
Abstract
Background
The blue-crowned laughingthrush (Garrulax courtoisi) is a critically endangered songbird endemic to Wuyuan, China, with population of ~323 individuals. It has attracted widespread attention, but the lack of a published genome has limited research and species protection.
Results
We report two laughingthrush genome assemblies and reveal the taxonomic status of laughingthrush species among 25 common avian species according to the comparative genomic analysis. The blue-crowned laughingthrush, black-throated laughingthrush, masked laughingthrush, white-browed laughingthrush, and rusty laughingthrush showed a close genetic relationship, and they diverged from a common ancestor between ~2.81 and 12.31 million years ago estimated by the population structure and divergence analysis using 66 whole-genome sequencing birds from eight laughingthrush species and one out group (Cyanopica cyanus). Population inference revealed that the laughingthrush species experienced a rapid population decline during the last ice age and a serious bottleneck caused by a cold wave during the Chinese Song Dynasty (960–1279 AD). The blue-crowned laughingthrush is still in a bottleneck, which may be the result of a cold wave together with human exploitation. Interestingly, the existing blue-crowned laughingthrush exhibits extremely rich genetic diversity compared to other laughingthrushes. These genetic characteristics and demographic inference patterns suggest a genetic heritage of population abundance in the blue-crowned laughingthrush. The results also suggest that fewer deleterious mutations in the blue-crowned laughingthrush genomes have allowed them to thrive even with a small population size. We believe that cooperative breeding behavior and a long reproduction period may enable the blue-crowned laughingthrush to maintain genetic diversity and avoid inbreeding depression. We identified 43 short tandem repeats that can be used as markers to identify the sex of the blue-crowned laughingthrush and aid in its genetic conservation.
Conclusions
This study supplies the missing reference genome of laughingthrush, provides insight into the genetic variability, evolutionary potential, and molecular ecology of laughingthrush and provides a genomic resource for future research and conservation.
Similar content being viewed by others


Background
Laughingthrush (Garrulax) is one of the most common genera (46 species) in the Timaliinae family and is widely distributed in Southeast Asia. Three quarters of the species are found in China [1]. Among them, the blue-crowned laughingthrush (Garrulax courtoisi, GCO) is endemic to China and only exists in Wuyuan, Jiangxi Province. It has a critically endangered (CR) status on the Redlist of China’s Biodiversity and was updated to the most critical class, I (Endangered rating, “I” means critically endangered), in the National Key Protective Species List [2], as well as on the International Union for Conservation of Nature (IUCN) Red List [3] (https://www.iucnredlist.org/).
A mitochondrial-based phylogenetic relationship study revealed a close genetic relationship between GCO and white-browed laughingthrush (Garrulax sannio, GSA) [4]. GCO is similar in body size and reproductive habits to GSA but has a different appearance. The top of GCO’s head is dark blue with black sides and chin. During the breeding season, GCO is bright yellow from the throat to the abdomen, while GSA has brown feathers (Fig. 1a). GCO is a collective nesting and cooperative breeding bird living in old-growth trees and bushes near villages. As a cooperative breeding laughingthrush, GCO offspring have a communal breeding behavior in which offspring act as helpers. This means that the offspring are breeding helpers that stay in the family group and assist their parents in caring for raising the next generation of nestlings [4]. This breeding behavior can be compared to how older human children might help their parents take care of younger children.

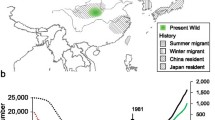
Comparative genomic analysis of the genome assemblies. a Image of GCO (left) and GSA (right). b Distribution of GCO in China. The magenta point represents current habitat of GCO in the Wuyuan area. The grayish blue indicates extinct GCO in Simiao area. c Genomic landscape of the GCO and GSA. The dark red numbers indicate the following: (1) Synteny blocks between the two species. (2) GC content (%). (3)–(8) Represent density of tandem repeats, TEprotein, Transposon, gene, INDELs, and SNPs within a 10-kb sliding window. (9) Assembled scaffolds, green for GCO and light yellow for GSA. The numbers outside the circle indicate the scaffold number of the assembled genome. The unit of (2) is the percentage, the scale of (3)–(8) is the frequency, and the unit (9) is Mb. The density distribution of Ks (d) and Ka/Ks (e) values. f A phylogentic tree was built on the 2391 shared single-copy gene families. Acanthisitta chloris (ACS) was the outgroup. The abbreviations of species correspond to Additional file 1: Table S13. The numerical value beside each node shows the estimated divergence time. The red and green numbers represent significantly expanded and contracted gene families, respectively (left). The classified homologous genes (right). Cre., Cretaceous; Pal., Paleocene; Eoc., Eocene; Oli., Oligocene; Mio., Miocene; Pli., Pliocene; Ple., Pleistocene
For several hundred years, GCO was distributed in Simao, Yunnan Province, and Wuyuan, Jiangxi Province (Fig. 1b). The populations of GCO in both areas were apparently eliminated in the late nineteenth century, but some wild individuals in Wuyuan were rediscovered approximately 100 years later, which verified that GCO had not gone extinct in the wild [5,6,1c; Additional file 1: Table S5). These data indicate high completeness of the genomes of the two species. Repetitive sequences in GCO and GSA account for 18.74% and 16.55% of their genomes, respectively (Additional file 1: Figs. S1, S2 and Tables S6, S7). These values are smaller than the California condor [20] and chicken (galGal6/GRCg6a), and may reflect a slightly expanded assembled genome of the former (Fig. 1c). We predicted 20,819 and 18,367 protein-coding genes for GCO and GSA (Fig. 1c; Table 1 and Additional file 1: Table S8) with high confidence based on homology protein map** and ab initio predictions. The distribution of gene features is consistent with six other homologous species, and the gene numbers averaged higher (23.6% and 13.4%) than these species (Additional file 1: Table S9). More than 90.9% of genes in the two genomes were functionally annotated and 74.6% of them were collectively annotated in SwissProt (http://www.uniprot.org/), KEGG (http://www.genome.jp/kegg/), and InterPro (https://www.ebi.ac.uk/interpro/) (Additional file 1: Fig. S3 and Table S10). Annotation of noncoding RNA of the two species revealed similar copy numbers for each genome (Additional file 1: Table S11). The two assemblies will provide the necessary basic conditions for subsequent research and protection for Garrulax species (Additional file 1: Method S1 and S2 for details).
Comparative genomic analysis
We identified 14,496 homologous genes accounting for 69.63% and 78.92% of the annotated genes of GCO and GSA, respectively, suggesting a high degree of conserved synteny between these two species. Unexpectedly, the variants of GCO were significantly more numerous than those of GSA (Fig. 1c). Some inversions and transpositions were also observed between long scaffolds in the two genomes (Additional file 1: Fig. S4). Non-synonymous mutation/synonymous mutation (Ka/Ks) analysis between GCO and GSA showed that 111 genes were positively selected (Ka/Ks>1, Fig. 1d, e) in GCO, of which 85 annotated genes (Additional file 1: Table S12) were mainly involved in the transcriptional regulation of blood circulation (ASPH, C3AR1, CAV3, P2RY2, PTGER2, HCN4, and SMTNL1) and adipose differentiation (FABP3, PTGER2, PPARGC1A, APOC3, and CAV3; Additional file 1: Fig. S5). We identified orthologous genes among GCO, GSA, and 25 other avian species covering ten bird orders (Additional file 1: Table S13). A total of 2319 1:1:1 single-copy orthologous genes and 4541 N:N:N orthologous genes were identified among them (Fig. 1f). We obtained 38 unique orthologous gene families, including 81 genes, of which HXK2, MyH6, MyH7, and HKDC1 are involved in the ATP metabolic process. In GSA, there are 18 unique orthologous gene families, including 60 genes, of which, PAK3, SCN2A, and NAE1 are involved in the neuron apoptotic process. The lineages to which the two species belong were estimated and found to have diverged from the collared flycatcher (Ficedula albicollis) ~31.47–81.42 Mya. GCO experienced the largest gene family expansion with 1698 expanded and 1114 contracted gene families, while GSA has 540 expanded and 890 contracted gene families (Additional file 1: Table S14). There are 29 genes with significant gain and loss in GCO, but only six in GSA (Fig. 1f).
Population structure of Garrulax species
We sequenced 66 birds including eight Garrulax species and a Cyanopica cyanus with an average coverage depth of ~21.41× (16.62–27.89×; Fig. 1b and Tables S15, S16). After excluding potential related individuals (Additional file 1: Fig. S6 and Table S17), 58 birds were clearly located at nine independent branches, which reflected distinct genetic differentiation among them (Fig. 2a; Additional file 1: Figs. S7, S8). In comparison, GCO has the closest genetic relationship with the black-throated laughingthrush (Garrulax chinensis, GCH), while GSA is close to the masked laughingthrush (Garrulax perspicillatus, GPE). This was simultaneously revealed by principal component analysis (PCA), maximum-likelihood (ML) phylogenies, and the ancestor consanguinity component (Fig. 2b; Additional file 1: Fig. S9). The red-tailed laughingthrush (Trochalopteron milnei, TMI), Chinese Hwamei (Garrulax canorus, GCA), and mustached laughingthrush (Garrulax cineraceus, GCI) are composed of the same ancestral lineages. The rusty laughingthrush (Garrulax berthemyi, GBE), GPE, and GSA comprised another lineage, while GCO and GCH showed a mixed lineage (Fig. 2c). Unexpectedly, when K = 7 (optimal K value; the minimum CV-error; Fig. 2c; Additional file 1: Figs. S9, S10), there was almost no separation of the ancestral lineage between GSA and GPE. The phylogenetic relationship of the eight Garrulax species based on single-nucleotide polymorphisms (SNP) further showed the relatively close genetic relationship associated with a more recent divergence time (~2.81–3.75 Mya; Fig. 2d; Additional file 1: Fig. S11).

Phylogenetic relationship population inference. a Neighbor-joining (NJ) phylogenetic tree of the nine species with 58 individuals. b Principal component analysis of 58 individuals with the first and second principal components being visualized. Color codes for different species are as in a. c Ancestry of Garrulax species. K represents assumptive ancestral lineage. Each vertical line represents one species, and each color represents one ancestral lineage. The length of each colored segment in each vertical indicates proportion of individual genome inferred from ancestral populations. Species are separated by black dotted lines. Abbreviations of species are given in Additional file 1: Table S15. d Estimation of the divergence time among eight Garrulax species. Values in square brackets indicate the divergence time ranges. Dark red nodes represent time calibration points. e Demographic inference based on the Pairwise Sequentially Markovian Coalescent (PSMC) model. The five-colored lines depict the estimated effective population size of the five species dating to roughly 10,000 years ago. The gray background colors indicate glacial periods. f, g showed effective population size in the last 10,000 years for GCO and GSA inferred via PopSizeABC. A 90% confidence interval is indicated by dotted lines. The gray shaded area represents the Song Dynasty period from 960 to 1279 AD
Gene flow among Garrulax species
We considered the large amount of similarities and overlaps in the food and habitats of Garrulax species, which may reflect potential gene flow among them. We confirmed the existence of some migration events among Garrulax using Treemix when the migration events ranged from one to five. Interestingly, GCO and GSA/GPE showed highest migration weight, which indicates the existence of strong gene flow among them. The D-statistics also detected evidence of gene flow between GCO and GSA/GPE, and between GCO and GCA/GBE (Additional file 1: Fig. S12, Table S18a, b). In addition, GCI showed weak gene flow with GPE, GCO, and GSA. We also verified gene flow among other Garrulax species, especially between GCH and GSA/GBE (Additional file 1: Table S18c). These results reflect a common phenomenon: various degrees of gene flow exist among closely related species in some genera, such as the Bos genus [26].
Demographic history in Garrulax species
Admixture and ABBA-BABA analysis showed that GCH, GCO, GBE, GPE, and GSA have a close genetic relationship and frequent gene flow. Therefore, we estimated the historical effective population size of these five species over the past 10,000–1 Mya. We found two different ancient demographic fluctuation patterns, GSA and GPE showed a continuous decline during penultimate glacial and the last glacial period, while GCO, GCH, and GBE showed another similar fluctuation pattern, in which the effective population size increased slowly during the penultimate glacial period and declined rapidly during the last glacial period (Fig. 2e). We found that the effective population size of GCO was higher than that of other Garrulax species until 10,000 years ago. The GCO population has decreased dramatically in the last 1000 years (Fig. 2f). Notably, the demographic pattern showed that GCO was almost extinct during the past ~200 years, which is consistent with past survey results [9], and this result was also supported by smc++ analysis (Additional file 1: Fig. S13). However, the GSA population also experienced a sharp decline, and then a rapid increase, over the last 1000 years (Fig. 2g).
In contrast to its current endangered status, GCO had a large effective population size before 10,000 years ago that was higher than other Garrulax species (Fig. 2e, Additional file 1: Fig. S14). However, both GCO and GSA fluctuated significantly during the most recent 10,000 years (Fig. 2f, g), and the two species sharply declined during the Chinese Song Dynasty (960–1279 AD). Subsequently, GSA gradually recovered to previous levels, whereas GCO maintained a small population size. This situation may be caused by low fecundity, a longer generation interval, or frequent human activities within the habitat of GCO. Monitoring data showed similar reproductive performance between GCO and GSA, including similar clutch size (2–4 vs. 3–4 eggs) and incubation period (12–14 vs. 15–17 days). However, the ratio of successful nesting appeared to be significantly different between GCO and GSA. GSA and GCO averaged 73.3% and 25% a year, respectively. In some years, we did not observe successful maturation of offspring in the GCO population throughout the year, which most likely resulted from frequent human disturbance [9, 27, 28]. The survival rate of juvenile GCO is low [27], but they exhibit cooperative breeding behavior that helps increase the survival rate of nestlings.
Abundant genetic diversity in endangered blue-crowned laughingthrush
Because of their small population size, threatened and endangered species often suffer from a lack of genetic diversity, potentially leading to inbreeding depression and reduced adaptability [29]. Previous studies have found significant differences in the genetic diversity between GCO and GSA [14, 30]. To confirm whether the genetic diversity of GSA is higher than that of GCO, we calculated nucleotide diversity (π) of the two species using SNPs detected from their respective reference genomes. Unexpectedly, GCO showed a significantly higher π than that of GSA at population (3.79×10−5 vs. 0.16×10−5) and individual (2.7×10−3 vs. 0.3×10−3; t-test P = 5.67×10−6) levels, respectively (Fig. 3a, b; Additional file 1: Figs. S15–S17). We also found long runs of homozygosity (LROH, π < 1.0×10−4) in the genomes of the two species, and the homozygous regions of GSA seem to be longer than those of GCO (Additional file 1: Figs. S16 and S17, P = 9.93×10−7). GCO showed shorter linkage disequilibrium (LD) extension (Fig. 3c) and run of homozygosity (ROH) lengths in different fragments (Fig. 3d), which further indicated a higher degree of inbreeding in GSA than in GCO. We also detected the genetic diversity among eight Garrulax species using the SNPs based on GSA reference. GCO had the richest genetic diversity, while the other seven Garrulax species showed very low genetic diversity (Additional file 1: Table S19). In comparison to other Garrulax species, GCO had the smallest inbreeding coefficient (−0.22 vs. −0.08; P = 8.5×10−3; Additional file 1: Fig. S18), highest π (1.56×10−4 vs. 4.78×10−5; P = 3.7×10−5; Additional file 1: Figs. S19–S21), shortest LD extension, and fewest ROH fragments (Additional file 1: Fig. S22) among the eight Garrulax species. However, GSA had the highest inbreeding levels (Additional file 1: Fig. S23).

Genetic diversity analysis. Nucleotide diversity estimated based on the respective reference genomes of GCO (a) and GSA (b). c The LD decay of GCO and GSA. The black dotted line indicates the mean length of genome segments when r2=0.3. d The ROH of GCO and GSA. The x-axis denotes four lengths of ROH segments: <100 kb, 100–500 kb, 500–1000 kb, and >1000 kb. The y-axis denotes the number of different ROH segments in their categories. e The proportion of deleterious mutations in GCO and GSA. f Histogram distribution of published genome-wide estimates of 186 species (167 animals, 15 plants, 3 fungi, and 1 protist taxa), with the position of GCO and GSA heterozygosity rates indicated by asterisks
Small populations are more likely to accumulate strong deleterious variants [29]. We predicted the degree of variants, and GCO showed a significantly lower proportion of homozygous strong deleterious variants than GSA (P < 0.004), whereas there was no difference in heterozygous variants (Fig. 3e). In contrast, GCO had a significantly higher proportion of slightly deleterious variants than GSA (P < 9.2×10−4; Additional file 1: Fig. S24 and Table S20). To further explore the endangered status of the two species among other biological species, we compared GCO and GSA to 186 species globally (Additional file 2: Table S21), including mammals, birds, marine organisms, invertebrates, and plants confronted with different degrees of threat. We found that the nucleotide diversity of GCO is higher than most LC species, while GSA is close to the CR crested ibis (Nipponia nippon), which is contrary to normal perception (Fig. 3f). These data suggest that the genomic status of GCO does not correspond to its endangered survival status.
Obvious selective sweeps occurred in the endangered blue-crowned laughingthrush
In consideration of the critically endangered status of GCO and its cooperative breeding behavior, we hypothesized that the GCO genome may retain footprints reflecting these characteristics. Hence, we conducted FST analysis between GCO and other laughingthrush species. After long-term natural selection, homozygous regions related to the above characteristics may remain in the GCO genome. Therefore, we carried out zHp analysis within the GCO population. Overlap windows of zHp and FST were used to explore the putative selective sweeps in GCO (Fig. 4 and Additional file 1: Fig. S25). Taking experiential top 1% (FST = 0.52 and zHp = −3.14) as selective sweeps thresholds, we identified 42 overlap** windows, of which, two windows including regions of 9.2–9.24 Mb on Scaffold 65 (FST = 0.53, zHp = −7.10) and 20.08–20.12 Mb on Scaffold 131 (FST = 0.55, zHp = −8.27) had the lowest zHp values. A total of 22 overlap** genes were identified from the 42 windows (Fig. 4a; Additional file 1: Table S22), of which, two windows including Bdkrb2 and CYP26A1 with the lowest zHp and highest FST were selected because they had lower π and Tajima’s D values with an obviously selective sweep in GCO (Fig. 4b, c).

Selective sweeps of GCO. a The Manhattan plots of Z-transformed average pooled heterozygosity in GCO (zHp), and average fixation index (FST) between GCO and seven other Garrulax species. The shadows identified by FST and zHp represent the regions with the extremum of the two statistics. Nucleotide diversity and Tajima’s D values with non-overlap** 10-kb sliding windows on Scaffold 65: 8.7–9.74 Mb (b) and Scaffold 131: 19.58–20.62 Mb (c)
Sex confirmation and marker-assisted breeding based on the short tandem repeat profiling of blue-crowned laughingthrush
Using genomic loci as trace to reduce inbreeding in endangered species is often used in the protection and rescue of endangered animals, as well as for sex identification [31]. In contrast to sexually dimorphic birds, the sexes of GCO are difficult to separate. We used a depth-based strategy to identify scaffolds representing the sex chromosomes of GCO and GSA. In GCO, 15 and 10 scaffolds were identified as Z- and W-related chromosomes (Additional file 1: Table S23). For GSA, 24 and five scaffolds were regarded as Z- and W-related chromosomes, respectively (Additional file 1: Table S24). We then checked the sex of all GCO and GSA individuals and found four females and ten males in GCO, and four females and seven males in GSA, which was identical to sample records (Additional file 2: Tables S23, S24).
Due to the abundant number of short tandem repeats (STR) and a high degree of variants among individuals, we detected a genome-wide STRs for GCO. We identified 233,287 STRs in the whole-genome of GCO, which the minor STR alleles exhibited a 17.35-bp difference from their major alleles. We then focused on the 1021 STRs with characteristics of a 4-bp repeat unit, allele types greater than four, and non-missing in 14 GCO individuals. Among these, 218 STRs with abundant polymorphism information content (PIC) were located in the 32-annotated-gene region (Additional file 1: Table S25) and can be used as identification of kinship in GCO. Besides, 1688 of 3941 STRs on the W-related scaffolds were identified with characteristics of homozygote in females and deletion in males, 43 of them were surrounded by 12 genes, which can be used as sex identification markers (Additional file 1: Table S26).
Discussion
The assembled genomes of GCO in CR state and GSA in LC state reported here provide insights into the evolutionary history of Garrulax species and a basis for future genetic and genome-wide studies, not only in the laughingthrush but also in other songbirds. These genomes are typical for avian species: total length, synteny, repeat burden, and GC content (Fig. 1c; Table 1). They are similar to those of previously sequenced bird genomes [32], even those of distant relatives [33]. Here, we noticed that the variants (SNPs and INDELs) in the GCO genome are much higher than those of GSA (Fig. 1c), which suggests an abundant demographic history of GCO before they became endangered. A high level of historical abundance was also found in the California condor [20]. This conclusion is consistent with demographic inferences based on several methods for eight Garrulax species, which showed that GCO maintained the highest effective population size before the last glaciation and kept a dramatically small number of wild populations after the Chinese Song Dynasty (960–1279 AD). Coincidentally, China experienced a period containing several cold intervals during the Song Dynasty [34, 35], which may have decreased avian population sizes because of reduced food supplies or poor adaptation to the cold conditions. The bottleneck events occurred almost simultaneously in the Garrulax species and remind us that there may have been parallel evolution among laughingthrush species due to their similar body size, biology, and overlap** habitats. Parallel evolution is common in birds, such as ptilopody in chickens and pigeons [36]. In addition to their similar dietary habits, GCO and GSA also show similar reproductive habits, like cooperative breeding. The economy and culture flourished during the Chinese Song Dynasty, which stimulated the prevalence of pet aviculture. One aviculture expression, called “Liu Niao,” translates to the behavior of people admiring pet birds in cages. Evidence for this is provided in the form of delicate birdcages and realistic bird paintings [37, 38], as well as in the excavation of cultural relics and research findings on the characteristics and trade of the thrush [39, 40]. In addition, with a colder climate and humans migrating southward, the habitats of GCO were likely affected, and some suitable habitats were probably destroyed [41]. Although laughingthrush were continuously captured and kept as pets [40], their population size recovered after suffering from a short-term bottleneck, which may be attributed to rising temperatures and strong reproductive performance, such as GCA and GSA [42]. However, since GCO persistently maintains a small population size, another reason may be human overhunting due to their desirable ornamental appearance compared to other laughingthrush. More than 400 GCO were poached from 1987 to 1992 and trafficked to various countries [13]. Fortunately, the Wuyuan National Nature Reserve has been established as a protected area to provide a better habitat for GCO and other CR species.
Small populations are more likely to experience genetic drift, accumulation of deleterious mutations, and reduced genetic diversity. They are especially vulnerable to extinction due to deterministic and stochastic threats [29]. For a species that was nearly extinct in the wild, inconsistent with its CR status, the GCO has unexpectedly higher genome-wide diversity than other laughingthrush. The geographical distance between Simao, a previous habitat of GCO, and the existing habitat of Wuyuan (more than 2300 km away), the migratory ability of GCO [4, 9, 27], and the results of rich genetic diversity suggest that GCO was once widely distributed in Southern China, and there may have been substantial gene exchange among different groups. Both a sustained bottleneck and a strong, yet brief, bottleneck can lead to the reduction of genetic diversity [44, 45]. After experiencing a long-term bottleneck, GCO still has substantial genetic diversity compared to the LC Garrulax species, which suggests that GCO had a large population size before it became endangered. In addition, the demographic inference results showed that GCO was abundant about 1000 years ago, which also supports the conclusion that GCO was once widely distributed in Southern China. In contrast, other laughingthrush birds lack migratory ability [42] (https://species.sciencereading.cn/). This phenomenon means a lack of genetic exchange among different groups that may lead to a decline in genetic diversity, even in laughingthrush birds widely distributed in China. These facts may help explain why the genetic diversity of GCO is significantly higher than that of other laughingthrushes, and also suggests that the effective population size of GCO was large until 1000 years ago. Decreased selection intensity and increased genetic drift can be caused by several generations of inbreeding in a small population, and result in a lower π, higher LD decay, longer ROHs, and more deleterious mutations [46]. However, our results support the opposite conclusion, which is similar to results on the California condor (Gymnogyps californianus) [20] and the island fox (Urocyon littoralis) [30]. The endangered GCO showed the smallest inbreeding coefficient, highest π, shortest LD extension, and fewest ROH fragments among eight Garrulax species. Compared to most other LC species, GCO have a higher heterozygosity rate. Such anomalies may be attributed to the communal breeding behavior in which GCO offspring act as helpers [4], which effectively avoids inbreeding. According to the field tracking observations and breeding experience with captive wildlife in Nanchang and Hong Kong, the lifespan of GCO is about 20–30 years, while other Garrulax species, such as GCA and GSA, have an average lifespan of 8–15 years. The longer generation times of GCO might slow the loss of genetic diversity. The length of time needed to reach maturity and the cooperative pattern of reproduction are important questions that would benefit from additional research.
Consistent with the genetics of kākāpō and island fox [30, 47], we found GCO accumulates more slightly deleterious variants and fewer strongly deleterious variants compared to GSA. Species in the CR state for a long time, such as GCO and island fox, that permanently maintain small population sizes tend to accumulate fewer strongly deleterious variants, and this reduces the risk of inbreeding depression [30]. For these species, longer generation times and reduced inbreeding will persistently eliminate strongly deleterious mutations. Our research showed that the critically endangered GCO currently fits this survival model.
Since GCO has the characteristics of being extremely endangered with a long lifespan, cooperative reproduction, and poor environmental adaptability, it is possible that these characteristics may leave genetic footprints on the GCO genome. Human tourism and hunting activities have an impact on GCO’s nesting and reproduction, and cause behaviors such as nest abandonment [48]. Multiple genes were selected in GCO after long-term natural selection, which are well-characterized in humans but not in birds. For example, previous studies showed that the Bdkrb2 gene is related to human anxiety [49], which suggests an association between Bdkrb2 and the sensitivity of GCO. According to field observations, GCO is very alert and sensitive and maintains a relatively long distance from humans, even when living in old-growth trees and bushes near villages that often have high levels of human activity. It is difficult to view GCO at close range or to observe them at any distance. As a famous caged Garrulax [40] species, GCO has a beautiful birdsong and bright feathers which has made it a desirable target for human capture, especially during the Song Dynasty. Consequently, humans capturing them and natural selection may have long subjected GCO to anxiety behavior. We detected a strong selected gene, CYO26A1, which is related to reproductive performance. The activity of CYP26A1 is important for maintaining pregnancy, and it may affect embryo implantation by regulating the differentiation and maturation of uterine dendritic cells. Thus, inhibiting the expression of CYP26A1 will lead to pregnancy failure [50]. To ensure the birth and survival of nestlings, the competitive behavior among adult birds during cooperative breeding is significantly lower than that during the non-breeding period [4], which may be related to the CYP26A1 gene in GCO. These genes are understood in humans, but it is unclear what the functions of these genes are in birds; thus, studies on model species such as zebra finches may reveal more about the roles of these genes.
Genoty** is a common way of identifying the sex of non-dimorphic birds. We identified scaffolds representing sex chromosomes and selected 43 sex-linked STRs to identify the sex of the GCO samples. These will be used to reconstruct the pedigree relationships of wild GCO. Then, we will establish individual identities to avoid losing genetic diversity and assist in increasing the GCO population size. This strategy has been successful in increasing the population size of crested ibis [14, 56]. The homozygous SNP ratios were less than 0.0008%, which reflected the accuracy of genome assemblies. Completeness of the two genomes were assessed based on 248 core genes in the Core Eukaryotic Genes Map** Approach (CEGMA) program [57] and 2586 conserved genes in the BUSCO program [58] with default parameters. CEGMA assessment combined with tblastn v2.2.26 [59], genewise v2.4.1 [60], and geneid v1.4.5 software (https://genome.crg.es/software/geneid/) to evaluate the completeness of assembled genomes. BUSCO assessment based on tblastn, Augustus v3.3.3 [61], and hmmer [62] to assess the integrity of assembled genomes. Results showed that over 84% of 248 core genes and over 97% of 2586 avian orthologous genes were found in the Garrulax genomes, also reflecting its high quality. In addition, the Program to Assemble Spliced Alignments (PASA) tool was used to assemble transcripts based on RNA-seq reads that were aligned to the two Garrulax genome assemblies.
Repeat elements
Repeat elements occupy a major proportion of the nuclear DNA in most eukaryotic genomes. Repeat sequence annotation methods were divided into two types: homologous sequence alignment and de novo prediction (Additional file 1: Method S2). We first customized a de novo repeat library of the genome using RepeatModeler, which can automatically execute two de novo repeat finding programs. The remaining consensus sequences were used as the library in RepeatMasker [63] to identify and cluster repetitive elements. The Tandem Repeats Finder (TRF) package v4.09 [64] was used to identify tandem repeat sequences in the Garrulax genomes.
Gene prediction and functional annotation
After the masking of repeats, we used a combination of homology-based prediction, de novo prediction, and RNA-Seq-assisted prediction to predict structural annotation of the Garrulax genomes (Additional file 1: Method S2). De novo prediction was performed using Augustus, GlimmerHMM v3.0.4 [65], and SNAP v2013.11.29 [66]. For homologous annotation, the Garrulax genome sequences were compared to the protein-coding sequences of homologous species (Struthio camelus, Nipponia nippon, Anas platyrhynchos, Gallus gallus, Meleagris gallopavo, and Anser cygnoides) by BLAST [67] and genewise [60] to predict the gene structure. The transcript isoforms of Garrulax were mapped to the genome and then assembled by PASA v2.4.1 [68]. EVidenceModeler v1.1.1 (EVM; http://evidencemodeler.sourceforge.net/) was used to integrate these gene models from the above methods. The predicted protein sequences were compared with several public databases (SwissProt, NR, Pfam, KEGG, and InterPro) to further detect the function of the protein-coding genes in Garrulax.
Comparative genome analysis
The genome synteny between GCO and GSA was evaluated by the One step MCScanX method from TBtools and visualized through the Advanced Circos method [69]. Ks values between the two species were calculated using Simple Ka/Ks Calculator method in TBtools. Protein and CDS sequences of the other 25 species were downloaded from NCBI (Additional file 1: Table S13). The 25 species selected cover the main families of birds to show the taxonomic position of Garrulax species in birds. OrthoFinder v2.4.1 [70] was used to analyze single-copy homologous gene families of the 27 bird species after extracting the longest transcripts and removing the transcripts if their length was less than 150 bp. The command -m MFP -bb 1000 -bnni -redo from IQ-tree v2.0.6 [71] was used to construct a species phylogenetic tree based on single-copy homologous gene families after aligning by MUSCLE v3.8.1551 [72] and BMGE [73] software. The divergence time of the 27 bird species was estimated by mcmctree program implemented in PAML v4.9 [74]. Four calibration times obtained from the TimeTree (http://www.timetree.org/) website were set for dating analysis: 26~35 million years ago (Mya) for Anser cygnoides and Anas platyrhynchos, 33.2~42.3 Mya for Coturnix japonica and Gallus gallus, 7~19.2 Mya for Pygoscelis adeliae and Pygoscelis antarcticus, 72~90 Mya for Egretta garzetta and Columba livia. The contraction and expansion of gene families were estimated using CAFE v4.2.1 [75].
Genome resequencing
Quality testing included (i) analysis of DNA purity and integrity by agarose gel electrophoresis, (ii) Nanodrop detection of the purity of DNA (OD260/280 ratio), and (iii) qubit quantification of the DNA concentration ratio. The qualified DNA samples of 66 birds were sequenced via Illumina NovaSeq platform with an average coverage depth of ~21.41×. Clean data were obtained after the following processing from raw data: (i) removing adapter paired-end reads, (ii) removing the paired-end read if the N content length exceeds 10% in one single-end read, (iii) deleting the paired-end read if the low-quality (Q≤5) base number exceeds 50% in one single-end read. After processing, base quality Q20 and Q30 were 96.82% and 92.03% respectively with an average of 29.56 Gb per sample. The cleaned fastq files were used to detect variants.
Variant detection
We used the higher-quality genome assembly of GSA as a reference to detect variants of 66 birds following the steps: (i) constructing reference genome index by BWA; (ii) aligning the cleaned fastq files of 66 individuals using BWA; (iii) sorting the aligned bam files using SAMtools; (iv) RealignerTargetCreator and IndelRealigner functions in GATK v4.0.2.1 (https://gatk.broadinstitute.org/hc/en-us) were used to mark PCR duplication and re-align; (v) HaplotypeCaller in GATK method was used to detect variants. A total of 126,883,783 variants were acquired after filtration with the criteria of QD < 2.0, FS > 20.0, MQ < 40.0, SOR > 3.0, MQRankSum < −2.0 and ReadPosRankSum < −5.0; (vi) 79,518,440 variations were detected by Platypus v0.8.1 [76]; (vii) 47,617,703 variants from (v) and (vi) intersection were used as the source to detect variants again utilizing Platypus. A total of 13,651,674 variants including 11,105,087 SNPs and 2,546,587 InDels were obtained after hard filtration again using the above criterion. The assembled GCO genome was used as a reference to detect variants of 14 GCO under the same strategy and 9,398,622 variants were detected after filtering.
Identification of scaffolds from chromosomes Z and W
We used SAMtools to count the depth of each individual and to identify sex-related scaffolds that belong to chromosomes Z and W by comparing the read depth for each scaffold in ten males and four females. We then downloaded most high-quality avian sex chromosomes from the NCBI database (Additional file 1: Table S27) and then combined these into a synthetic reference genome. The sex-related scaffolds of GCO and GSA were consistently split into non-overlap** short reads with 300 bp length and aligned to the synthetic reference genome using BWA. SAMtools was then used to extract reads of Map** Quality > 30 for screening and classification. We discarded reads that aligned on different chromosomes or multi-reads aligned on the same chromosome and kept all results with the same map** values or one result with the peak score. The scaffolds with a BWA map** rate > 20% were considered sex scaffolds. The sex-related scaffolds of GCO and GSA were aligned to the synthetic genome using minimap2 [77]. Finally, the sex-related scaffolds were verified according to the proportion of aligned reads or the results of minmap2. We identified 15 scaffolds in GCO, which belonged to chromosome Z with a total length of 86.74 Mb, and 10 scaffolds, which belonged to chromosome W with a total length of 28.67 Mb (Additional file 2: Table S23). For GSA, there were 24 scaffolds that belonged to chromosome Z with a total length of 72.59 Mb, and five scaffolds belonged to chromosome W with a total length of 18.61 Mb (Additional file 2: Table S24).
Genetic structure and divergent time estimation analysis among laughingthrush species
A phylogenetic tree was constructed to explore relationships among the nine species. We extracted SNPs from predicted CDS regions and removed SNPs with LD (r2) > 0.5. A new dataset of 78,947 SNPs was generated and used for analysis. We set an ultrafast bootstrap approximation (UFBoot = 2000) to assess branch supports [78] and found an optimization model (TVM + F + ASC + G4) using the ModelFinder method [79]. The maximum-likelihood (ML) tree between individuals was estimated through IQ-tree software, and we visualized the tree using Figtree v1.4.4 (http://tree.bio.ed.ac.uk/software/figtree/). The statistics PI_HAT and DST were calculated by the command --genome from PLINK v1.9 [80]. Of which, PI_HAT means shared proportion of identity-by-descent (IBD) and was calculated by formula PI_HAT = PIBD=2 + 0.5*PIBD=1 and DST was calculated by DST = (IBS2 + 0.5*IBS1)/(IBS0 + IBS1 + IBS2). In this equation, IBS means identity-by-state, IBS0, IBS1, and IBS2 represent the number of IBS 0, 1, and 2 non-missing variants respectively. According to ML-tree and estimated genetic relationship among the 66 individuals. Several individuals appeared to be related (Additional file 1: Table S17 and Fig. S6). The individuals with lower sequencing quality among these related individuals were removed and a total of 58 individuals remained and used to reconstruct ML-tree. The IBS distance matrix among individuals was estimated by command --pairwise-distance from PLINK, visualized by pheatmap package in R language, and we constructed a neighbor joining (NJ) tree using SplitsTree v4.16.1 [81].
We randomly selected five individuals from each Garrulax species to establish an accurate taxonomy and divergence time of eight Garrulax species. To reduce computation time, 43,475 SNPs in predicted CDS regions with LD (r2) < 0.5 were reserved. Then, divergence times among Garrulax species were estimated by SNAPP software from BEAST v2.6.4 [1: Fig. S20, Table S20).
Deleterious mutation analysis
Frameshift, stop gain, stop loss, and splicing mutations are considered to be strong deleterious variants, while missense and synonymous mutations are slight deleterious variants. We analyzed the deleterious mutations in the two species following steps: (i) constructing genome database of GCO and GSA through ANNOVAR [91] and converting gff3 to gtf format through gffread v0.12.4 software [92]; (ii) converting gtf to genePred format by gtftoGenepred; (iii) transcripts in fasta format obtained from retrieve_seq_from_fasta.pl algorithm in ANNOVAR; (iv) using table_annovar.pl in ANNOVAR algorithm to annotate variations of the two species. We then counted the number of each mutation type and visualized them by in-house R scripts.
Genome-wide selection analysis
FST between ten GCO and 47 other Garrulax individuals was performed with sliding non-overlap** 40-kb windows across the genome. For each window with the number of SNPs more than 10, we also calculated zHp through formula [93]:
where nNAJ and nMIN represent the frequency of primary and secondary alleles of each SNP respectively, μ and σ represent mean and standard deviation of Hp. We calculated Tajima’s D and π values with non-overlap** 10-kb sliding windows via VCFtools on regions of extending 500 kb on both sides of the two windows.
Identification of sex-related scaffolds and genome-wide STRs
To distinguish the sex of GCO more easily, we identified the sex-related scaffolds for further research based on the sequencing depth through depth function of SAMtools. To detect STRs, we defined STRs using Tandem Repeat Finder [94] by parameters of Match = 2, Mismatch = 7, Delta = 7, PM = 80, PI = 10, Minscore = 30, and MaxPeriod = 6. Then the whole-genome STRs of GCO were detected by GangSTR [95].
Availability of data and materials
Genomic data of blue-crowned laughingthrush and white-browed laughingthrush were uploaded to the National Genomics Data Center (NGDC) under accession number PRJCA005228 [96]. The raw sequencing data of 66 birds is available from NGDC under accession number PRJCA005263 [97]. Analytical pipelines and code are available on the Zenodo (doi: 10.5281/zenodo.6570622) [98].
Abbreviations
- GCO :
-
Garrulax courtoisi
- CR:
-
Critically endangered
- IUCN:
-
International Union for Conservation of Nature
- GSA :
-
Garrulax sannio
- LC:
-
Least concern
- CEGMA:
-
Core Eukaryotic Genes Map** Approach
- PCA:
-
Principal component analysis
- ML:
-
Maximum-likelihood
- GCA :
-
Garrulax canorus
- GCI :
-
Garrulax cineraceus
- GBE :
-
Garrulax berthemyi
- SNP:
-
Single-nucleotide polymorphisms
- LD:
-
Linkage disequilibrium
- ROH:
-
Run of homozygosity
- STR:
-
Short tandem repeats
- PIC:
-
Polymorphism information content
- CCY:
-
Cyanopica cyana
- CEGMA:
-
Core Eukaryotic Genes Map** Approach
- TRF:
-
Tandem Repeats Finder
- NJ:
-
Neighbor joining
- PC:
-
Principal components
References
Zheng Z. The evolutionary of Chinese Garrulax and comparison among different habitats. Curr Zool. 1982;03:4–9.
China NFaGAo. Official release of the updated list of wild animals under Special State Protection in China; 2021.
Cai T, Cibois A, Alstrom P, Moyle RG, Kennedy JD, Shao S, et al. Near-complete phylogeny and taxonomic revision of the world's babblers (Aves: Passeriformes). Mol Phylogenet Evol. 2019;130:346–56.
Liu D, Wu Z, Wang X, Huang H, Li D. Cooperative breeding behavior of captive blue-crowned laughingthrush (Garrulax courtoisi). Chin Wildlife. 2016;37(3):228–33.
Wilkinson R, He F, Gardner L, Wirth R. A highly threatened bird—Chinese yellow-throated laughing thrushes in China and in zoos. Int Zoo News. 2004;51(8):456–69.
Borrell O. A short history of the heude museum "Musee Heude": its botanist and plant collectors. J Hong Kong Branch Royal Asiatic Soc. 1991;31:183–91.
He F, ** Z. Garrulax galbanus courtoisi in Wuyuan, Jiangxi Province. Chin J Zool. 2002;38(5).
Hong Y, He F, Wirth R, Melville D, Zheng P, Wang X, et al. Little-known oriental bird: Courtois's laughingthrush; 2003.
He F, Lin J, Wen C, Shi Q, Huang H, Cheng S, et al. A preliminary study on the biology Garrulax courtoisi in Wuyuan. Chin J Zool. 2017;52(1):167–75.
Liao W, Hong Y, Yu S, Ouyang X, He G. Research on the breeding ecology of Garrulax galbanus courtoisi in Wuyuan and its relationship between villages. Acta Agric Univ Jiangxiensis. 2007;29(005):837–50.
Hong Y, Yu S, Liao W. Study on breeding habitat of Garrulax galbanus courtoisi in Wuyuan. Acta Agric Univ Jiangxiensis. 2006;28(006):907–11.
Chen G, Zheng C, Wan N, Liu D, Fu V, Yang X, et al. Low genetic diversity in captive populations of the critically endangered Blue-crowned Laughingthrush (Garrulax courtoisi) revealed by a panel of novel microsatellites. Peer J. 2019;7:e6643.
Li N, Huang X, Yan Q, Zhang W, Wang Z. Save China's blue-crowned laughingthrush. Science. 2021;373(6551):171.
Feng S, Fang Q, Barnett R, Li C, Han S, Kuhlwilm M, et al. The genomic footprints of the fall and recovery of the crested ibis. Curr Biol. 2019;29(2):340–349 e347.
Li S, Li B, Cheng C, **ong Z, Liu Q, Lai J, et al. Genomic signatures of near-extinction and rebirth of the crested ibis and other endangered bird species. Genome Biol. 2014;15(12):557.
Guang X, Lan T, Wan Q-H, Huang Y, Li H, Zhang M, et al. Chromosome-scale genomes provide new insights into subspecies divergence and evolutionary characteristics of the giant panda. Sci Bull. 2021;66(19):2002–13.
Lynch M, Conery J, Burger R. Mutation accumulation and the extinction of small populations. Am Nat. 1995;146(4):489–518.
Rogers RL, Slatkin M. Excess of genomic defects in a woolly mammoth on Wrangel island. PLoS Genet. 2017;13(3):e1006601.
van der Valk T, Diez-Del-Molino D, Marques-Bonet T, Guschanski K, Dalen L. Historical genomes reveal the genomic consequences of recent population decline in eastern gorillas. Curr Biol. 2019;29(1):165–70.
Robinson JA, Bowie RCK, Dudchenko O, Aiden EL, Hendrickson SL, Steiner CC, et al. Genome-wide diversity in the California condor tracks its prehistoric abundance and decline. Curr Biol. 2021;31:2939–46.
Diez-Del-Molino D, Sanchez-Barreiro F, Barnes I, Gilbert MTP, Dalen L. Quantifying temporal genomic erosion in endangered species. Trends Ecol Evol. 2018;33(3):176–85.
Hohenlohe PA, Funk WC, Rajora OP. Population genomics for wildlife conservation and management. Mol Ecol. 2021;30(1):62–82.
Qu Y, Chen C, Chen X, Hao Y, She H, Wang M, et al. The evolution of ancestral and species-specific adaptations in snowfinches at the Qinghai–Tibet Plateau. P Natl Acad Sci USA. 2021;118(13).
Lai YT, Yeung CKL, Omland KE, Pang EL, Hao Y, Liao BY, et al. Standing genetic variation as the predominant source for adaptation of a songbird. P Natl Acad Sci USA. 2019;116(6):2152–7.
Daccord N, Celton JM, Linsmith G, Becker C, Choisne N, Schijlen E, et al. High-quality de novo assembly of the apple genome and methylome dynamics of early fruit development. Nat Genet. 2017;49(7):1099–106.
Wu D, Ding X, Wang S, Wojcik J, Zhang Y, Tokarska M, et al. Pervasive introgression facilitated domestication and adaptation in the Bos species complex. Nat Ecol Evol. 2018;2(7):1139–45.
Liu D. Molecular biological methods for sex determination of Garrulax courtoisi: Jiangxi Agricultural University; 2017.
Zhu F, Zhou C, Yang Z, Li X. Observation on the reproductive behavior of the White-browed Laughingthrush in Nanchong, Sichuan. Chin J Zool. 2010;4:6.
Frankham R. Conservation genetics. Annu Rev Genet. 1995;29:305–27.
Robinson JA, Brown C, Kim BY, Lohmueller KE, Wayne RK. Purging of strongly deleterious mutations explains long-term persistence and absence of inbreeding depression in island foxes. Curr Biol. 2018;28(21):3487–94.
Kohn MH, Murphy WJ, Ostrander EA, Wayne RK. Genomics and conservation genetics. Trends Ecol Evol. 2006;21(11):629–37.
Jarvis ED, Mirarab S, Aberer AJ, Li B, Houde P, Li C, et al. Whole-genome analyses resolve early branches in the tree of life of modern birds. Science. 2014;346(6215):1320–31.
Prum RO, Berv JS, Dornburg A, Field DJ, Townsend JP, Lemmon EM, et al. A comprehensive phylogeny of birds (Aves) using targeted next-generation DNA sequencing. Nature. 2015;526(7574):569–73.
Zhu K. A preliminary study on Chinese climate change during the last five thousand years. China Sci. 1973;2:15–38.
Zheng X. Chinese ancient economic center migrate southward and economic research during Tang and Song dynasties: Yuelu Press; 2003.
Bortoluzzi C, Megens HJ, Bosse M, Derks MFL, Dibbits B, Laport K, et al. Parallel Genetic Origin of Foot Feathering in Birds. Mol Biol Evol. 2020;37(9):2465–76.
Lin C. Ripe fruit attracts birds. Chinese Song Dynasty.
Zhao J. Sketch of rare birds. Chinese Song Dynasty.
Fan Z, Song Y. The biology characteristic and trade of thrush. Chinese Journal of Wildlife. 2000;6:3.
Chen S. Looking at the development of bird-raising culture and Sino-foreign exchanges during Song Dynasty from the perspective of bird food jar fonud in "Nanhai No. 1". China Ports. 2021.
Yao W. Grand Chinese Song Dynasty moved southward: Henan Literature and Art Publishing House; 2010.
Liu P, Li N, Zhang J, Qin X, Lou Y, Sun Y. Research status on the ecology of laughingthrushes in China. Chin J Zool. 2018;53(2):10.
Wang S, **e Y. China species red list. Bei**g: Science Press; 2009. p. 2.
Fan L, Zheng H, Milne RI, Zhang L, Mao K. Strong population bottleneck and repeated demographic expansions of Populus adenopoda (Salicaceae) in subtropical China. Ann Bot. 2018;121(4):665–79.
Hu Y, Thapa A, Fan H, Ma T, Wu Q, Ma S, et al. Genomic evidence for two phylogenetic species and long-term population bottlenecks in red pandas. Sci Adv. 2020;6(9):eaax5751.
Slatkin M. Linkage disequilibrium--understanding the evolutionary past and map** the medical future. Nat Rev Genet. 2008;9(6):477–85.
Dussex N, Van Der Valk T, Morales HE, Wheat CW, Díez-del-Molino D, Von Seth J, et al. Population genomics of the critically endangered kākāpō. Cell Genomics. 2021;1(1):100002.
Zheng G. A checklist on the classification and distribution of the birds of China (Third Edition): Science Press; 2017.
Rouhiainen A, Kulesskaya N, Mennesson M, Misiewicz Z, Sipila T, Sokolowska E, et al. The bradykinin system in stress and anxiety in humans and mice. Sci Rep. 2019;9(1):19437.
Gu AQ, Li DD, Wei DP, Liu YQ, Ji WH, Yang Y, et al. Cytochrome P450 26A1 modulates uterine dendritic cells in mice early pregnancy. J Cell Mol Med. 2019;23(8):5403–14.
Modi A, Vai S, Caramelli D, Lari M. The Illumina Sequencing Protocol and the NovaSeq 6000 System. Methods Mol Biol. 2021;2242:15–42.
Compeau PE, Pevzner PA, Tesler G. How to apply de Bruijn graphs to genome assembly. Nat Biotechnol. 2011;29(11):987–91.
Ruan J, Li H. Fast and accurate long-read assembly with wtdbg2. Nat Methods. 2020;17(2):155–8.
Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One. 2014;9(11):e112963.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–60.
Li H. A statistical framework for SNP calling, mutation discovery, association map** and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27(21):2987–93.
Parra G, Bradnam K, Korf I. CEGMA. a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics. 2007;23(9):1061–7.
Simao FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31(19):3210–2.
Altschul SF, Boguski MS, Gish W, Wootton JC. Issues in searching molecular sequence databases. Nat Genet. 1994;6(2):119–29.
Birney E, Clamp M, Durbin R. GeneWise and Genomewise. Genome Res. 2004;14(5):988–95.
Stanke M, Steinkamp R, Waack S, Morgenstern B. AUGUSTUS: a web server for gene finding in eukaryotes. Nucleic Acids Res. 2004;32:W309–12.
Meng X, Ji Y. Modern computational techniques for the HMMER sequence analysis. ISRN Bioinform. 2013;2013:252183.
Tarailo-Graovac M, Chen N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics. 2009;4:4–10.
Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27(2):573–80.
Majoros WH, Pertea M, Salzberg SL. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics. 2004;20(16):2878–9.
Leskovec J, Sosic R. SNAP: a general purpose network analysis and graph mining library. ACM T Intel Syst Tec. 2016;8(1).
Kent WJ. BLAT--the BLAST-like alignment tool. Genome Res. 2002;12(4):656–64.
Haas BJ, Delcher AL, Mount SM, Wortman JR, Smith RK Jr, Hannick LI, et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 2003;31(19):5654–66.
Chen C, Chen H, Zhang Y, Thomas HR, Frank MH, He Y, et al. TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol Plant. 2020;13(8):1194–202.
Emms D, Kelly S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 2019;20(1):238.
Nguyen L, Schmidt H, von Haeseler A, Minh B. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015;32(1):268–74.
Edgar R. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;5:113.
Criscuolo A, Gribaldo S. BMGE (Block Map** and Gathering with Entropy): a new software for selection of phylogenetic informative regions from multiple sequence alignments. BMC Evol Biol. 2010;10:210.
Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24(8):1586–91.
Hahn M, De Bie T, Stajich J, Nguyen C, Cristianini N. Estimating the tempo and mode of gene family evolution from comparative genomic data. Genome Res. 2005;15(8):1153–60.
Rimmer A, Phan H, Mathieson I, Iqbal Z, Twigg S, Consortium W, et al. Integrating map**-, assembly- and haplotype-based approaches for calling variants in clinical sequencing applications. Nat Genet. 2014;46(8):912–8.
Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34(18):3094–100.
Hoang D, Chernomor O, von Haeseler A, Minh B, Vinh L. UFBoot2: improving the ultrafast bootstrap approximation. Mol Biol Evol. 2018;35(2):518–22.
Kalyaanamoorthy S, Minh B, Wong T, von Haeseler A, Jermiin L. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 2017;14(6):587–9.
Chang C, Chow C, Tellier L, Vattikuti S, Purcell S, Lee J. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4:7.
Huson D, Bryant D. Application of phylogenetic networks in evolutionary studies. Mol Biol Evol. 2006;23(2):254–67.
Bouckaert R, Heled J, Kuhnert D, Vaughan T, Wu CH, **e D, et al. BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS Comput Biol. 2014;10(4):e1003537.
Rambaut A, Drummond AJ, **e D, Baele G, Suchard MA. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst Biol. 2018;67(5):901–4.
Price A, Patterson N, Plenge R, Weinblatt M, Shadick N, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38(8):904–9.
Pickrell J, Pritchard J. Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet. 2012;8(11):e1002967.
Li H, Durbin R. Inference of human population history from individual whole-genome sequences. Nature. 2011;475(7357):493–6.
Boitard S, Rodriguez W, Jay F, Mona S, Austerlitz F. Inferring population size history from large samples of genome-wide molecular data - an approximate Bayesian computation approach. PLoS Genet. 2016;12(3):36.
Terhorst J, Kamm JA, Song YS. Robust and scalable inference of population history from hundreds of unphased whole genomes. Nat Genet. 2017;49(2):303–9.
Patterson N, Moorjani P, Luo Y, Mallick S, Rohland N, Zhan Y, et al. Ancient admixture in human history. Genetics. 2012;192(3):1065–93.
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo M, et al. The variant call format and VCFtools. Bioinformatics. 2011;27(15):2156–8.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164.
Pertea G, Pertea M. GFF Utilities: GffRead and GffCompare. F1000Research. 2020;9.
Rubin C, Zody M, Eriksson J, Meadows J, Sherwood E, Webster M, et al. Whole-genome resequencing reveals loci under selection during chicken domestication. Nature. 2010;464(7288):587–91.
Gary B. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;2:573–80.
Mousavi N, Shleizer-Burko S, Yanicky R, Gymrek M. Profiling the genome-wide landscape of tandem repeat expansions. Nucleic Acids Res. 2019;47(15):e90.
Chen H, Huang M, Liu D, Tang H, Zheng S, Ouyang J et al. Genomic signatures and evolutionary history of critically endangered Garrulax courtoisi and other Garrulax species. https://ngdc.cncb.ac.cn/gwh/. 2022.
Chen H, Huang M, Liu D, Tang H, Zheng S, Ouyang J et al. Genomic signatures and evolutionary history of critically endangered Garrulax courtoisi and other Garrulax species. https://ngdc.cncb.ac.cn/gsa/. 2022.
Chen H, Huang M, Liu D, Tang H, Zheng S, Ouyang J, et al. Genomic signatures and evolutionary history of critically endangered Garrulax courtoisi and other Garrulax species. 2022. https://doi.org/10.5281/zenodo.6570622.
Acknowledgements
We thank Nanchang Zoo for assistance in providing samples.
Funding
This study was supported by National Natural Science Foundation of China (31760625) and Science and Technology Research Project of Jiangxi Provincial Education Department (GJJ190581).
Author information
Authors and Affiliations
Contributions
X.Y., J.H., J.R., and D.L. conceptualized and organized the research. D.L. conducted an investigation and sample collection. X.Y., J.R., and J.H. designed the study. J.H. provided funding support. H.C. and M.H. performed the bioinformatics, population genetics, and evolutionary history analyses. H.Z., Y.O., H.T., S.Z., L.W., K.L., Y.G., Y.W.(1), Y.W.(2), Y.X., T.L., Y.H., and R.X. performed experiments and assisted in the analysis. H.C., M.H., and D.L. wrote the draft manuscript. X.Y. and H.C. revised the paper. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Fig. S1.
The genome annotation pipeline of the GCO and GSA. Fig. S2. Distribution of the divergence rate of each type of TE in the GCO and GSA genome. Fig. S3. Venn diagram of gene function annotation results of different protein database. Fig. S4. Syntenic blocks shared between the GCO and GSA scaffolds. Grey lines connect matched gene pairs. Fig. S5. Enrichment analysis of positive selection genes. Fig. S6. The ML tree was constructed based on 66 individuals. Fig. S7. The heatmap of identity-by-state distance from 58 individuals. Fig. S8. The ML tree of the 58 individuals. Fig. S9. ADMIXTURE analysis with ancestral lineages K from 2 to 9. Fig. S10. CV-error of ADMIXTURE analysis. Fig. S11. The phylogenetic relationships among Garrulax species constructed by SNAPP software. Fig. S12. Treemix phylogeny of Garrulax species with TMI as root. Fig. S13. The population inference estimated by smc++. Fig. S14. Population inference in GCO. Fig. S15. Nucleotide polymorphism (π) of each individual from GCO and GSA. Fig. S16. Nucleotide polymorphism (π) of ten GCO individuals. The grey shadow means homozygosis gap region. Fig. S17. Nucleotide polymorphism (π) of 11 GSA individuals. Fig. S18. Distribution of inbreeding coefficient (F) in eight Garrulax species. Fig. S19. Distribution of π value for the eight Garrulax species. Fig. S20. The scope of π value for eight Garrulax species. Fig. S21. The scope of heterozygosity rate value for eight Garrulax species. Fig. S22. LD pattern (left) and ROH number (right) for each Garrulax species based on GSA genome. Fig. S23. The scope of FROH for eight Garrulax species. Fig. S24. Comparison missense and synonymous mutations between GCO and GSA. Fig. S25. Distributions of heterozygosity Hp and zHp. Table S1. Summary of the sequencing data for the de novo genomes. Table S2. Assembly statistics of the GCO and GSA genomes. Table S3. Base contents of the assembled genomes. Table S4. Read coverage of genome assemblies. Table S5. The completeness of the two assembled genomes was assessed by CEGMA and BUSCO. Table S6. Prediction of repeat elements in the two assembled genomes. Table S7. Categories of repeat elements. Table S8. Prediction of gene structure in two genomes. Table S9. Gene structure of genomes of Garrulax species and other avians. Table S10. Functional annotation of the predicted protein-coding genes in the Garrulax genome assemblies. Table S11. Statistics of noncoding RNAs of the GCO and GSA genomes. Table S12. Positive selection genes (PSGs) identified in GCO and GSA. Table S13. The information of 25 species download from NCBI database. Table S14. Expanded and contracted gene families identified by CAFE. Table S15. Sequencing data quality of 66 individuals. Table S16. Detailed sampling information of 66 sequenced individuals. Table S17. Pair individuals with closely relationship of 66 specimens. Table S18. Evidence of gene flow between Garrulax species. Table S19. Genetic diversity of eight Garrulax species based on a reference genome of white-browed laughingthrush. Table S20. Deleterious variants of blue-crowned laughingthrush and white-browed laughingthrush. Table S22. The overlap** windows identified by top 1% FST and zHp value. Table S25. The 218 STRs makersof blue-crowned laughingthrush. Table S26. The 43 STRs were identified on W-related scaffolds to be used as sex confirmation. Table S27. The Z and W chromosomes of 31 avian.
Additional file 2: Table S21.
The heterozygosity rate of GCO, GSA, and the other 186 species. Table S23. Scaffolds belong to chromosome Z and W and the depth information of GCO. Table S24. Scaffolds belong to chromosome Z and W and the depth information of GSA.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Chen, H., Huang, M., Liu, D. et al. Genomic signatures and evolutionary history of the endangered blue-crowned laughingthrush and other Garrulax species. BMC Biol 20, 188 (2022). https://doi.org/10.1186/s12915-022-01390-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12915-022-01390-4