
Abstract
Background
Circular RNAs (circRNAs) are differentially expressed between normal and cancerous tissues, contributing to tumor initiation and progression. However, comprehensive landscape of dysregulated circRNAs across cancer types remains unclear.
Methods
In this study, we conducted Ribo-Zero transcriptome sequencing on tumor tissues and their adjacent normal samples including glioblastoma, esophageal squamous cell carcinoma, lung adenocarcinoma, thyroid cancer, colorectal cancer, gastric cancer and hepatocellular carcinoma. CIRCexplorer2 was employed to identify circRNAs and dysregulated circRNAs and genes were determined by DESeq2 package. The expression of hsa_circ_0072309 (circLIFR) was measured by reverse transcription and quantitative real-time PCR, and its effect on cell migration was examined by Transwell and wound healing assays. The role of circLIFR in tumor metastasis was evaluated via mouse models of tail-vein injection and spleen injection for lung and liver metastasis, respectively.
Results
Distinct circRNA expression signatures were identified among seven types of solid tumors, and the dysregulated circRNAs exhibited cancer-specific expression or shared common expression signatures across cancers. Bioinformatics analyses indicated that aberrant expression of host genes and/or RNA-binding proteins contributed to circRNA dysregulation in cancer. Finally, circLIFR was experimentally validated to be downregulated in six solid tumors and to significantly inhibit cell migration in vitro and tumor metastasis in vivo.
Conclusions
Our results provide a comprehensive landscape of differentially expressed circRNAs in solid tumors and highlight that circRNAs are extensively involved in cancer pathogenesis.
Similar content being viewed by others

Background
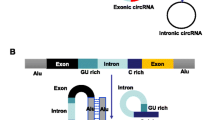
Circular RNAs (circRNAs) are a class of single-stranded endogenous RNAs with covalently closed-loop structures generated by back-splicing [1]. According to the origin of sequences, circRNAs are classified into three major groups, namely exonic circRNA (EcRNA), exon-intron circRNA (EIciRNA) and intronic circRNA (ciRNA). The biogenesis of circRNA is tightly regulated by cis-acting elements and trans-acting proteins. Moreover, a single gene locus could produce multiple circRNA isoforms by alternative circularization [2]. Recently, circRNAs have emerged as essential regulators in various biological processes through diverse mechanisms such as regulating transcription, sponging microRNA (miRNA), acting as RNA-binding protein (RBP) decoy, and translating into protein [1, 3].
Increasing evidence has revealed that circRNAs are commonly dysregulated in cancer and exert crucial functions during tumor initiation and development. For instance, circ-SMO is significantly upregulated in glioblastoma (GBM) and activates the Hedgehog pathway to promote tumorigenesis through encoding a novel protein SMO-193a.a. [24], suggesting their potential functions in the maintenance of basal cellular processes. Moreover, these ubiquitous genes were predominantly enriched in biological processes and signaling pathways that are fundamental to cells, such as RNA splicing and transporting (Additional file 4: Fig. S1B-C). We also identified 1765 tissue-specific protein-coding genes (TSI ≥ 0.85, Additional file 4: Fig. S1D and Additional file 6: Table S9) and further confirmed their gene expression profiles in normal tissue from the GTEx database (Additional file 4: Fig. S2). Functional enrichment analysis of these tissue-specific genes demonstrated tissue-specific biological processes and signaling pathways (Additional file 4: Fig. S1E-F and Fig. S3). For instance, the genes specific to the brain were enriched in brain-specific biological processes and signaling pathways, such as synapse, learning, cognition and axon guidance (Additional file 4: Fig. S1E-F). Collectively, these results confirmed the reliability of our sample preparation and data analysis.
Considering that most circRNAs were discovered at low frequency and expressed at low levels, 1762 circRNAs with high abundance (median RPM expression value ≥0.1 in at least one tissue) were retained to obtain a high-confidence profile. As a result, 30 circRNAs with TSI ≤ 0.3 were defined as the ubiquitous circRNAs (Fig. 2A and Additional file 7: Table S10), indicating their important functions in fundamental cellular processes. In addition, we found 439 circRNAs with TSI ≥ 0.85 and defined them as tissue-specific circRNAs (Fig. 2B and Additional file 8: Table S11), implying their tissue-specific functions. Moreover, we found that the brain exhibited the highest number of tissue-specific circRNAs while the intestine exhibited the least (Fig. 2B and Additional file 8: Table S11). For instance, we identified that hsa_circ_0000497 was exclusively highly expressed in the brain (Fig. 2E), which was further supported by circAtlas data (Additional file 4: Fig. S4). However, the functions of these ubiquitous and tissue-specific circRNAs remain elusive.

Tissue-specific expression patterns of circRNAs in human normal tissues. A Heatmap of ubiquitously expressed circRNAs. B Heatmap of tissue-specifically expressed circRNAs. C The number of circRNAs produced from tissue-specific genes. D The Pearson correlation coefficients of tissue-specific circRNAs with host gene expression. E The tissue specificity of hsa_circ_0000497 was governed by the brain-specific host gene SLAIN1. F The Pearson correlation coefficients of tissue-specific circRNAs with RBP expression. G Expression of RBFOX1, hsa_circ_0070057 and its host gene SEPT11 in different tissues. H Correlation of hsa_circ_0070057 and RBFOX1 expression. I eCLIP-seq read density for RBFOX1 within introns flanking the hsa_circ_0070057 back-splicing site. The vertical red lines indicate the GCATG binding motifs of RBFOX1
Tissue-specific expression of host genes and/or RBPs contributes to the tissue specificity of circRNAs
The tissue-specific expression signatures of circRNAs may be the consequence of their biogenesis. Given that most circRNAs were processed from the primary transcripts transcribed from gene loci, we initially examined whether both circRNA and its host gene showed the same tissue specificity and found that only 135 (30.75%) circRNAs were produced from tissue-specific host genes (Fig. 2C and Additional file 4: Fig. S5A). Pearson correlation analysis further supported that circRNAs derived from tissue-specific genes exhibited a strong correlation with their host genes (Fig. 2D), implying that the expression of these circRNAs highly depend on their host gene expression. For example, SLIAN1 is a microtubule plus-end tracking protein exclusively expressed in the brain to regulate microtubule dynamics and promote axonal development [25], so hsa_circ_0000497 derived from the SLAIN1 gene was also highly expressed in the brain (Fig. 2E and Additional file 4: Fig. S5B-D). Therefore, tissue-specific expression of host genes contributes to the tissue specificity of these tissue-specific circRNAs.
It was worth noting that 304 (69.25%) tissue-specific circRNAs were derived from nontissue-specific genes, implying that other factors contribute to their tissue specificity. As RBPs are known to play a role in circRNA biogenesis, we speculated that tissue-specific RBPs may regulate the tissue specificity of circRNAs. According to a previous study [6], 215 RBPs (Additional file 2: Table S2) were selected to calculate their TSI, and 8 RBPs (Additional file 3: Table S12) with TSI ≥ 0.85 were identified as tissue-specific. Next, we analyzed correlations between 304 circRNAs and 8 RBPs, and found that 155 (35.31%) circRNAs showed a strong correlation with these RBPs (Pearson correlation coefficient ≥ 0.7, Fig. 2F), indicating that the tissue specificity of these circRNAs may be regulated by tissue-specific RBPs. Among these RBPs, RBFOX1 was found to be exclusively expressed in the brain (TSI = 1) (Fig. 2G), consistent with a previous study [26]. Therefore, we chose RBFOX1 as an example to investigate its potential regulatory mechanisms on the biogenesis of tissue-specific circRNAs. First, we found that brain-specific hsa_circ_0070057 had a strong association with RBFOX1 expression in the brain, not with the expression of its host gene (Fig. 2G-H), Furthermore, eCLIP-seq data of RBFOX1 [21] demonstrated that RBFOX1 indeed bound to the flanking introns of hsa_circ_0070057 (Fig. 2I), suggesting that RBFOX1 facilitates the biogenesis of this circRNA. In addition, brain-specific hsa_circ_0073539 may also be regulated by QKI (TSI = 0.86 in brain) which is a well-known factor in circRNA biogenesis (Additional file 4: Fig. S6) [27], not by its host gene MAN2A1. Together, tissue-specific expression of RBPs may play an important role in the biogenesis of tissue-specific circRNAs.
Expression patterns of differentially expressed circRNAs in cancer
Emerging evidence has reported that circRNAs are frequently dysregulated in cancer. To depict the abnormal expression of circRNAs in cancer, we compared the global circRNA abundance in tumors and their paired normal samples, and found the downregulation of global circRNA abundance in almost all cancer types (Fig. 3A) under the comparable sequencing depths (Additional file 4: Fig. S7A and Additional file 9: Table S13). As shown in Fig. 3B, we also identified the differentially expressed circRNAs in each type of cancer, and the detailed upregulated and downregulated circRNAs are listed in Additional file 10: Table S14. Interestingly, GBM (n = 603) showed the most differentially expressed circRNAs while GC (n = 123) had the least (Additional file 4: Fig. S7B). Next, we investigated whether these differentially expressed circRNAs were cancer-specific. As expected, each cancer exhibited unique patterns of differentially expressed circRNAs (Fig. 3B-C). Among the 1209 dysregulated circRNAs, 962 (79.57%) were found to be differentially expressed in only one cancer type (Fig. 3C and Additional file 10: Table S14), implying their cancer-specific roles in tumorigenesis.

Characterization of differentially expressed circRNAs in seven solid tumors. A Total circRNA abundance in tested cancers. B Profiles of differentially expressed circRNAs identified in this study. The blue box indicates the unique circRNA expression signatures of each cancer. C The number of cancer-specific differentially expressed circRNAs. D List of differentially expressed circRNAs shared by at least two types of cancers. E Heatmap of twenty-five differentially expressed circRNAs shared by at least four types of cancers. The asterisk indicates circRNA expression is significantly altered. Red and green colors indicate upregulation and downregulation, respectively. F The number of differentially expressed circRNAs that exhibited consistent or inconsistent expression changes in at least two cancer types. G Expression alteration of RBPs associated with circRNA biogenesis in GBM. H eCLIP-seq read density for RBFOX1 within introns flanking hsa_circ_0111312 back-splicing site. The vertical red box indicates the GCATG binding motifs of RBFOX1
In addition, 247 differentially expressed circRNAs were dysregulated in at least two cancer types (Fig. 3D and Additional file 10: Table S14). Moreover, 25 circRNAs were found to be differentially expressed in at least four types of cancers (Fig. 3E and Additional file 10: Table S14). For instance, hsa_circ_0072309 was consistently downregulated in our tested tumor samples except for GBM (Fig. 3E), highlighting its pivotal role in tumorigenesis. Of 247 circRNAs, the expression of 187 circRNAs exhibited consistent changes across different tumors (Fig. 3F and Additional file 10: Table S14), implying their common functions during tumorigenesis. For instance, hsa_circ_0077837 was significantly downregulated in CRC, ESCC, LUAD, GC and HCC (Fig. 3E and Additional file 4: Fig. S8A), indicating its universal tumor-suppressive role in cancer, which was recently proven in HCC [28]. In addition, abnormal expression of 60 circRNAs exhibited inconsistent tendencies among cancer types (Fig. 3F and Additional file 10: Table S14), suggesting their diverse roles in different cancers. For example, we found that hsa_circ_0001681 was elevated in 4 cancer types (CRC, LUAD, GC, THCA) but dramatically downregulated in GBM (Fig. 3E and Additional file 4: Fig. S8B). Consistent with our findings, hsa_circ_0001681 was previously reported to be upregulated and exert an oncogenic function in THCA [29], but to be downregulated and play a tumor-suppressive role in renal cell carcinoma [30], Therefore, such dysregulated circRNAs may exert their different functions in a cancer-type context.
Abnormal expression of the host genes and/or RBPs contribute to circRNA dysregulation in cancer
Of note, the prevalence of differential expression profiles of circRNAs in cancer further raised the question of how these circRNAs were dysregulated. Vo et al. reported that the dysregulation of some circRNAs in prostate cancer was predominantly caused by aberrant expression of their parental genes [7]. In our study, an average of 41.84% (ranging from 33.50 to 57.81%) of differentially expressed circRNAs were explained by differentially expressed genes (Additional file 4: Fig. S9). For instance, hsa_circ_0099329 and its host gene PPFIA2 were exclusively downregulated in GBM (Additional file 4: Fig. S10A). In addition, we also observed that the alterations of some circRNAs (ranging from 42.19 to 66.50%) were inconsistent with their host genes (Additional file 4: Fig. S9), suggesting the involvement of other mechanisms in circRNA dysregulation.
Given that 66.50% of dysregulated circRNAs in GBM were derived from genes without significant changes in expression, we focused on GBM to explore whether the dysregulated circRNAs were associated with RBPs. We first examined the expression patterns of 215 RBPs involving deaminase, endonuclease, helicase and splicing factor in GBM (Additional file 11: Table S15). As shown in Fig. 3G, these RBPs such as well-characterized PTBP1 [31] and ESRP1 [32] were generally observed to be altered in GBM. Of these, PTBP1 was elevated in GBM, suggesting its potential to regulate biogenesis of some circRNAs, which wa supported by a previous report [31]. In addition, ESRP1 was decreased in GBM, implying that the downregulation of circRNAs in GBM might be affected by this RBP.
We quantified the correlations between dysregulated RBPs and dysregulated circRNAs from nondysregulated genes, and found that 174 (28.86%) circRNAs showed a strong correlation with RBPs (Pearson correlation coefficient ≥ 0.7), implying that the dysregulation of these circRNAs might be regulated by RBPs. Intriguingly, among these dysregulated RBPs, we noticed that RBFOX1, a neuron-specific splicing factor involved in neurodevelopmental and neuropsychiatric disorders [26, 33], was the most significantly downregulated RBP in GBM in our study (Fig. 3G and Additional file 4: Fig. S10B) and in the GEPIA database. Therefore, we examined the differentially expressed circRNAs associated with RBFOX1 and found that downregulation of hsa_circ_0111312 in GBM was strongly correlated with decreased RBFOX1 expression in GBM (Additional file 4: Fig. S10C), independent of expression of its host gene RASAL2 (Additional file 4: Fig. S10D). Furthermore, eCLIP data of RBFOX1 [21] confirmed the strong binding of RBFOX1 to the flanking introns of hsa_circ_0111312 (Fig. 3H), suggesting the involvement of RBFOX1 in circRNA biogenesis. Therefore, aberrant expression of RBPs in GMB can contribute to a subset of circRNA dysregulation.
Characterization of circLIFR in cancer cells
According to our bioinformatics analysis, hsa_circ_0072309 exhibited high abundance in normal tissues and was downregulated in our tested tumors except for GBM. Since hsa_circ_0072309 is derived from exons 2–5 of the LIFR gene (Fig. 4A), we termed it as circLIFR. First, we designed the F1/R1 divergent primers (F1 primer crossing the junction site) to conduct RT-qPCR, and the results confirmed that circLIFR was indeed downregulated in our tested tumors, except for GBM, compared with their adjacent normal tissues (Fig. 4B and Additional file 4: Fig. S11A), implying that circLIFR could play a tumor-suppressive role in solid tumors. To further characterize circLIFR, we performed PCR with divergent (red) and convergent primers (blue) (Fig. 4A) using cDNA and genomic DNA (gDNA) as templates. As shown in Fig. 4C, the expected bands were observed using convergent primers from both cDNA and gDNA templates, but there was no band using gDNA as a template and the divergent primers, confirming that circLIFR was a circular RNA produced by back-splicing, which was further validated by RNase R digestion treatment (Fig. 4D) and Sanger sequencing in different cancer cells (Fig. 4E and Additional file 4: Fig. S11B). To detect the cellular distribution of circLIFR, we isolated the cytoplasmic and nuclear fractions of cells to measure circLIFR levels by RT-qPCR, and found that circLIFR was predominantly located in the cytoplasm (Fig. 4F and Additional file 4: Fig. S11C), which was confirmed by FISH experiments (Fig. 4G and Additional file 4: Fig. S11D). We also evaluated circLIFR stability after actinomycin D treatment and found that circLIFR was more stable than its linear transcript LIFR mRNA (Fig. 4H). Collectively, these results demonstrated that circLIFR was a bona fide circRNA, predominantly distributed in the cytoplasm and significantly downregulated in our tested tumors except for GBM.

Characterization of circLIFR in solid tumors. A Schematic illustration showing circLIFR is derived from exons 2–5 of LIFR mRNA. The F1/R1 primers were used to measure circLIFR by RT-qPCR. DP, divergent primers; CP, convergent primers. B circLIFR expression measured by RT-qPCR in ESCC, HCC and their matched adjacent normal tissues, normalized to U6 RNA levels. N, normal tissues; T, tumor tissues. Data represent the mean ± S.D., and the p-value was determined by two-tailed paired Student’s t-test. C The presence of circLIFR was validated in KYSE150 cells by RT-PCR. Divergent primers were used to amplify circLIFR from cDNA, but not from genomic DNA. LIFR mRNA linear transcripts were used as a negative control. cDNA, complementary DNA; gDNA, genomic DNA. D Total RNA from KYSE150 cells with or without RNase R treatment was subjected to RT-PCR. E Agarose gel electrophoresis and Sanger sequencing of RT-PCR products of circLIFR in KYSE30 and SK-Hep-1 cells. F RT-qPCR analyses of circLIFR after cytoplasmic/nuclear fractionation of KYSE30 and SK-Hep-1 cells. β-actin mRNA and U6 RNA represent cytoplasmic and nuclear RNAs, respectively. Western blotting confirmed the efficiency of nuclear/cytoplasmic isolation. Data are shown as the mean ± SD. G Fluorescence in situ hybridization of circLIFR in KYSE30 and SK-Hep-1 cells. 18S and U6 RNA represent cytoplasmic and nuclear RNAs, respectively. H circLIFR and LIFR mRNA levels in KYSE30 and SK-Hep-1cells without or with actinomycin D treatment
circLIFR inhibits cell migration in vitro and tumor metastasis in vivo
circLIFR was reported to affect cellular functions in breast cancer [34], bladder cancer [35], renal carcinoma [36] and gastric cancer [37]. However, there are no reports of circLIFR in ESCC, HCC, THCA and CRC. To investigate the biological functions of circLIFR in these tumors, we first measured circLIFR levels in cancer cell lines (Additional file 4: Fig. S12A) and selected 4 cancer cells with low circLIFR expression (KYSE30 and KYSE150 esophageal cancer cells, SK-Hep-1 liver cancer cells, HCT-116 colorectal cancer cells) to construct stable cells overexpressing circLIFR. As shown in Fig. 5A and Additional file 4: Fig. S12B, circLIFR was correctly circularized and successfully expressed in these cells. Transwell migration and wound healing assays showed that overexpression of circLIFR apparently inhibited cell migration and cell motility (Fig. 5B-C and Additional file 4: Fig. S12C-D). Next, we sought to evaluate the function of circLIFR in vivo. To this end, we conducted a tail-vein injection mouse model using circLIFR-overexpressing KYSE30 cells to check its role in lung metastasis. The results demonstrated that after ectopic expression of circLIFR in esophageal cancer cells, tumor metastasis was significantly inhibited compared to that of control cells in lung metastasis models (Fig. 5D). To further evaluate the function of circLIFR, we injected circLIFR-overexpressing SK-Hep-1 cells into the spleen, and observed that circLIFR dramatically repressed tumor metastasis to mouse liver (Fig. 5E). Together, circLIFR significantly inhibited cell migration in vitro and tumor metastasis in vivo.

circLIFR inhibits cell migration in vitro and tumor metastasis in vivo. A Agarose gel electrophoresis and Sanger sequencing of semi-quantitative RT-PCR products demonstrated that circLIFR was correctly circularized and successfully overexpressed in KYSE30 and SK-Hep-1 cells. B Transwell migration assays showed that circLIFR overexpression inhibited the migratory ability of KYSE30 and SK-Hep-1 cells. C Wound healing assays showed that circLIFR overexpression inhibited cell migration in both KYSE30 and SK-Hep-1 cells. D Decreased tumor metastasis formed in the lungs of mice via tail vein injection of circLIFR-overexpressing KYSE30 cells, as indicated by representative bioluminescent images of lung metastasis at 6 weeks (left) and H&E staining of lung metastatic lesions (right) of mice for each group (n = 5 mice/group). E Decreased tumor metastasis formed in the livers of mice via spleen injection of circLIFR-overexpressing SK-Hep-1 cells, as indicated by representative bioluminescent images of liver metastasis at 8 weeks (left) and H&E staining of liver metastatic lesions (right) of mice for each group (n = 5 mice/group). F Volcano plot of differentially expressed genes affected by circLIFR in KYSE30 cells. G,H Gene Ontology (G) and GSEA analysis (H) showed that dysregulated genes upon circLIFR overexpression was involved in cell adhesion. NES, normalized enrichment score; FDR, false discovery rate. p-values are calculated by permutation test. I,J Quantification of metastasis-related genes by RT-qPCR in KYSE30 control cells and circLIFR-overexpressing cells. Data represented mean ± S.D.; the p-values were determined by a two-tailed unpaired Student’s t-test
To explore the underlying mechanism of circLIFR, we performed RNA-seq for transcriptome analyses. As shown in Fig. 5F, ectopic expression of circLIFR affected the expression levels of 448 genes, of which 292 genes were upregulated and 156 genes were downregulated, including epithelial-mesenchymal transition (EMT) marker genes (Additional file 12: Table S16). Gene Ontology analyses indicated that these differentially expressed genes were significantly enriched in cell adhesion, extracellular matrix organization and cell-substrate junction assembly (Fig. 5G), which was further confirmed by GSEA analysis results (Fig. 5H). Finally, the expression change of a subset of metastasis-related genes affected by circLIFR was validated by RT-qPCR (Fig. 5I-J). Therefore, circLIFR inhibits tumor metastasis by regulating a subset of cell adhesion and EMT-related genes.
Discussion
Accumulating evidence has demonstrated that circRNAs are frequently altered in cancer and play indispensable roles during cancer initiation and progression. Recently, several studies have attempted to present the landscape of circRNAs in tumor tissues and cancer cell lines, shedding light on the importance of circRNAs [6, 7, 38, 39]. However, the lack of matched normal tissues in these studies cannot depict the dysregulated circRNAs across cancer types. Therefore, we collected 122 tumor samples of seven types of malignancies (GBM, ESCC, LUAD, HCC, GC, THCA and CRC) and their matched normal tissues to conduct Ribo-zero RNA-seq, which allows us to systematically profile circRNAs in normal tissues and their matched tumors without bias.
From these samples, we identified 59,056 circRNAs, most of which were originated from exons (n = 59,020). Interestingly, we found that 182 circRNAs harbored a complete CDS as they originated from both the CDS and UTR, indicating their potential for translation into intact peptides. Consistent with previous studies [3, 40], we found that the majority of circRNAs were expressed at low levels. However, we identified 22 circRNAs with much higher abundance than their cognate linear transcripts (Additional file 3: Table S6), suggesting their important roles in basic cellular functions. Although a single gene locus can produce multiple circRNAs via alternative circularization, this gene shows a tendency to express a predominant circRNA isoform (Fig. 1G-H). This could be explained by our analysis that the introns flanking circularized exons of the predominant circRNA isoform are much longer and have more repetitive elements (Fig. 1I-J). In addition, we identified 30 circRNAs that were ubiquitous across our tested tissues, and 439 circRNAs were tissue-specific, indicating their general and tissue-specific cellular functions (Additional file 7: Table S10 and Additional file 8: Table S11).
Compared with normal tissues, the global abundance of circRNAs was remarkably decreased in tumors (Fig. 3A), which may be caused by inefficient back-splicing events or the dose dilution of nascent circRNAs in the rapid division of tumor cells [41, 42]. This was further supported by the fact that the total abundance of circRNAs was significantly elevated when inhibiting cell proliferation [7]. Moreover, we identified 1209 differentially expressed circRNAs in cancers (Additional file 10: Table S14), of which 962 (79.57%) were cancer-specific, while 247 were dysregulated in at least two cancer types. Of the 247 noncancer-specific circRNAs, 187 circRNAs were consistently altered in tumors, while 60 circRNAs exhibited distinct expression patterns in different cancer types, indicating that consistently altered circRNAs exert a common role in cancers, while inconsistently altered circRNAs may play distinct functions in different cancers. For instance, hsa_circ_0001681 was elevated in papillary thyroid cancer and promoted tumor growth by sponging miR-198 [29], while downregulation of circRAPGEF5 in renal cell carcinoma inhibited cancer progression by sequestering miR-27a-3p [30], supporting its diverse roles in cancers.
Given that some circRNAs originating from cancer tissues may enter the circulatory system and be stably present in sera [43, 44], cancer-specific dysregulated circRNAs could serve as biomarkers for cancer diagnosis and prognosis. For instance, the EML4-ALK fusion gene produces a novel F-circEA in non-small cell lung cancer (NSCLC), and the existence of F-circEA in sera makes it a promising liquid biopsy biomarker for monitoring the EML4-ALK fusion gene [45]. Moreover, circRNAs driving tumor progression could be therapeutic targets. For example, circPRKCI was demonstrated to be a proto-oncogenic circular RNA in lung adenocarcinoma tissues, and the intratumor injection of cholesterol-conjugated siRNA specifically targeting circPRKCI inhibited tumor growth in a patient-derived lung adenocarcinoma xenograft model [46]. Therefore, elucidation of the biological functions and clinical relevance of such dysregulated circRNAs is helpful to identify new diagnostic biomarkers and therapeutic targets.
Our study provided abundant dysregulated circRNAs to be experimentally validated for their roles in tumorigenesis. Here we focused on hsa_circ_0072309 (circLIFR) because of its consistent decrease in LUAD, ESCC, CRC, GC, HCC and THCA. First, we confirmed the downregulation of circLIFR in these six types of cancers by RT-qPCR, supporting the robustness of our pipeline for transcriptome analyses. Combined with recent findings about circLIFR in breast cancer [34], renal carcinoma [36] and bladder cancer [35], downregulation of circLIFR seems to be general in cancers, suggesting its important tumor-suppressive roles in cancer pathology. Indeed, circLIFR was reported to repress the progression of breast cancer [34], renal carcinoma [36] and GC [37]. In esophageal cancer and liver cancer models, we verified that circLIFR inhibited cell migration in vitro and tumor metastasis in vivo. Therefore, circLIFR plays a pivotal role during tumorigenesis. To explore the effects of circLIFR on gene expression, we performed RNA-seq on circLIFR-overexpressing KYSE30 cells and revealed that circLIFR significantly altered the expression patterns of metastasis-related genes (such as cell adhesion molecules). Mechanistically, circLIFR was reported to act as a sponge of miR-492 in breast cancer [34] and miR-100 in renal carcinoma [36] to inhibit tumor progression. In bladder cancer, circLIFR interacted with the MSH2 protein to positively modulate cisplatin-sensitivity through the MutSα/ATM-p73 axis [35]. However, the detailed underlying mechanism of circLIFR in esophageal cancer is still vague and needs further investigation.
In addition, we characterized some tissue-specific circRNAs in our tested normal tissues and cancer-specific dysregulation of circRNAs in tumors. Given that circRNA biogenesis is regulated by cis-acting elements and trans-acting proteins, we analyzed the factors to determine circRNA expression. As shown in Fig. 2C and Additional file 4: Fig. S5A, we found that the tissue-specific expression of the host genes contributes to 30.75% of tissue-specific circRNAs, while aberrant expression of the host gene contributes to approximately 41.84% of dysregulated circRNAs, indicating that the expression levels of host genes are an important factor for circRNA expression. Moreover, we identified that 35.31% of tissue-specific circRNAs and 28.86% of dysregulated circRNAs in GBM were highly correlated with RBPs, which was further supported by published eCLIP-seq data (Fig. 2I and Fig. 3H), demonstrating pivotal roles in RBPs in circRNA biogenesis. Because we found a strong association between dysregulated RBPs (such as PTBP1, RBFOX1 and ESRP1) and the expression of a subset of circRNAs in GBM, it is important to dissect how these RBPs affect circRNA biogenesis in the future.
Conclusions
In this study, we identified the expression landscape of circRNAs in paired normal-tumor clinical samples from seven types of malignancies. The dysregulated circRNAs exhibited cancer-specific expression or shared common expression signatures across cancers, which could be regulated by the expression of host genes and RBPs. Finally, we experimentally validated that circLIFR was downregulated and played a tumor-suppressive role in tumors. Collectively, this study provides the comprehensive profiles of differentially-expressed circRNAs in cancers and highlights the important roles of circRNAs in cancer biology.
Availability of data and materials
All data and materials obtained and/or analyzed in this study were available from the corresponding authors upon a reasonable request.
Abbreviations
- circRNA:
-
Circular RNA
- RNA-seq:
-
Transcriptome sequencing
- EcRNAs:
-
Exonic circRNAs
- EIciRNAs:
-
Exon-intron circRNAs
- ciRNAs:
-
Intronic circRNAs
- miRNA:
-
microrna
- RBP:
-
RNA-binding protein
- SMO:
-
Smoothened, frizzled class receptor
- GBM:
-
Glioblastoma
- IGF2BP3:
-
Insulin-like growth factor 2 mRNA binding protein 3
- CCLE:
-
Cancer Cell Line Encyclopedia
- rRNA:
-
Ribosomal RNA
- ESCC:
-
Esophageal squamous cell carcinoma
- LUAD:
-
Lung adenocarcinoma
- THCA:
-
Thyroid cancer
- CRC:
-
Colorectal cancer
- GC:
-
Gastric cancer
- HCC:
-
Hepatocellular carcinoma
- LIFR:
-
Leukemia inhibitory factor receptor alpha
- RT-qPCR:
-
Quantitative real-time PCR
- PE150:
-
Paired-end 150 bp
- RPM:
-
Reads per million mapped reads
- FPKM:
-
Fragments per kilobase per million
- TSI:
-
Tissue specificity index
- GO:
-
Gene Ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- GSEA:
-
Gene Set Enrichment Analysis
- eCLIP-seq:
-
Enhanced UV crosslinking and immunoprecipitation followed by sequencing
- RBFOX1:
-
RNA binding protein, fox-1 homolog (C. elegans) 1
- QKI:
-
QKI, KH domain containing
- RNA binding:
-
RT-PCR: reverse transcription PCR
- FBS:
-
Fetal bovine serum
- FISH:
-
Fluorescence in situ hybridization
- DAPI:
-
4′,6-diamidino-2-phenylindole
- CDS:
-
Coding sequence
- UTR:
-
Untranslated region
- HIPK3:
-
Homeodomain interacting protein kinase 3
- GTEx:
-
Genotype-Tissue Expression
- SLAIN1:
-
SLAIN motif family, member 1
- PPFIA2:
-
PTPRF interacting protein alpha 2
- PTBP1:
-
Polypyrimidine tract binding protein 1
- ESRP1:
-
Epithelial splicing regulatory protein 1
- GEPIA:
-
Gene Expression Profiling Interactive Analysis
- cDNA:
-
Complementary DNA
- gDNA:
-
Genomic DNA
- EMT:
-
Epithelial-mesenchymal transition
- RAPGEF5:
-
Rap guanine nucleotide exchange factor 5
- EML4:
-
Echinoderm microtubule associated protein like 4
- ALK:
-
Anaplastic lymphoma receptor tyrosine kinase
- NSCLC:
-
Non-small cell lung cancer
- PRKCI:
-
Protein kinase C iota
References
Li J, Sun D, Pu W, Wang J, Peng Y. Circular RNAs in cancer: biogenesis, function, and clinical significance. Trends Cancer. 2020;6:319–36.
Zhang XO, Dong R, Zhang Y, Zhang JL, Luo Z, Zhang J, et al. Diverse alternative back-splicing and alternative splicing landscape of circular RNAs. Genome Res. 2016;26:1277–87.
Chen LL. The expanding regulatory mechanisms and cellular functions of circular RNAs. Nat Rev Mol Cell Biol. 2020;21:475–90.
Wu X, **ao S, Zhang M, Yang L, Zhong J, Li B, et al. A novel protein encoded by circular SMO RNA is essential for Hedgehog signaling activation and glioblastoma tumorigenicity. Genome Biol. 2021;22:33.
Hanniford D, Ulloa-Morales A, Karz A, Berzoti-Coelho M, Moubarak R, Sánchez-Sendra B, et al. Epigenetic silencing of CDR1as drives IGF2BP3-mediated melanoma invasion and metastasis. Cancer Cell. 2020;37:55–70.e15.
Ruan H, **ang Y, Ko J, Li S, **g Y, Zhu X, et al. Comprehensive characterization of circular RNAs in ~ 1000 human cancer cell lines. Genome Med. 2019;11:55.
Vo JN, Cieslik M, Zhang Y, Shukla S, **ao L, Zhang Y, et al. The landscape of circular RNA in cancer. Cell. 2019;176:869–81.e13.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20.
Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21.
Glazar P, Papavasileiou P, Rajewsky N. circBase: a database for circular RNAs. RNA. 2014;20:1666–70.
Zhang XO, Wang HB, Zhang Y, Lu X, Chen LL, Yang L. Complementary sequence-mediated exon circularization. Cell. 2014;159:134–47.
Gao Y, Zhang J, Zhao F. Circular RNA identification based on multiple seed matching. Brief Bioinform. 2018;19:803–10.
Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12:357–60.
Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 2015;33:290–5.
Yanai I, Benjamin H, Shmoish M, Chalifa-Caspi V, Shklar M, Ophir R, et al. Genome-wide midrange transcription profiles reveal expression level relationships in human tissue specification. Bioinformatics. 2005;21:650–9.
Ludwig N, Leidinger P, Becker K, Backes C, Fehlmann T, Pallasch C, et al. Distribution of miRNA expression across human tissues. Nucleic Acids Res. 2016;44:3865–77.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550.
Yu GC, Wang LG, Han YY, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–7.
Gebauer F, Schwarzl T, Valcárcel J, Hentze M. RNA-binding proteins in human genetic disease. Nat Rev Genet. 2021;22:185–98.
Mukaka MM. Statistics Corner: A guide to appropriate use of Correlation coefficient in medical research. Malawi Med J. 2012;24:69–71.
Li J, Choi PS, Chaffer CL, Labella K, Hwang JH, Giacomelli AO, et al. An alternative splicing switch in FLNB promotes the mesenchymal cell state in human breast cancer. Elife. 2018;7:e37184.
Liang D, Wilusz J. Short intronic repeat sequences facilitate circular RNA production. Genes Dev. 2014;28:2233–47.
Zheng Q, Bao C, Guo W, Li S, Chen J, Chen B, et al. Circular RNA profiling reveals an abundant circHIPK3 that regulates cell growth by sponging multiple miRNAs. Nat Commun. 2016;7:11215.
Eisenberg E, Levanon E. Human housekee** genes, revisited. Trends Genet. 2013;29:569–74.
van der Vaart B, Franker MAM, Kuijpers M, Hua SS, Bouchet BP, Jiang K, et al. Microtubule plus-end tracking proteins SLAIN1/2 and ch-TOG promote axonal development. J Neurosci. 2012;32:14722–9.
Zhang CL, Zhang Z, Castle J, Sun SY, Johnson J, Krainer AR, et al. Defining the regulatory network of the tissue-specific splicing factors Fox-1 and Fox-2. Genes Dev. 2008;22:2550–63.
Conn SJ, Pillman KA, Toubia J, Conn VM, Salmanidis M, Phillips CA, et al. The RNA binding protein quaking regulates formation of circRNAs. Cell. 2015;160:1125–34.
Chen F, He L, Qiu LM, Zhou Y, Li ZL, Chen G, et al. Circular RNA circEPB41L2 functions as tumor suppressor in hepatocellular carcinoma through sponging miR-590-5p. Cancer Manag Res. 2021;13:2969–81.
Liu W, Zhao J, ** M, Zhou M. circRAPGEF5 contributes to papillary thyroid proliferation and metastatis by regulation miR-198/FGFR1. Mol Ther Nucleic Acids. 2019;14:609–16.
Chen Q, Liu T, Bao Y, Zhao T, Wang J, Wang H, et al. CircRNA cRAPGEF5 inhibits the growth and metastasis of renal cell carcinoma via the miR-27a-3p/TXNIP pathway. Cancer Lett. 2020;469:68–77.
Li H, Shen SY, Ruan XL, Liu XB, Zheng J, Liu YH, et al. Biosynthetic circRNA_001160 induced by PTBP1 regulates the permeability of BTB via the CircRNA_001160/miR-195-5p/ETV1 axis. Cell Death Dis. 2019;10:960.
Zeng KX, He BS, Yang BB, Xu T, Chen XX, Xu M, et al. The pro-metastasis effect of circANKS1B in breast cancer. Mol Cancer. 2018;17:160.
Fogel BL, Wexler E, Wahnich A, Friedrich T, Vijayendran C, Gao FY, et al. RBFOX1 regulates both splicing and transcriptional networks in human neuronal development. Hum Mol Genet. 2012;21:4171–86.
Yan L, Zheng M, Wang H. Circular RNA hsa_circ_0072309 inhibits proliferation and invasion of breast cancer cells via targeting miR-492. Cancer Manag Res. 2019;11:1033–41.
Zhang H, **ao X, Wei W, Huang C, Wang M, Wang L, et al. CircLIFR synergizes with MSH2 to attenuate chemoresistance via MutSα/ATM-p73 axis in bladder cancer. Mol Cancer. 2021;20:70.
Chen T, Shao S, Li W, Liu Y, Cao Y. The circular RNA hsa-circ-0072309 plays anti-tumour roles by sponging miR-100 through the deactivation of PI3K/AKT and mTOR pathways in the renal carcinoma cell lines. Artif Cells Nanomed Biotechnol. 2019;47:3638–48.
Guo X, Qin M, Hong H, Xue X, Fang J, Jiang L, et al. Circular RNA hsa_circ_0072309 inhibits the proliferation, invasion and migration of gastric cancer cells via inhibition of PI3K/AKT signaling by activating PPARγ/PTEN signaling. Mol Med Rep. 2021;23:349.
**a S, Feng J, Chen K, Ma Y, Gong J, Cai F, et al. CSCD: a database for cancer-specific circular RNAs. Nucleic Acids Res. 2018;46:D925–D29.
Feng J, Chen W, Dong X, Wang J, Mei X, Deng J, et al. CSCD2: an integrated interactional database of cancer-specific circular RNAs. Nucleic Acids Res. 2022;50:D1179–D83.
Salzman J, Gawad C, Wang PL, Lacayo N, Brown PO. Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS One. 2012;7:e30733.
Zhang Y, Xue W, Li X, Zhang J, Chen S, Zhang JL, et al. The biogenesis of nascent circular RNAs. Cell Rep. 2016;15:611–24.
Bachmayr-Heyda A, Reiner AT, Auer K, Sukhbaatar N, Aust S, Bachleitner-Hofmann T, et al. Correlation of circular RNA abundance with proliferation--exemplified with colorectal and ovarian cancer, idiopathic lung fibrosis, and normal human tissues. Sci Rep. 2015;5:8057.
Memczak S, Papavasileiou P, Peters O, Rajewsky N. Identification and characterization of circular RNAs as a new class of putative biomarkers in human blood. PLoS One. 2015;10:e0141214.
Li Y, Zheng Q, Bao C, Li S, Guo W, Zhao J, et al. Circular RNA is enriched and stable in exosomes: a promising biomarker for cancer diagnosis. Cell Res. 2015;25:981–4.
Tan S, Gou Q, Pu W, Guo C, Yang Y, Wu K, et al. Circular RNA F-circEA produced from EML4-ALK fusion gene as a novel liquid biopsy biomarker for non-small cell lung cancer. Cell Res. 2018;28:693–5.
Qiu M, **a W, Chen R, Wang S, Xu Y, Ma Z, et al. The circular RNA circPRKCI promotes tumor growth in lung adenocarcinoma. Cancer Res. 2018;78:2839–51.
Acknowledgements
We would like to thank West China Biobanks, Department of Clinical Research Management, Department of Surgery, West China Hospital of Sichuan University for providing the tissue specimens, especially Ms. Weiwei Tan and Jialei ** for their help during this study.
Funding
This work was supported by National Natural Science Foundation of China (92159302, 91859203, 82100119 and 81871890) and National Key R&D Program of China (2017YFA0504304).
Author information
Authors and Affiliations
Contributions
WL and YP conceived the study and designed the experiments. WRL analyzed the results; CW, ST, WRL, and JC collected and prepared specimens. ST, JKZ, XX, YM, JL and JH performed the experiments. CW, WRL, ST, ZZ and YZ wrote the manuscript. WL and YP supervised this study. All the authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The study was approved by the Ethics Committee of West China Hospital of Sichuan University. All patients provided written informed consent for genomic analysis of their cancer as well as participation in the study.
Consent for publication
The authors approved the publication of this manuscript.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Clinical characteristics of the patients enrolled in this study.
Additional file 2: Table S2.
The information of 215 RBPs that associated with circRNA biogenesis.
Additional file 3: Table S3.
Information of primers and probes used in this study. Table S4. The distribution of putative spliced length of exonic circRNAs. Table S5. Exon numbers of identified exonic circRNAs. Table S6. The circRNAs expressed higher than its host genes. Table S7. The number of circRNAs produced from one gene. Table S12. Information of tissue-specific RBPs.
Additional file 4: Figure S1.
Characterization of protein-coding gene profiles in normal tissues. Figure S2. Bioinformatics validation of tissue-specifically expressed genes using the GTEx dataset. Gene expression was measured in transcripts per million (TPM). Figure S3. The genes with tissue-specific expression patterns were generally enriched in the physiological processes (A) and signaling pathways (B) that were specific to the corresponding tissue. Figure S4. The tissue-specific expression patterns of circRNAs were confirmed by the circAtlas database. Figure S5. The tissue specificity of a subset of circRNAs was governed by their cognate genes. Examples of tissue-specific circRNAs corresponding to their host gene expression. B-D Bioinformatics validation of brain-specifically expressed SLAIN1 using FANTOM5 (B), GTEx (C) and HPA (D) datasets. Figure S6. Brain-specifically expressed hsa_circ_0073539 was regulated by the brain-specific splicing factor QKI. A Brain-specific expression of hsa_circ_0111312 was independent of its host gene (MAN2A1) expression. B The Pearson correlation coefficients between brain-specific hsa_circ_0073539 and the splicing factor QKI. Figure S7. The distribution of uniquely mapped reads (A) and the number of identified upregulated and downregulated circRNAs (B) in each cancer sample. The p-value was calculated using the Wilcoxon rank-sum test, ns denotes no significance, * denotes p < 0.05, ** denotes p < 0.01, *** denotes p < 0.001. Figure S8. Expression patterns of dysregulated circRNAs in cancer. A,B The expression profiles of hsa_circ_0077837 (A) and hsa_circ_0001681 (B) in different cancers. The p-value was calculated using the Wilcoxon rank-sum test, ns denotes no significance, * denotes p < 0.05, ** denotes p < 0.01, *** denotes p < 0.001. Figure S9. Comparison of expression changes between differentially expressed circRNAs and their parental genes among different cancers. Based on circRNA fold-change versus its linear transcript fold-change, circRNAs were divided into four groups. Figure S10. Aberrant expression of circRNAs in cancers may be regulated by the host genes or RBPs. A Expression of hsa_circ_0099329 and its host gene PPFIA2 in tested cancers. B The splicing factor RBFOX1 was exclusively downregulated in GBM. C The Pearson correlation coefficients between RBFOX1 and hsa_circ_0111312 in GBM. The p-value was calculated using the Wilcoxon rank-sum test, ns denotes no significance, * denotes p < 0.05, ** denotes p < 0.01, *** denotes p < 0.001. D Expression of hsa_circ_0111312 and its host gene RASAL2 in tested cancers. Figure S11. circLIFR expression in solid tumors and cells. A Quantification of circLIFR by RT-qPCR in solid tumors (LUAD, CRC, GC, THCA, GBM) and their matched adjacent normal tissues, and circLIFR expression was normalized to U6 mRNA levels. N, normal tissues; T, tumor tissues. B Identification of the circularization site of circLIFR in KYSE150 and HCT-116 cells by RT-PCR and Sanger sequencing. C,D Cellular distribution of circLIFR in KYSE150 and HCT-116 cells, determined by cell nucleus/cytoplasm fractionation and RT-qPCR analyses (C) and fluorescence in situ hybridization (D). β-actin mRNA and U6 RNA represent cytoplasmic and nuclear RNAs, respectively. Western blotting confirmed the efficiency of nuclear/cytoplasmic isolation. Data are shown as the mean ± SD. Figure S12. Effects of circLIFR on cell migration and motility in KYSE150 and HCT-116 cells.
Additional file 5: Table S8.
The expression profiles of 1590 protein-coding genes that ubiquitously expressed in normal tissues.
Additional file 6: Table S9.
The expression profiles of 1765 tissue-specific protein-coding genes.
Additional file 7: Table S10.
The expression profiles of 30 circRNAs that ubiquitously expressed in normal tissues.
Additional file 8: Table S11.
The expression profiles of 439 tissue-specific circRNAs.
Additional file 9: Table S13.
The detailed map** statistics of samples used in this study.
Additional file 10: Table S14.
The expression profiles of 1209 differentially expressed circRNAs identified in this study.
Additional file 11: Table S15.
The differential expression profiles of 215 RBPs that associated with circRNA biogenesis in GBM.
Additional file 12: Table S16.
The differentially expressed protein-coding genes identified in circLIFR-overexpressing KYSE30 cells.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wang, C., Liu, WR., Tan, S. et al. Characterization of distinct circular RNA signatures in solid tumors. Mol Cancer 21, 63 (2022). https://doi.org/10.1186/s12943-022-01546-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-022-01546-4