu Y, Zhao Y, Bi Z, Yao Y, Liu Q, et al. m(6)A methylation controls pluripotency of porcine induced pluripotent stem cells by targeting SOCS3/JAK2/STAT3 pathway in a YTHDF1/YTHDF2-orchestrated manner. Cell Death Dis. 2019;10(3):171. https://doi.org/10.1038/s41419-019-1417-4.
Luo H, Liu W, Zhang Y, Yang Y, Jiang X, Wu S, et al. METTL3-mediated m(6)A modification regulates cell cycle progression of dental pulp stem cells. Stem Cell Res Ther. 2021;12(1):159. https://doi.org/10.1186/s13287-021-02223-x.
Luo H, Wang C, Liu M, Yin B, Peng A, Huang D, et al. Inhibition of SOX9 promotes inflammatory and immune responses of dental pulp. J Endod. 2018;44(5):792–9. https://doi.org/10.1016/j.joen.2018.02.004.
Wu Y, **e L, Wang M, **ong Q, Guo Y, Liang Y, et al. Mettl3-mediated m(6)A RNA methylation regulates the fate of bone marrow mesenchymal stem cells and osteoporosis. Nat Commun. 2018;9(1):4772. https://doi.org/10.1038/s41467-018-06898-4.
Gronthos S, Mankani M, Brahim J, Robey PG, Shi S. Postnatal human dental pulp stem cells (DPSCs) in vitro and in vivo. Proc Natl Acad Sci U S A. 2000;97(25):13625–30. https://doi.org/10.1073/pnas.240309797.
Li B, Yu F, Wu F, Hui T, Liao X, et al. EZH2 impairs human dental pulp cell mineralization via the Wnt/β-catenin pathway. J Dent Res. 2018;97(5):571–9. https://doi.org/10.1177/0022034517746987.
Yin B, Yu F, Wang C, Li B, Liu M, Ye L. Epigenetic control of mesenchymal stem cell fate decision via histone methyltransferase Ash1l. Stem cells (Dayton, Ohio). 2019;37(1):115–27. https://doi.org/10.1002/stem.2918.
** H, Ying X, Que B, Wang X, Chao Y, Zhang H, et al. N(6)-methyladenosine modification of ITGA6 mRNA promotes the development and progression of bladder cancer. EBioMedicine. 2019;47:195–207. https://doi.org/10.1016/j.ebiom.2019.07.068.
Huang H, Weng H, Sun W, Qin X, Shi H, Wu H, et al. Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol. 2018;20(3):285–95. https://doi.org/10.1038/s41556-018-0045-z.
Wang H, Wang M, Cheng Q. Capturing the alternative cleavage and polyadenylation sites of 14 NAC genes in Populus using a combination of 3′-RACE and high-throughput sequencing. Molecules (Basel, Switzerland). 2018;23(3):608. https://doi.org/10.3390/molecules23030608.
Kiss DL, Oman KM, Dougherty JA, Mukherjee C, Bundschuh R, Schoenberg DR. Cap homeostasis is independent of poly(A) tail length. Nucleic Acids Res. 2016;44(1):304–14. https://doi.org/10.1093/nar/gkv1460.
Guo W, Zhang L, Christopher DM, Teng ZQ, Fausett SR, Liu C, et al. RNA-binding protein FXR2 regulates adult hippocampal neurogenesis by reducing Noggin expression. Neuron. 2011;70(5):924–38. https://doi.org/10.1016/j.neuron.2011.03.027.
Yoon KJ, Ringeling FR, Vissers C, Jacob F, Pokrass M, Jimenez-Cyrus D, et al. Temporal control of mammalian cortical neurogenesis by m(6)A methylation. Cell. 2017;171(4):877-89.e17. https://doi.org/10.1016/j.cell.2017.09.003.
Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell. 2012;149(7):1635–46. https://doi.org/10.1016/j.cell.2012.05.003.
Sheng R, Wang Y, Wu Y, Wang J, Zhang S, Li Q, et al. METTL3-mediated m(6) A mRNA methylation modulates tooth root formation by affecting NFIC translation. J Bone Miner Res Off J Am Soc Bone Miner Res. 2021;36(2):412–23. https://doi.org/10.1002/jbmr.4180.
Cai W, Ji Y, Han L, Zhang J, Ni Y, Cheng Y, et al. METTL3-dependent glycolysis regulates dental pulp stem cell differentiation. J Dent Res. 2021;101:220345211051594. https://doi.org/10.1177/00220345211051594.
Yao Y, Bi Z, Wu R, Zhao Y, Liu Y, Liu Q, et al. METTL3 inhibits BMSC adipogenic differentiation by targeting the JAK1/STAT5/C/EBPβ pathway via an m(6)A-YTHDF2-dependent manner. FASEB J Off Publ Feder Am Soc Exp Biol. 2019;33(6):7529–44. https://doi.org/10.1096/fj.201802644R.
Swanson WB, Gong T, Zhang Z, Eberle M, Niemann D, Dong R, et al. Controlled release of odontogenic exosomes from a biodegradable vehicle mediates dentinogenesis as a novel biomimetic pulp cap** therapy. J Control Release Off J Control Release Soc. 2020;324:679–94. https://doi.org/10.1016/j.jconrel.2020.06.006.
Song M, Kim S, Kim T, Park S, Shin KH, Kang M, et al. Development of a direct pulp-cap** model for the evaluation of pulpal wound healing and reparative dentin formation in mice. J Vis Exp JoVE. 2017;2017(119):54973. https://doi.org/10.3791/54973.
Huang M, Xu S, Liu L, Zhang M, Guo J, Yuan Y, et al. m6A methylation regulates osteoblastic differentiation and bone remodeling. Front Cell Dev Biol. 2021;9:783322. https://doi.org/10.3389/fcell.2021.783322.
Liu XM, Qian SB. Linking m(6)A to Wnt signaling. EMBO Rep. 2020;21(4):e50097. https://doi.org/10.15252/embr.202050097.
Sun Z, Su Z, Zhou Z, Wang S, Wang Z, Tong X, et al. RNA demethylase ALKBH5 inhibits TGF-β-induced EMT by regulating TGF-β/SMAD signaling in non-small cell lung cancer. FASEB J Off Publ Feder Am Soc Exp Biol. 2022;36(5):e22283. https://doi.org/10.1096/fj.202200005RR.
Tucker AS, Matthews KL, Sharpe PT. Transformation of tooth type induced by inhibition of BMP signaling. Science (New York, NY). 1998;282(5391):1136–8. https://doi.org/10.1126/science.282.5391.1136.
Warren SM, Brunet LJ, Harland RM, Economides AN, Longaker MT. The BMP antagonist noggin regulates cranial suture fusion. Nature. 2003;422(6932):625–9. https://doi.org/10.1038/nature01545.
Groppe J, Greenwald J, Wiater E, Rodriguez-Leon J, Economides AN, Kwiatkowski W, et al. Structural basis of BMP signalling inhibition by the cystine knot protein Noggin. Nature. 2002;420(6916):636–42. https://doi.org/10.1038/nature01245.
Qin W, Yang F, Deng R, Li D, Song Z, Tian Y, et al. Smad 1/5 is involved in bone morphogenetic protein-2-induced odontoblastic differentiation in human dental pulp cells. J Endod. 2012;38(1):66–71. https://doi.org/10.1016/j.joen.2011.09.025.
Jani P, Zhang H, Benson MD, Qin C. Noggin inhibition of mouse dentinogenesis. J Oral Biosci. 2020;62(1):72–9. https://doi.org/10.1016/j.job.2019.11.001.
Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, et al. N(6)-methyladenosine modulates messenger RNA translation efficiency. Cell. 2015;161(6):1388–99. https://doi.org/10.1016/j.cell.2015.05.014.
Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505(7481):117–20. https://doi.org/10.1038/nature12730.
Boo SH, Kim YK. The emerging role of RNA modifications in the regulation of mRNA stability. Exp Mol Med. 2020;52(3):400–8. https://doi.org/10.1038/s12276-020-0407-z.
Nicholson AL, Pasquinelli AE. Tales of detailed Poly(A) tails. Trends Cell Biol. 2019;29(3):191–200. https://doi.org/10.1016/j.tcb.2018.11.002.
Kojima S, Gendreau KL, Sher-Chen EL, Gao P, Green CB. Changes in poly(A) tail length dynamics from the loss of the circadian deadenylase Nocturnin. Sci Rep. 2015;5:17059. https://doi.org/10.1038/srep17059.
Slobodin B, Bahat A, Sehrawat U, Becker-Herman S, Zuckerman B, Weiss AN, et al. Transcription dynamics regulate Poly(A) tails and expression of the RNA degradation machinery to balance mRNA levels. Mol Cell. 2020;78(3):434-44.e5. https://doi.org/10.1016/j.molcel.2020.03.022.
Liu N, Dai Q, Zheng G, He C, Parisien M, Pan T. N(6)-methyladenosine-dependent RNA structural switches regulate RNA–protein interactions. Nature. 2015;518(7540):560–4. https://doi.org/10.1038/nature14234.
Li E, Wei B, Wang X, Kang R. METTL3 enhances cell adhesion through stabilizing integrin β1 mRNA via an m6A-HuR-dependent mechanism in prostatic carcinoma. Am J Cancer Res. 2020;10(3):1012–25.
Chen Y, Peng C, Chen J, Chen D, Yang B, He B, et al. WTAP facilitates progression of hepatocellular carcinoma via m6A-HuR-dependent epigenetic silencing of ETS1. Mol Cancer. 2019;18(1):127. https://doi.org/10.1186/s12943-019-1053-8.
Du H, Zhao Y, He J, Zhang Y, ** H, Liu M, et al. YTHDF2 destabilizes m(6)A-containing RNA through direct recruitment of the CCR4-NOT deadenylase complex. Nat Commun. 2016;7:12626. https://doi.org/10.1038/ncomms12626.
Liu J, Gao M, Xu S, Chen Y, Wu K, Liu H, et al. YTHDF2/3 are required for somatic reprogramming through different RNA deadenylation pathways. Cell Rep. 2020;32(10):108120. https://doi.org/10.1016/j.celrep.2020.108120.
Liu Y, Nie H, Liu H, Lu F. Poly(A) inclusive RNA isoform sequencing (PAIso-seq) reveals wide-spread non-adenosine residues within RNA poly(A) tails. Nat Commun. 2019;10(1):5292. https://doi.org/10.1038/s41467-019-13228-9.
Lim J, Kim D, Lee YS, Ha M, Lee M, Yeo J, et al. Mixed tailing by TENT4A and TENT4B shields mRNA from rapid deadenylation. Science (New York, NY). 2018;361(6403):701–4. https://doi.org/10.1126/science.aam5794.
Acknowledgements
This work was supported by the Guangdong Basic and Applied Basic Research Foundation (2021A1515012475, 2021A1515111059), the National Natural Science Foundation of China (81900989, 52072167) and the Guangzhou Basic and Applied Basic Research Foundation (SL2023A04J00332).
Author information
Authors and Affiliations
Contributions
LHY contributed to conception, design, data acquisition, analysis, and interpretation, drafted and critically revised the manuscript; LWJ contributed to data acquisition and analysis, critically revised the manuscript; ZYC contributed to design, data acquisition, critically revised the manuscript; ZYL contributed to data analysis, and interpretation, critically revised the manuscript; WJR and WRL contributed to data interpretation, critically revised the manuscript; SLQ contributed to conception, design, critically revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All experimental procedures were approved by the Ethics Committees of Stomatological Hospital, Southern Medical University (ethical code 2019(16)) and written informed consent was obtained from every participant.
Consent for publication
All authors had final approval of the submitted versions and read the journal’s authorship statement.
Competing interests
The authors declare no competing financial interests and no conflicts of interest for this work.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Additional Table 1:
The sequence of primers used in qPCR. Additional Figure 1: KEGG enrichment analysis of m6A-tagged transcripts and the relative expression of m6A-related genes during DPSC mineralization. Additional Figure 2: H&E staining of the composites of β-TCP/HA scaffolds and DPSCs. Additional Figure 3: Volcano plots of differentially expressed genes in OM-DPSCs after METTL3 inhibition. Additional Figure 4: Immunofluorescence staining of METTL3 and NOG in DPSCs. Additional Figure 5: Immunofluorescence staining of p-Smad3 and Smad in the composites of β-TCP/HA scaffolds with METTL3-interfering DPSCs.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Luo, H., Liu, W., Zhou, Y. et al. Stage-specific requirement for METTL3-dependent m6A modification during dental pulp stem cell differentiation. J Transl Med 20, 605 (2022). https://doi.org/10.1186/s12967-022-03814-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-022-03814-9