
Abstract
Background
Acute lung injury (ALI) is a devastating clinical disorder with a high mortality rate, and there is an urgent need for more effective therapies. Fibroblast growth factor 18 (FGF18) has potent anti-inflammatory properties and therefore has become a focus of research for the treatment of lung injury. However, the precise role of FGF18 in the pathological process of ALI and the underlying mechanisms have not been fully elucidated.
Methods
A mouse model of ALI and human umbilical vein endothelial cells (HUVEC) stimulated with lipopolysaccharide (LPS) was established in vivo and in vitro. AAV-FGF18 and FGF18 proteins were used in C57BL/6J mice and HUVEC, respectively. Vascular cell adhesion molecule-1 (VCAM-1), intercellular adhesion molecule-1 (ICAM-1), interleukin-6 (IL-6), tumor necrosis factor-alpha (TNF-α), and p65 protein levels were determined by western blotting or immunofluorescent staining. Afterward, related inhibitors were used to explore the potential mechanism by which FGF18 relieves inflammation.
Results
In this study, we found that FGF18 was significantly upregulated in LPS-induced ALI mouse lung tissues and LPS-stimulated HUVECs. Furthermore, our studies demonstrated that overexpressing FGF18 in the lung or HUVEC could significantly alleviate LPS-induced lung injury and inhibit vascular leakage.
Conclusions
Mechanically, FGF18 treatment dramatically inhibited the NF-κB signaling pathway both in vivo and in vitro. In conclusion, these results indicate that FGF18 attenuates lung injury, at least partially, via the NF-κB signaling pathway and therefore may be a potential therapeutic target for ALI.
Similar content being viewed by others


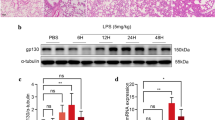
Introduction
Acute lung injury (ALI) is a life-threatening medical condition associated with high morbidity and mortality rates [1], which always characterized by widespread lung inflammation and loss of endothelial integrity [2, 3]. Unfortunately, there is currently no effective pharmacological treatment for ALI. Lipopolysaccharide (LPS) is commonly used to induce an immune response in endothelial cells and is widely employed in mouse models to induce ALI [4, 5]. The development of acute pulmonary inflammation is closely tied to the activation of the mitogen-activated protein kinase (MAPK) and nuclear transcription factor-kappa B (NF-κB) signaling pathways [6, 7]. Abnormal NF-κB activation is a defining feature of ALI [4A, B). RT-PCR further verified the success of FGF18 knockout. (Supplementary Fig. 4B). WT and FGF18+/− mice were subject to LPS administration for 12 h and then euthanized to get lung tissues. In addition, we found that the deletion of FGF18 exacerbated lung injury and inflammatory cell recruitment (Fig. 4C). Moreover, FGF18 deficiency significantly aggravated the elevated levels of VCAM-1, ICAM-1, IL-6, and TNF-α, compared to levels in WT LPS-treated mice (Fig. 4D). Meanwhile, more cell adhesion molecules with less VE-cadherin were also observed in FGF18+/− mice (Fig. 4E, Supplementary Fig. 3B). Taken together, these results indicate that FGF18 ablation aggravates LPS-induced lung inflammation and injury.

FGF18 deletion exacerbates LPS-induced lung injury. (A) FGF18+/+ and FGF18+/− mice were subjected to western blotting analysis. The expression of FGF18 was detected. (n = 5 per group). (B) Immunofluorescent staining of FGF18 (red) and DAPI (blue) in FGF18+/+ and FGF18+/− mice were detected. (n = 5 per group, Scale bar = 150 μm). (C) HE staining in lung tissues of FGF18+/+ and FGF18+/− mice after PBS or LPS injection for 12 h (n = 5 per group). (D) Western blotting was performed and quantitatively analyzed to determine the protein levels of VCAM-1, ICAM-1, IL-6, and TNF-α in the lungs from FGF18+/+ and FGF18+/− mice in the presence of LPS (n = 5 per group). (E) Immunofluorescent staining of ICAM-1/VCAM-1 (red) and DAPI (blue) in FGF18+/+ and FGF18+/− mice were detected. (n = 5 per group, Scale bar = 150 μm)
The knockdown of FGF18 exacerbates LPS-induced HUVEC injury
To further verify the function of FGF18 in LPS treated HUVEC, western blotting analysis showed that FGF18 was significantly decreased after treated with si-FGF18 (Fig. 5A, Supplementary Fig. 4A). To explore the function of FGF18 in vitro, we found that the protein level of VCAM-1, ICAM-1, IL-6, and TNF-α was further increased in HUVECs transfected with si-FGF18 (Fig. 5B), and the mRNA level of ICAM-1 and IL-6 was also increased (Supplementary Fig. 1G, H), suggesting that deletion of FGF18 could exacerbate LPS-induced HUVEC damage. Moreover, we found that the elevated level of ICAM-1 and VCAM-1 (Fig. 5C, D) and decreased level of VE-cadherin (Supplementary Fig. 2B) under LPS conditions were further increased or decreased by si-FGF18 treatment as measured by immunofluorescent staining, respectively. Collectively, these data provide further evidence that knocking down FGF18 could aggravate LPS-induced HUVEC injury.

The knockdown of FGF18 exacerbates LPS-induced HUVEC injury. (A) Western blotting of FGF18 in HUVECs was detected. (n = 5 per group). (B) HUVECs were subjected to western blotting analysis. The expression of VCAM-1, ICAM-1, IL-6, and TNF-α were detected. (n = 5 per group). (C) Immunofluorescent staining of ICAM-1 (red) and DAPI (blue) in HUVECs were detected. (n = 3 per group, Scale bar = 50 μm). (D) Immunofluorescent staining of VCAM-1 (red) and DAPI (blue) in HUVECs were detected. (n = 3 per group, Scale bar = 50 μm)
FGF18 promotes lung repair and attenuates HUVEC injury by inhibiting the NF-κB pathway
LPS treatment caused significant activation of the NF-κB pathway by enhancing the phosphorylation of IκBα and NF-κB p65 in HUVECs and reversed by FGF18 treatment (Fig. 6A). Meanwhile, western blotting analyses revealed that IκBα and NF-κB p65 phosphorylation was significantly decreased in the lungs of AAV-FGF18-treated mice than in the lungs of control mice (Fig. 6B), and higher in FGF18+/− mice than in lungs of WT mice (Supplementary Fig. 3A). Additionally, results from in vitro experiments showed that si-FGF18 treatment increased the phosphorylation of IκBα and NF-κB p65 (Fig. 6C), indicating that FGF18 knockdown enhanced the activation of the NF-κB signaling pathway. Moreover, a nuclear-cytoplasmic separation experiment demonstrated that FGF18 treatment reduced the entry of NF-κB p65 into the nucleus after LPS stimulation, while si-FGF18 treatment increased the nuclear translocation of NF-κB p65 (Fig. 6D, E). These findings suggest that FGF18 plays a direct protective role against LPS-induced endothelial impairment by inhibiting the activation and nuclear translocation of NF-κB p65. To clarify whether FGF18 protects endothelial cell by inhibiting the NF-κB pathway, we used NF-κB p65 inhibitors JSH-23 and Bay11-7082, which inhibits the expression of p-p65 [19, 20]. The results showed that TNF-α and IL-6 were enriched after si-FGF18 treatment, and this phenomenon was alleviated by the treatment of JSH-23 and Bay11-7082 (Fig. 7A, B), suggesting that FGF18 could suppress the TNF-α and IL-6 expression via p65. In addition, JSH-23 and Bay11-7082 treatment eliminated the ICAM-1 and VCAM-1 exacerbations induced by si-FGF18 (Fig. 7C, D).

FGF18 promotes lung repair and attenuates HUVEC injury by inhibiting the NF-κB pathway. (A) HUVECs were subjected to western blotting analysis. The expression of p-p65, p65, p-IκBα, and IκBα were detected. (n = 5 per group). (B) C57BL/6J mice were treated with AAV-FGF18 and AAV-LacZ in the presence of LPS and then subjected to western blotting analysis. (n = 5 per group). (C) HUVECs were subjected to western blotting analysis. The expression of p-p65, p65, p-IκBα, and IκBα were detected. (n = 5 per group). (D) Immunofluorescent staining of p65 (red) and DAPI (blue) in HUVECs were detected. (n = 5 per group, Scale bar = 50 μm). (E) Immunofluorescent staining of p65 (red) and DAPI (blue) in HUVECs were detected. (n = 5 per group, Scale bar = 50 μm)

FGF18 promotes HUVECs repair by inhibiting NF-κB p65 activation. (A) HUVECs transfected with si-FGF18 were then treated with JSH-23 (20 µM) for 3 h, HUVECs were subjected to western blotting analysis. The expression of TNF-α and IL-6 were detected. (n = 5 per group). (B) HUVECs transfected with si-FGF18 were then treated with Bay11-7082 (10 µM) for 3 h, HUVECs were subjected to western blotting analysis. The expression of TNF-α and IL-6 were detected. (n = 5 per group). (C) Immunofluorescent staining of ICAM-1/VCAM-1 (red) and DAPI (blue) in HUVECs were detected. (n = 3 per group, Scale bar = 50 μm). (D) Immunofluorescent staining of ICAM-1/VCAM-1 (red) and DAPI (blue) in HUVECs were detected. (n = 3 per group, Scale bar = 50 μm)
Overall, the data indicate that FGF18 ameliorates lung injury in LPS-induced ALI by inhibiting the NF-κB signaling pathway and protecting against endothelial impairment.
FGF18 is consistent with MAPK kinase inhibitors in reducing the activation of NF-κB pathway
MAPK kinases were widely reported to regulate the inflammation-related signaling pathway. Because of the tight association between the MAPK and NF-κB signaling pathways, a large number of studies have shown that MAPK kinase inhibitors could attenuate phosphorylation of NF-κB p65 [19,20,21]. In the inhibitors group, mice received SB203580 by intraperitoneal injection (Fig. 8A). Moreover, the other two groups of mice underwent gavage of SP600125 and U0126 (Fig. 8B). HE staining revealed that the AAV-FGF18 group and MAPK kinase inhibitors could alleviate lung injury and inflammatory cell aggregation (Fig. 8C). In addition, LPS stimulation enhanced the phosphorylation of IκBα and p65, but this activation was significantly ameliorated by FGF18 and MAPK kinase inhibitors treatment, indicating that FGF18 inhibited LPS-induced activation consistent with MAPK kinase inhibitors in ALI mice (Fig. 8D). Overall, these findings confirm that FGF18 exhibits a comparable protective effect against LPS-induced lung injury as MAPK kinase inhibitors.

FGF18 inhibits NF-κB pathway activation in ALI mice in line with MAPK kinase inhibitors. (A, B) A schematic diagram demonstrates the animal experiment design. (C) HE staining in lung tissues of MAPK kinase inhibitors-treated mice in the presence of AAV-FGF18 after LPS injection for 12 h. (n = 5 per group, Scale bar = 50 μm). (D) C57BL/6J mice were treated with AAV-FGF18 and MAPK kinase inhibitors in the presence of LPS and then subjected to western blotting analysis. (n = 5 per group)
The in vitro experiments conducted in HUVECs demonstrated that FGF18, as well as MAPK kinase inhibitors (SP600125 and SB203580), and si-Erk could alleviate the phosphorylation of IκBα and p65 (Supplementary Fig. 7A). This indicates that FGF18 and MAPK kinase inhibitors have a similar effect in inhibiting the activation of the NF-κB signaling pathway. Furthermore, western blotting analysis confirmed that pretreatment with FGF18 and the inhibition of MAPK kinases reduced the nuclear localization of IκBα and NF-κB p65 induced by LPS (Supplementary Fig. 7B). This suggests that FGF18 and MAPK kinase inhibitors can prevent the translocation of NF-κB p65 into the nucleus. Immunofluorescent staining further supported these findings by demonstrating that FGF18 and MAPK kinase inhibitors could alleviate the entry of NF-κB p65 into the nucleus (Supplementary Fig. 7C). Intriguingly, the combination of FGF18 and MAPK kinase inhibitors could further inhibit the NF-κB pathway (Supplementary Fig. 8A). Overall, these results provide strong evidence that the therapeutic effect of FGF18 is consistent with that of MAPK kinase inhibitors in inhibiting the NF-κB signaling pathway. This suggests that FGF18 may serve as a novel approach for the treatment of acute lung injury.
Discussion
ALI and ARDS are the two main causes of acute lung failure, which is characterized by high morbidity and mortality and for which effective therapeutic strategies are lacking. Thus, identifying novel treatments for ALI is urgently needed. Several lines of evidence show that FGFs play a central role in pulmonary inflammation. Dhlamini, Q et al. found that FGF1 alleviates LPS-induced ALI via suppression of inflammation and oxidative stress [22]. Tichelaar JW et al. confirmed that FGF7 improved survival during ALI in adult mouse lungs after short-term expression [23]. Wang Q et al. demonstrated that the anti-inflammatory effect of FGF10 on NF-κB signaling was mediated through the regulation of oxidative stress [24].
It is well-realized that FGF18 is released by interstitial cells and possibly endothelial cells in the lung and is known to drive cell migration [25], especially for endothelial cells [26]. FGF18 transgene induction also enhanced the expression of other genes that may be involved in angiogenesis, including endothelial cell growth and differentiation factor Wnt2 [27]. Interestingly, previous findings identified FGF18 as a likely important player in the control of alveolar angiogenesis [18], an event that is an absolute requirement for alveolarization and is compromised in bronchopulmonary dysplasia. However, the role of FGF18 in the pathological development of ALI has not been reported. In this study, we revealed that FGF18 protects against pulmonary injury by inhibiting the NF-κB pathway both in vivo and in vitro (Fig. 6). FGF18 inhibits nuclear accumulation of NF-κB p65 and thereby, alleviates cellular inflammation and pulmonary repair. These findings provide new clues and ideas for develo** potential methods to treat ALI and promote pulmonary repair.
Our previous study demonstrated that FGF18 plays a protective role in the liver, especially in liver fibrosis and hepatic ischemia-reperfusion [28, 29]. In the present study, we found that FGF18 was increased upon LPS stimulation, and FGF18 treatment could decrease the phosphorylation of IκBα, and this effect was also correlated to a parallel decrease in the nuclear translocation of the NF-κB p65 as confirmed by immunofluorescence and western blotting analysis. On the contrary, reducing the expression of FGF18 in vivo and in vitro exacerbates lung injury and endothelial cell damage, respectively (Fig. 4). Our work showed that the elevated p-IκBα and p-p65 expression in LPS-treated HUVECs was largely reversed by FGF18 treatment along with alleviated HUVEC injury (Fig. 6). This is the first time for us to explore the relationship between FGF18 and the NF-κB pathway in the context of acute lung injury.
Double immunofluorescent staining indicated that FGF18 was mainly co-localized with CD34 expression, suggesting that endothelial cells are the main source of FGF18 in mice lungs. Consistently, FGF18 was upregulated in LPS-treated HUVECs (Fig. 1). Lung vascular leakage in response to an unchecked cytokine storm generated by the activation of innate immune cells is a hallmark of sepsis-induced inflammatory injury [30,31,32]. The vascular leakage in the lungs is the result of endothelial barrier breakdown and the change level of cell adhesion molecules [33, 34]. Endothelial permeability is normally tuned by the interaction of VE-cadherin in endothelial monolayers [35, 36]. Multiple studies have demonstrated that modifications of FGFs are important for the structure of endothelial cells [37]. Furthermore, aberrant NF-κB activation contributes to the development of vascular leakage [38], among other inflammatory disorders, by mediating the transcription of proinflammatory cytokines such as TNF-α, IL-6, and IL-1β, which in turn enhance the inflammatory response. Here, FGF18 pretreatment significantly suppressed LPS-induced phosphorylation of IκBα and NF-κB p65 in mice, consistent with MAPK kinase inhibitors (Fig. 7).
Hyper-phosphorylation of MAPK molecules can lead to the activation of NF-κB and the subsequent production of inflammatory molecules [19,20,21]. The MAPK and NF-κB pathways have been identified as important targets in LPS-induced ALI. In our study, it was observed that FGF18 significantly attenuated the phosphorylation of NF-κB p65 in a dose-dependent manner, although the specific data was not shown. This finding is consistent with previous studies that have shown MAPK kinase inhibitors can alleviate p65 phosphorylation [39, 40]. Based on these observations, it is reasonable to speculate that FGF18 may affect the NF-κB pathway in a similar manner to MAPK kinase inhibitors. The potential mechanism of FGF18 inhibits p-IκBα and p-p65 may involve several aspects. Firstly, FGF18 may interfere with the activity of kinases involved in the NF-κB signaling pathway, such as the IKK complex (IκB kinase), reduces the phosphorylation of IκBα, thereby inhibiting the activation and intranuclear transfer of NF-κB. Secondly, FGF18 may indirectly affect the NF-κB pathway by affecting the upstream signals of NF-κB activation, such as those of inflammatory factors (TNF-α or IL-1β). Further investigation is still required to fully understand the underlying mechanisms and confirm the specific interactions between FGF18 and the NF-κB pathway.
It appears that our study demonstrated the beneficial effects of FGF18 in alleviating LPS-induced inflammation and preventing endothelial cell leakage in the lung. The overexpression of FGF18 suppressed NF-κB activation, as evidenced by decreased levels of ICAM-1/VCAM-1 and increased levels of VE-cadherin. On the other hand, FGF18 knockdown in mouse models and HUVECs exacerbated endothelial cell leakage. These findings highlight a previously unknown role of FGF18 in regulating the activation of vascular endothelial cells and shed light on the role and mechanism of FGF18 in the pathophysiology of ALI. Consequently, targeting FGF18 may represent a promising therapeutic strategy for the treatment of ALI. It is important to note that FGF18 as a therapeutic agent for ALI would need to be further investigated in preclinical and clinical studies before it can be considered for clinical use.
Conclusion
In conclusion, these data suggest that FGF18 is associated with ALI and that FGF18 effectively protects against ALI by inhibiting NF-κB mediated by p65 activation. This study enriched the regulatory mechanism of NF-κB in sepsis-associated ALI, suggesting that FGF18 may be a therapeutic pathway for ALI. Considering that inflammation is involved in the pathological processes of many diseases, and that FGF18 effectively inhibits the NF-κB pathway in LPS-induced ALI model, we can further validate its therapeutic potential in many inflammation-related diseases.
Data availability
All data generated and analyzed during the study are included in the published article and can be shared upon request.
References
Meyer NJ, Gattinoni L, Calfee CS. Acute respiratory distress syndrome. Lancet. 2021;398:622–37.
Li W, Long L, Yang X, Tong Z, Southwood M, King R, Caruso P, Upton PD, Yang P, Bocobo GA, et al. Circulating BMP9 protects the pulmonary endothelium during inflammation-induced Lung Injury in mice. Am J Respir Crit Care Med. 2021;203:1419–30.
Wu J, Deng Z, Sun M, Zhang W, Yang Y, Zeng Z, Wu J, Zhang Q, Liu Y, Chen Z, et al. Polydatin protects against lipopolysaccharide-induced endothelial barrier disruption via SIRT3 activation. Lab Invest. 2020;100:643–56.
Li J, Lu K, Sun F, Tan S, Zhang X, Sheng W, Hao W, Liu M, Lv W, Han W. Panaxydol attenuates ferroptosis against LPS-induced acute lung injury in mice by Keap1-Nrf2/HO-1 pathway. J Transl Med. 2021;19:96.
Fukatsu M, Ohkawara H, Wang X, Alkebsi L, Furukawa M, Mori H, Fukami M, Fukami SI, Sano T, Takahashi H, et al. The suppressive effects of mer inhibition on inflammatory responses in the pathogenesis of LPS-induced ALI/ARDS. Sci Signal. 2022;15:eabd2533.
Shen W, Gan J, Xu S, Jiang G, Wu H. Penehyclidine hydrochloride attenuates LPS-induced acute lung injury involvement of NF-kappaB pathway. Pharmacol Res. 2009;60:296–302.
Kim BW, More SV, Yun YS, Ko HM, Kwak JH, Lee H, Suk K, Kim IS, Choi DK. A novel synthetic compound MCAP suppresses LPS-induced murine microglial activation in vitro via inhibiting NF-kB and p38 MAPK pathways. Acta Pharmacol Sin. 2016;37:334–43.
**ao K, He W, Guan W, Hou F, Yan P, Xu J, Zhou T, Liu Y, **e L. Mesenchymal stem cells reverse EMT process through blocking the activation of NF-κB and hedgehog pathways in LPS-induced acute lung injury. Cell Death Dis. 2020;11:863.
Wang L, Cao Y, Gorshkov B, Zhou Y, Yang Q, Xu J, Ma Q, Zhang X, Wang J, Mao X, et al. Ablation of endothelial Pfkfb3 protects mice from acute lung injury in LPS-induced endotoxemia. Pharmacol Res. 2019;146:104292.
Shen H, Wu N, Wang Y, Han X, Zheng Q, Cai X, Zhang H, Zhao M. JNK inhibitor SP600125 attenuates Paraquat-Induced Acute Lung Injury: an in vivo and in Vitro Study. Inflammation. 2017;40:1319–30.
Xu X, Zhi T, Chao H, Jiang K, Liu Y, Bao Z, Fan L, Wang D, Li Z, Liu N, Ji J. ERK1/2/mTOR/Stat3 pathway-mediated autophagy alleviates traumatic brain injury-induced acute lung injury. Biochim Biophys Acta Mol Basis Dis. 2018;1864:1663–74.
Krick S, Grabner A, Baumlin N, Yanucil C, Helton S, Grosche A, Sailland J, Geraghty P, Viera L, Russell DW et al. Fibroblast growth factor 23 and Klotho contribute to airway inflammation. Eur Respir J 2018, 52.
Kim YS, Hong G, Kim DH, Kim YM, Kim YK, Oh YM, Jee YK. The role of FGF-2 in smoke-induced emphysema and the therapeutic potential of recombinant FGF-2 in patients with COPD. Exp Mol Med. 2018;50:1–10.
Charoenlarp P, Rajendran AK, Iseki S. Role of fibroblast growth factors in bone regeneration. Inflamm Regen. 2017;37:10.
Yang Y, Zhu X, Jia X, Hou W, Zhou G, Ma Z, Yu B, Pi Y, Zhang X, Wang J, Wang G. Phosphorylation of Msx1 promotes cell proliferation through the Fgf9/18-MAPK signaling pathway during embryonic limb development. Nucleic Acids Res. 2020;48:11452–67.
Li XG, Song X, Wang JY, Sun CH, Li ZQ, Meng LL, Chi SH. Fibroblast growth factor 18 alleviates hyperoxia-induced lung injury in mice by adjusting oxidative stress and inflammation. Eur Rev Med Pharmacol Sci. 2021;25:1485–94.
Sun K, Sun J, Yan C, Sun J, Xu X, Shi J. Sympathetic neurotransmitter, VIP, Delays Intervertebral Disc Degeneration via FGF18/FGFR2-Mediated activation of akt signaling pathway. Adv Biol (Weinh) 2023:e2300250.
Franco-Montoya ML, Boucherat O, Thibault C, Chailley-Heu B, Incitti R, Delacourt C, Bourbon JR. Profiling target genes of FGF18 in the postnatal mouse lung: possible relevance for alveolar development. Physiol Genomics. 2011;43:1226–40.
Plastira I, Bernhart E, Joshi L, Koyani CN, Strohmaier H, Reicher H, Malle E, Sattler W. MAPK signaling determines lysophosphatidic acid (LPA)-induced inflammation in microglia. J Neuroinflammation. 2020;17:127.
Muscella A, Vetrugno C, Cossa LG, Marsigliante S. TGF-β1 activates RSC96 Schwann cells migration and invasion through MMP-2 and MMP-9 activities. J Neurochem. 2020;153:525–38.
Yang CM, Luo SF, Hsieh HL, Chi PL, Lin CC, Wu CC, Hsiao LD. Interleukin-1beta induces ICAM-1 expression enhancing leukocyte adhesion in human rheumatoid arthritis synovial fibroblasts: involvement of ERK, JNK, AP-1, and NF-kappaB. J Cell Physiol. 2010;224:516–26.
Dhlamini Q, Wang W, Feng G, Chen A, Chong L, Li X, Li Q, Wu J, Zhou D, Wang J, et al. FGF1 alleviates LPS-induced acute lung injury via suppression of inflammation and oxidative stress. Mol Med. 2022;28:73.
Tichelaar JW, Wesselkamper SC, Chowdhury S, Yin H, Berclaz PY, Sartor MA, Leikauf GD, Whitsett JA. Duration-dependent cytoprotective versus inflammatory effects of lung epithelial fibroblast growth factor-7 expression. Exp Lung Res. 2007;33:385–417.
Wang Q, Shi Q, Liu L, Qian Y, Dong N. FGF10 mediates protective anti-oxidative effects in particulate matter-induced lung injury through Nrf2 and NF-κB signaling. Ann Transl Med. 2022;10:1203.
Antoine M, Wirz W, Tag CG, Gressner AM, Wycislo M, Müller R, Kiefer P. Fibroblast growth factor 16 and 18 are expressed in human cardiovascular tissues and induce on endothelial cells migration but not proliferation. Biochem Biophys Res Commun. 2006;346:224–33.
Yun EJ, Lorizio W, Seedorf G, Abman SH, Vu TH. VEGF and endothelium-derived retinoic acid regulate lung vascular and alveolar development. Am J Physiol Lung Cell Mol Physiol. 2016;310:L287–298.
Klein D, Demory A, Peyre F, Kroll J, Augustin HG, Helfrich W, Kzhyshkowska J, Schledzewski K, Arnold B, Goerdt S. Wnt2 acts as a cell type-specific, autocrine growth factor in rat hepatic sinusoidal endothelial cells cross-stimulating the VEGF pathway. Hepatology. 2008;47:1018–31.
Tong G, Chen Y, Chen X, Fan J, Zhu K, Hu Z, Li S, Zhu J, Feng J, Wu Z, et al. FGF18 alleviates hepatic ischemia-reperfusion injury via the USP16-mediated KEAP1/Nrf2 signaling pathway in male mice. Nat Commun. 2023;14:6107.
Tong G, Chen X, Lee J, Fan J, Li S, Zhu K, Hu Z, Mei L, Sui Y, Dong Y, et al. Fibroblast growth factor 18 attenuates liver fibrosis and HSCs activation via the SMO-LATS1-YAP pathway. Pharmacol Res. 2022;178:106139.
Friedrich EE, Hong Z, **ong S, Zhong M, Di A, Rehman J, Komarova YA, Malik AB. Endothelial cell Piezo1 mediates pressure-induced lung vascular hyperpermeability via disruption of adherens junctions. Proc Natl Acad Sci U S A. 2019;116:12980–5.
He H, Yang W, Su N, Zhang C, Dai J, Han F, Singhal M, Bai W, Zhu X, Zhu J et al. Activating NO-sGC crosstalk in the mouse vascular niche promotes vascular integrity and mitigates acute lung injury. J Exp Med 2023, 220.
Li Z, Yin M, Zhang H, Ni W, Pierce RW, Zhou HJ, Min W. BMX represses Thrombin-PAR1-Mediated endothelial permeability and vascular leakage during early Sepsis. Circ Res. 2020;126:471–85.
Wu B, Xu MM, Fan C, Feng CL, Lu QK, Lu HM, **ang CG, Bai F, Wang HY, Wu YW, Tang W. STING inhibitor ameliorates LPS-induced ALI by preventing vascular endothelial cells-mediated immune cells chemotaxis and adhesion. Acta Pharmacol Sin. 2022;43:2055–66.
Perkins TN, Oczypok EA, Milutinovic PS, Dutz RE, Oury TD. RAGE-dependent VCAM-1 expression in the lung endothelium mediates IL-33-induced allergic airway inflammation. Allergy. 2019;74:89–99.
Flemming S, Burkard N, Renschler M, Vielmuth F, Meir M, Schick MA, Wunder C, Germer CT, Spindler V, Waschke J, Schlegel N. Soluble VE-cadherin is involved in endothelial barrier breakdown in systemic inflammation and sepsis. Cardiovasc Res. 2015;107:32–44.
Pulous FE, Grimsley-Myers CM, Kansal S, Kowalczyk AP, Petrich BG. Talin-Dependent integrin activation regulates VE-Cadherin localization and endothelial cell barrier function. Circ Res. 2019;124:891–903.
Zhang HY, Su L, Huang B, Zhao J, Zhao BX, Zhang SL, Miao JY. N-benzyl-5-phenyl-1H-pyrazole-3-carboxamide promotes vascular endothelial cell angiogenesis and migration in the absence of serum and FGF-2. Acta Pharmacol Sin. 2011;32:209–16.
Hussman JP. Cellular and Molecular pathways of COVID-19 and potential points of therapeutic intervention. Front Pharmacol. 2020;11:1169.
Ma L, Sun P, Zhang JC, Zhang Q, Yao SL. Proinflammatory effects of S100A8/A9 via TLR4 and RAGE signaling pathways in BV-2 microglial cells. Int J Mol Med. 2017;40:31–8.
Jayakumar T, Hou SM, Chang CC, Fong TH, Hsia CW, Chen YJ, Huang WC, Saravanabhavan P, Manubolu M, Sheu JR, Hsia CH. Columbianadin Dampens in Vitro inflammatory actions and inhibits Liver Injury via Inhibition of NF-κB/MAPKs: impacts on (∙)OH radicals and HO-1 expression. Antioxid (Basel) 2021, 10.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Nature Science Foundation of China (82273560) and the Zhejiang Provincial Natural Science Foundation (LY22H110002).
Author information
Authors and Affiliations
Contributions
ZY-H, JD-D and HF-M carried out most of the in vitro experiments. ZY-H and JD-D drafted the manuscript. ZY-H, TP-X, Y-W and GX-S conceived, supervised the work and revised the manuscript. HC and JZ performed the in vivo studies. ZY-H, Y-W and LT-J discussed experimental designs and gave some revision of the article. All authors discussed the data, commented on the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The animal experiment protocol was reviewed and approved by the Wenzhou Medical University Institutional Animal Care and Use Committee.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Hu, Z., Dai, J., Xu, T. et al. FGF18 alleviates sepsis-induced acute lung injury by inhibiting the NF-κB pathway. Respir Res 25, 108 (2024). https://doi.org/10.1186/s12931-024-02733-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12931-024-02733-1