
Abstract
Background
Constitutional mismatch repair deficiency (CMMRD) is a rare autosomal recessive condition, which is caused by biallelic mutations in mismatch repair genes: MSH2, MLH1, MSH6, and PMS2.
Case presentation
We reported a unique case of an 11-year-old Chinese girl with colorectal polyposis and café-au-lait macules who had no obvious family history of Lynch syndrome-associated tumors, followed by brain gliomas and colorectal carcinoma five years later. The diagnosis of CMMRD was based on gene sequencing analysis showing a homozygous deletion NM_00535.5:c.1577delA (p.Asp526fs) in exon 11 of the PMS2 gene. Although the patient underwent surgery and radiation therapy, and close surveillances including radiological, endoscopic and hematological screening have been recommended, she died of the exacerbation of neurological symptoms at the age of 18.
Conclusions
We identified a novel homozygous deletion in the PMS2 gene in a CMMRD patient with complex clinical features.
Similar content being viewed by others

Background
Mismatch repair (MMR) proteins can correct nucleotide mismatches during DNA replication process and act to maintain genomic stability. Defective MMR system results in the persistence of point mutation or frame-shift mutation and microsatellite instability (MSI), which drives neoplastic changes. Constitutional mismatch repair deficiency (CMMRD) is a rare MMR deficiency-related syndrome, resulting from a homozygous mutation in four MMR genes: MSH2, MLH1, MSH6, and PMS2 [1]. This entity is characterized by a greater risk of develo** childhood and adolescence malignancies, compared to Lynch syndrome (LS) which is caused by a heterozygous mutation in MMR genes and presents with cancers in adult life [1]. Brain malignant tumors are found in over 50% of all CMMRD patients, followed by gastrointestinal tract tumors and hematological tumors. Café-au-lait macules (CALMs) are the most common non-neoplastic manifestations of CMMRD. The prognosis of CMMRD patients is extremely poor due to aggressive phenotypes of tumors, with a median survival period of fewer than 30 months after the first diagnosis. Therefore, there is a clinical imperative to identify CMMRD because close surveillance is thought to greatly reduce cancer morbidity and mortality. Genetic testing for the MMR genes contributes to a confirmed diagnosis and can provide personalized management programs for both patients and their relatives. However, the diagnosis is often delayed because of obscure disease-specific clinical features. In some instances, it is even not stated owing to the lack of disease awareness. We herein presented a unique case of CMMRD who had CALMs and metachronous gliomas and colorectal cancer (CRC). The diagnosis was established by gene sequencing analysis showing a novel homozygous mutation (p.Asp526fs) in PMS2.
Case presentation
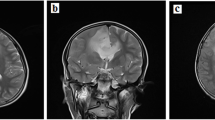
An 11-year-old girl was admitted to our hospital because of a polyp mass drop** out of the anus. Physical examination revealed multiple CALMs, mainly localized on the anterior abdominal wall (Fig. 1A) and lower back (Fig. 1B). The CAL lesions showed foci of skin hypopigmentation with irregular edges and within areas of dense freckles. She also had a hairy pigmented cutaneous nevi on her right forearm (Fig. 1C). Colonoscopy revealed two large polyps in rectus and anus. The pathological results of endoscopically-resected specimen were tubular adenomas with low-grade dysplasia. At the age of 16 she had a severe headache and brain MRI showed a right temporal-occipital ring-enhancing lesion of about 3.5 × 4.5 cm in size (Fig. 1D). She then underwent the gross total resection of the tumor and the pathological analysis demonstrated a glioblastoma multiforme (WHO grade II) and partial anaplastic astrocytomas (WHO grade III) (Fig. 1E). Given the suspected diagnosis of CMMRD, sequencing analysis of MMR genes was performed on both tumor and blood specimens. Other genes associated with childhood tumors were also analyzed, including TP53, BRAF, PTCH1, NF1, and APC, etc. The sequencing on genomic DNA demonstrated a homozygous deletion NM_00535.5:c.1577delA (p.Asp526fs) in exon 11 of the PMS2 gene (Table 1). All other exons and their flanking regions showed a normal sequence. Molecular testing for MSI was also performed on tumor tissue using the Promega MSI analysis system v1.2, which included fluorescently labeled primers for co-amplification of five mononucleotide repeat markers (BAT-25, BAT -26, NR-21, NR-24 and MONO-27). The result showed high-frequency microsatellite instability (MSI-H). Four months after the first surgery, MRI revealed vascular enhancement of nodular mass at the site of the previous surgery which suggested tumor relapse (Fig. 1F). She underwent second surgical resection of the tumor (Fig. 1G), followed by cranial radiotherapy and temozolomide chemotherapy based on the Children's Oncology Group study [2]. She tolerated radiation therapy and temozolomide very well. However, when she started on maintenance cycles of oral temozolomide, she had severe pneumonia. Serum lymphocyte subset analysis showed a decrease in the absolute numbers of CD4+ T cells. Determination of serum immunoglobulin showed her IgG, IgA and IgM levels were profoundly decreased 1.36 g/L (normal 7–16), 0.258 g/L (normal 0.7–4), and 0.29 g/L (normal 0.4–2.3), respectively. She was forced to stop temozolomide treatment due to possible immunodeficiency and IgG substitution was given subsequently for the treatment of severe lung infection. At the age of 17 she did the second colonoscopy which revealed multifocal CR cancer lesions (2 in the transverse colon and 1 in the rectum). The patient then underwent subtotal proctocolectomy and the pathological results showed moderately differentiated CRC (T3N1M0) (Fig. 1H). Immunohistochemistry (IHC) staining detected expression loss of PMS2 protein in both CRC and non-neoplastic tissue (Fig. 1I), while MHL1, MSH2 and MSH6 were all expressed (Additional file 1: Fig. S1). Bevacizumab and topotecan were given due to metastatic CRC and gliomas. The MRI finding suggested an aggressive tumor relapse (Fig. 1J, K) and the patient died of the exacerbation of neurological symptoms at the age of 18.

Photograph of the patient skin cafe´-au-lait macules located on (A) the anterior abdominal wall and (B) lower back and (C) a hairy pigmented cutaneous nevi located on the forearm. Pathologic findings showing (E) diffuse tumor cells, abundant focal cells with atypia, and vascular hyperplasia (HE, ×200); (G) diffuse tumor cells with different forms, multinucleated tumor cells, and focal interstitial small vessel hyperplasia (HE, ×200); (H) colon tubular adenomas, atypical tumor cells, deep-dyed big nucleolus and the mitosis were common (HE, ×200); (I) the expression loss of PMS2 proteins in both tumor and non-neoplastic tissue (IHC, ×200). MRI of the patient brain showing (D) a right temporal-occipital ring-enhancing lesion (arrow) of about 3.5 × 4.5 cm in size at the age of 16, (F) vascular enhancement of nodular mass (arrow) at the site of the previous surgery at the age of 16, (J, K) aggressive tumor relapse (arrow) at the age of 18
Discussion and conclusions
The rarity of CMMRD and its diverse tumor features pose both a diagnostic and managerial challenge. Our patient was first diagnosed as adenomatous polyps with low-grade dysplasia. Based on retrospective research on 288 pediatric patients with coloscopic-proved polyps, isolated juvenile polyps were the most frequent histopathological type while juvenile polyposis syndrome, Peutz-Jeghers syndrome, familial adenomatosis polyposis (FAP), and other cancer predisposition syndromes accounted for the rest kinds of tumorous polyps [7]. After the maternity confirmed, we have performed germline copy number aberrations (CNAs) analysis and determined that the loss of heterozygosity (LOH) has occurred newly in the patient. Sequencing data (FASTQ) of WBC samples collected from 68 healthy individuals were used to construct a "reference panel", which underwent the same experiment and analysis procedures to ensure that it was comparable to the patient's WBC sample. For each reference sample, targeted genomic regions on chromosome 7 were binned into 100 bp. We then applied Circular Binary Segmentation (CBS) on depth ratio between the patient sample and reference. The results showed that c.1577delA resides in the patient's LOH genomic region (Fig. 2A), and c.1621A>G and c.59G>A are located in the younger brother's LOH and diploid regions, respectively (Fig. 2B). These present the diagnostic challenges and difficulties in identifying familial PMS2 mutations.

Germline copy number aberrations (CNAs) and LOH events in the patient and younger brother. The PMS2 c.1577delA resides in the patient's LOH genomic region (A), and c.1621A>G and c.59G>A are located in the younger brother's LOH and diploid regions, respectively (B)
Close colonoscopic surveillance has shown vast advantages by decreasing the modality and mortality of CRC for patients with cancer-prone polyposis syndromes [8]. Generally, colonoscopic screening recommendations depend on risk stratification including the age when colorectum polyps develop, the interval at which polyps grow, the frequency and rate at which polyps may progress to cancer [8]. Those with likely or confirmed hereditary CRC syndromes belong to high-risk patients and should initiate screening earlier and undergo surveillance at shorter intervals than average or increased risk individuals. For CMMRD patients, due to the faster progression of polyp to cancer development, colonoscopy is recommended beginning at the first decade of life or earlier and with a shorter screening interval of 6 months [1]. Video-capsule endoscopy and double-balloon enteroscopy are also recommended to identify small bowel adenomas or cancers. Unfortunately, the patient didn’t comply with surveillance protocols until the development of left-side CRC. Except for the delay in making CMMRD diagnosis, her reluctance to undergo interval screening was the main reason.
Current knowledge of therapeutic strategies and their outcomes in CMMRD are limited because the related information comes from limited research of individual patients or small patient series. Due to the high mutation rate caused by diminished replication repair, rapid onset and frequent recurrence of cancers may occur despite standard-of-care treatment. Chemotherapy remains a frontline treatment based on various phenotypes of CMMRD cancer, but effective chemotherapy drugs are still lacking. This is due to severe drug toxicity in children, MMR deficient associated resistance to chemotherapeutics, and the ability to accelerate somatic mutations which cause an increased risk of secondary tumors. Recently, there is growing evidence to support immunotherapy in the treatment of CMMRD cancers. Immune checkpoint inhibitors, such as programmed cell death-1 blockers, have been found to produce a successful clinical response in CMMRD patients holding MSI-H/hypermutated tumors [9]. Hypermutated tumors lead to the production of truncated protein products termed neoantigens. The preliminary efficacy of neoantigen-based vaccines has been observed in LS patients that show neoantigen-specific immune responses [10]. This creates fair expectations on this class of agents as an effective therapeutic approach for CMMRD. More importantly, given that intensive surveillance may not guarantee detection at a curable stage, preventive treatment strategy represents a promising avenue for CMMRD management. Aspirin has been shown potential in CMMRD patients for chemoprevention against MSI-H CRC, ovarian cancer, and T cell lymphoma [11]. The underlying mechanism is explained by nitric oxide-mediated apoptosis of cells of an MSI phenotype while further clarity is required [12].
Taken together, the diagnosis and treatments of a Chinese girl with CMMRD were outlined and the newly found mutation in PMS2 will update our understanding of genomic variants which contributes to enhanced patient management. Importantly, for pediatric patients with CR adenomas, especially those with CALMs, we recommended the integration of molecular (or genomic) testing into routine clinical care to identify CMMRD.
Availability of data and materials
The datasets generated during the current study are available from the China National Genomics Data Center https://ngdc.cncb.ac.cn/gsa-human/browse/HRA000532, and are also available from the corresponding author on reasonable request. The contact person is Li Ying, email: yingli0209@163.com. The below listed databases were used for data analysis (weblinks provided). PolyPhen2: http://genetics.bwh.harvard.edu/pph2/; ClinVar: https://www.ncbi.nlm.nih.gov/clinvar/; SNPs&GO: https://snps-and-go.biocomp.unibo.it/cgi-bin/snps-and-go/runpred.cgi.
Abbreviations
- CMMRD:
-
Constitutional mismatch repair deficiency
- CALMs:
-
Café-au-lait macules
- CRC:
-
Colorectal cancer
- LS:
-
Lynch syndrome
- MMR:
-
Mismatch repair
- MSI-H:
-
High-frequency microsatellite instability
- NF-1:
-
Neurofibromatosis type 1
- HE:
-
Hematoxylin and eosin staining
- IHC:
-
Immunohistochemistry
- LOH:
-
Loss of heterozygosity
- CNAs:
-
Copy number aberrations
References
Vasen HFA, Ghorbanoghli Z, Bourdeaut F, et al. Guidelines for surveillance of individuals with constitutional mismatch repair-deficiency proposed by the European Consortium “Care for CMMR-D” (C4CMMR-D). J Med Genet. 2014;51(5):283–93.
Cohen KJ, Pollack IF, Zhou T, et al. Temozolomide in the treatment of high-grade gliomas in children: a report from the Children’s Oncology Group. Neuro Oncol. 2011;13(3):317–23.
Wei C, Dayong W, Liqun J, **aoman W, Yu W, **aohong Q. Colorectal polyps in children: a retrospective study of clinical features and the value of ultrasonography in their diagnosis. J Pediatr Surg. 2012;47(10):1853–8.
Lobbous M, Bernstock JD, Coffee E, et al. An update on neurofibromatosis type 1-associated gliomas. Cancers. 2020;12(1):pii.E114.
Gould GM, Grauman PV, Theilmann MR, et al. Detecting clinically actionable variants in the 3’ exons of PMS2 via a reflex workflow based on equivalent hybrid capture of the gene and its pseudogene. BMC Med Genet. 2018;19(1):176.
Wimmer K, Kratz CP, Vasen HFA, et al. Diagnostic criteria for constitutional mismatch repair deficiency syndrome: suggestions of the European consortium “care for CMMRD” (C4CMMRD). J Med Genet. 2014;51(6):355–65.
Steinke V, Engel C, Büttner R, Schackert HK, Schmiegel WH, Prop** P. Hereditary nonpolyposis colorectal cancer (HNPCC)/Lynch syndrome. Dtsch Arztebl Int. 2013;110(3):32–8.
Huck MB, Bohl JL. Colonic polyps: diagnosis and surveillance. Clin Colon Rectal Surg. 2016;29(4):296–305.
Bouffet E, Larouche V, Campbell BB, et al. Immune checkpoint inhibition for hypermutant glioblastoma multiforme resulting from germline biallelic mismatch repair deficiency. J Clin Oncol. 2016;34(19):2206–11.
Schwitalle Y, Kloor M, Eiermann S, et al. Immune response against frameshift-induced neopeptides in HNPCC patients and healthy HNPCC mutation carriers. Gastroenterology. 2008;134(4):988–97.
Leenders EKSM, Westdorp H, Brüggemann RJ, et al. Cancer prevention by aspirin in children with Constitutional Mismatch Repair Deficiency (CMMRD). Eur J Hum Genet. 2018;26(10):1417–23.
McIlhatton MA, Tyler J, Burkholder S, et al. Nitric oxide-donating aspirin derivatives suppress microsatellite instability in mismatch repair-deficient and hereditary nonpolyposis colorectal cancer cells. Cancer Res. 2007;67(22):10966–75.
Acknowledgements
We would like to thank the patient and her family for their collaboration, and thank the Genetron Health, Inc for their contribution on next-generation sequencing and data analysis.
Funding
This work was supported by the Natural Science Foundation of Liaoning Province under Grant number 2019-ZD-0913. The funding body had no influence on study design, data analysis, interpretation of data and writing the manuscript.
Author information
Authors and Affiliations
Contributions
Material preparation, data collection and analysis were performed by SQT and XTW. The first draft of the manuscript was written by AXW and LY, and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Ethics Committee of the Second Hospital of Dalian Medical University, and written informed consent was obtained from all participants and the legal guardian of the participants under the age of 16.
Consent for publication
Written informed consent for publication of identifying images or other personal or clinical details was obtained from all participants and the legal guardian of the participants under the age of 18.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1. Fig. S1
: Positive immunohistochemistry for (A) MHL1, (B) MSH2 and (C) MSH6 in both tumor and non-neoplastic tissue (IHC, ×200).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Tan, S., Wu, X., Wang, A. et al. Diagnostic challenges in a CMMRD patient with a novel mutation in the PMS2 gene: a case report. BMC Med Genomics 14, 184 (2021). https://doi.org/10.1186/s12920-021-01031-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-021-01031-9