
Abstract
Background
The therapeutic armamentarium in multiple myeloma has been significantly broadened by proteasome inhibitors, highly efficient means in controlling of multiple myeloma. Despite the developments of therapeutic regimen in treatment of multiple myeloma, still the complete remission requires a novel therapeutic strategy with significant difference in outcomes. Proteasome inhibitors induce autophagy and ER stress, both pivotal pathways for protein homeostasis. Recent studies showed that the IRE1α-XBP1 axis of the unfolded protein response (UPR) is up-regulated in multiple myeloma patients. In addition, XBP1 is crucial for the maintenance of viability of acute lymphoblastic leukemia (ALL).
Results
We analyzed the efficacy of targeting IRE1α-XBP1 axis and autophagy in combination with proteasome inhibitor, ixazomib in treatment of multiple myeloma. In this present study, we first show that targeting the IRE1α-XBP1 axis with small molecule inhibitors (STF-083010, A106) together with the ixazomib induces cell cycle arrest with an additive cytotoxic effect in multiple myeloma. Further, we examined the efficacy of autophagy inhibitors (bafilomycin A, BAF and chloroquine, CQ) together with ixazomib in multiple myeloma and observed that this combination treatment synergistically reduced cell viability in multiple myeloma cell lines (viable cells Ixa: 51.8 ± 3.3, Ixa + BAF: 18.3 ± 7.2, Ixa + CQ: 38.4 ± 3.7) and patient-derived multiple myeloma cells (Ixa: 59.6 ± 4.4, Ixa + CQ: 7.0 ± 2.1). We observed, however, that this combined strategy leads to activation of stress-induced c-Jun N-terminal kinase (JNK). Cytotoxicity mediated by combined proteasome and autophagy inhibition was reversed by addition of the specific JNK inhibitor JNK-In-8 (viable cells: Ixa + BAF: 11.6 ± 7.0, Ixa + BAF + JNK-In-8: 30.9 ± 6.1).
Conclusion
In this study we showed that combined inhibition of autophagy and the proteasome synergistically induces cell death in multiple myeloma. Hence, we consider the implication of pharmaceutical inhibition of autophagy together with proteasome inhibition and UPR-directed therapy as promising novel in vitro treatment strategy against multiple myeloma.
Similar content being viewed by others

Introduction
Multiple myeloma (MM) is characterized by monoclonal proliferation of plasma cells mostly within the bones often leading to local destruction. Plasma cells play a crucial role in the mammalian immune response which diminishes in its specificity due to the monoclonality of MM cells, thereby leading to an acquired immunodeficiency [1, 2]. MM accounts for about 10% of all hematologic cancers, with a particularly high incidence in adults above 50 years of age [3]. MM cells depend on proteostasis for their survival because of their increased production and accumulation of non-functional immunoglobulin (Ig) [4, 5]. Proteostasis is a complex network with three major branches: the ubiquitin–proteasome system (UPS), the unfolded protein response (UPR) and autophagy. The UPS is the essential network responsible for degradation of unwanted proteins and is targeted by proteasome inhibitors, which are efficient and widely used for the treatment of MM [5,6,7,8,9].
The UPR mediates its function through three subordinated pathways, namely inositol-requiring enzyme 1 alpha (IRE1α), protein kinase RNA-like ER kinase (PERK) and activating transcription factor 6 (ATF6) [10]. The stress sensors IRE1α, PERK and ATF6 are dissociated from the endoplasmic reticulum (ER) chaperone heat shock 70 kDa protein 5/78 kDa glucose-regulated protein (HSPA5/GRP78) thereby activating the UPR pathway. Activated PERK causes phosphorylation of the eukaryotic translation initiation factor 2α (eIF2α) at serine 51. ATF6 in its active form invokes a variety of downstream genes involved in ER-associated degradation (ERAD). The most conserved UPR pathway is mediated by IRE1α and contains an endoribonuclease (RNAse) and a kinase domain. The RNAse domain reduces the ER stress load by splicing XBP1 mRNA through removal of a 26-nucleotide intron. IRE1α also governs microRNA biogenesis and degradation of specific mRNA during UPR activation by regulated IRE1-dependent decay (RIDD) [10,11,12,13]. Our recently published findings demonstrate that high risk subsets of acute lymphoblastic leukemia are vulnerable to IRE1α based therapy and that genetic and pharmacological inhibition of IRE1α negatively affects the survival of ALL cells [14, 15]. Other studies revealed the importance of the UPR in maintaining malignancy [16], and IRE1α–directed therapy has been successfully applied in triple-negative breast cancer and multiple myeloma [17, 18]. Treatment with the proteasome inhibitors bortezomib and ixazomib influences the originating microenvironment of MM cells (i.e. the bone marrow niche) through an up-regulation of the UPR providing a rationale for an additional UPR targeting in multiple myeloma [19, 20].
Autophagy is strongly activated as a response to proteasome inhibition and represents a strategy within the cell to circumvent drug-induced interruption of proteostasis. As an adaptive pathway it thereby supports the survival of malignant cells [21,22,23] and can be targeted by specific inhibitors. In our study, we treated MM cell lines KMS11 and RPMI-8226 with ixazomib in combination with UPR inhibitors leading to a substantial decrease of cell survival and proliferation. Addition of autophagy inhibitors as a third substance generated a significant increase of the strong cytotoxic effect. Attacking multiple myeloma cells by inhibition of the proteasome, the UPR and autophagy together is consecutively delineated as a promising in vitro treatment strategy in our study.
Material and methods
Human cell culture
Human cell lines KMS11 and RPMI-8226 were originally obtained from JCRB Cell Bank (Japanese Collection of Research Bank) and DSMZ (German Collection of Microorganisms and Cell Cultures), Braunschweig, Germany, respectively. The human mesenchymal stem cell line immortalized by expression of the telomerase reverse transcriptase gene (hMSC-TERT) was kindly provided by Dr. Rebecca Schneider-Kramann (RWTH Aachen). All cell lines were authenticated via STR profiling by Multiplexion (Heidelberg, Germany) [24]. All cell lines were cultured as described previously [15]. The inhibitory drugs comprising STF-083010, A106, bafilomycin A1 and chloroquine were purchased from Sigma Aldrich® and ixazomib (MLN97098) was kindly provided by Takeda® (Cambridge, MA, USA).
Colony formation assay
Primary bone marrow (BM) samples from patients with MM were provided by RWTH centralized BioMaterial Bank (cBMB). Mononuclear cells (MNCs) from these samples were isolated by gradient centrifugation with Ficoll paque (density 1.077 g/mL). The MNCs were cultured in Iscove´s Modified Dulbecco´s Medium (IMDM) with GlutaMAX® containing 10% fetal bovine serum (FBS, Gibco®), 100 IU/mL penicillin and 100 μg/mL streptomycin (Gibco®) were supplemented with 2 mM l-glutamine, 10−4 M 2-mercaptoethanol, 10 ng/mL (rh)IL-6, 10 ng/ml rhSCF, 10 ng/mL rhIL-3, 20 ng/mL G-CSF and 10 ng/mL FLT3-ligand (Immunotools®) in an incubator with a humidified atmosphere of 5% CO2 at 37 °C. For the human CFU assays, 80% methylcellulose without cytokines (Methocult, H4230, Stem Cell Technologies®), 20% IMDM, 10−4 M 2-mercaptoethanol, 2 mM l-glutamine were supplemented with 50 ng/ml rhSCF, 10 ng/ml rhIL-3, 10 ng/ml rhGM-CSF, 3 U/ml rhEPO (Immunotools®) and 1% penicillin/streptomycin. 5 \(\times\) 104 of pretreated BM MNCs/mL were incubated at 37 °C for 48 h with 5% CO2 for 14 days and colonies were counted using inverted light microscopy.
Flow cytometry
Cell viability was measured using propidium iodide (PI) staining (1 μg/mL of PI, Sigma Aldrich®). Apoptosis was measured using flow cytometric quantification of Annexin-V/PI fraction as described in Supplementary Material. Cell cycle was assayed by flow cytometric quantification of DNA content using PI staining. To synchronize the cell cycle progression at a specific phase of the cell cycle, cells were deprived of FBS in culture medium for 16 h and followed by culture conditions as described above with FBS to re-initiate their cell cycle. Cells were resuspended and permeabilized as described in Supplementary Material.
SDS-PAGE and western blot analysis
Cells were extracted, loaded and transferred as described in Supplementary Material. As primary antibodies, beta actin, P21 (Abcam®), PARP, p27, phospho-SAPK/JNK, BIM, Caspase-3, cleaved -caspase-3, Beclin-1, LC3A/B (all manufactured by Cell Signaling Technology®) were applied. The proteins were developed with PCA-ECL solution (100 mM Tris–HCL, pH 8.8, 2.5 mM luminol, 0.198 mM p-coumaric acid and 0.2% v/v hydrogen peroxide (Sigma Aldrich®). The protein bands were detected by enhanced chemiluminescence (ECL) detection system (Vilber®).
Quantitative real-time PCR
RNA isolation and cDNA generation was performed as described previously [15]. Quantitative real-time PCR was performed using the iTaq Universal SYBR Green supermix (Bio-Rad®). Gene expression assays (Applied Biosystems®; ABI7500 FAST real-time PCR) were performed according to manufacturer’s instructions. Endogenous expression of COX6B was used for normalization and relative quantification of target gene expression was calculated by the comparative threshold cycle method. Triplicates were measured for each tested condition. Quantitative data are expressed as mean ± SD. Differences between values obtained from each condition were considered statistically significant for values of p < 0.05. Primer sequences are listed in Supplementary Table 1.
Statistical analysis
Statistical analyses were performed using two-tailed student’s t test and ANOVA by GraphPad Prism (GraphPad Software, Inc.). The Bliss formula was used to analyze the efficacy of drug combination in comparison with monotherapy, using equation: Yab,P = (Ya + Yb)- (Ya × Yb), which defines the concepts of synergy, antagonism and additive effects between two inhibitory drugs [25].
Results
Inhibition of IRE1α sensitizes multiple myeloma cells to ixazomib under support of mesenchymal stromal cells (MSCs)
We investigated the capacity of bone marrow MSCs to modulate viability and proliferation of MM cells upon treatment with ixazomib and its combination with selective inhibitors of the IRE1α RNAse domain (STF-083010, A106). We employed hTERT-MSCs with a strong supportive capacity for hematopoesis as a representative model for human bone marrow derived MSCs. MSCs alone did not alter viability of MM cells but treatment with ixazomib supported survival and tumor cell growth compared to culture without MSC support. Interestingly, the combination of ixazomib with STF-083010 and A106, respectively, significantly reduced the viability of multiple myeloma cells even when protected by bone marrow MSCs. This implicates that inhibition of IRE1α in combination with ixazomib abrogates the supportive effect of the bone marrow microenvironment for the survival of MM cells (Fig. 1A, S1A). We furthermore investigated the efficacy of our drug combination in hypoxic conditions to mimic the hypoxic bone marrow niche and measured viability of MM cells with single and dual treatment under normoxic (20% O2) versus hypoxic (1% O2) conditions. The IRE1α inhibitor STF-083010 did not reduce cell viability in combination with ixazomib under hypoxic conditions (Figure S1B). We then screened the expression signature of major UPR genes, namely XBP1s, EIF2AK3 and ATF6, upon treatment with ixazomib as a single agent and in combination with STF-083010. Quantitative RT-PCR analysis showed that ixazomib treated cells only slightly increased mRNA expression of EIF2AK3 and ATF6 while the XBP1s mRNA level was strongly up-regulated upon treatment with ixazomib (Fig. 1B, C) suggesting that MM cells activate the IRE1α-XBP1 axis as an adaptive pathway shielding against ixazomib-induced cell death.

IRE1α inhibitors in combination with ixazomib have synergistic effects on cytotoxicity of multiple myeloma cells under support of mesenchymal stromal cell line (hMSC-TERT). A KMS11 and RPMI-8226 cells were treated with 10 nM ixazomib alone or combined with 30 μM STF-083010 for five days with or without support of immortalized stromal cells (hMSC-TERT), (n = 3). Cell viability were measured by PI staining. B Total RNA was extracted after 16 h of treatment as described above. B, C XBP1s, ATF6 and EIF2AK3 mRNA levels were assessed using RT-qPCR, (n = 3). P values were calculated by two-way analysis of variance (ANOVA) for A, B and one-way analysis for C. A P value of less than 0.05 was considered statistically significant for all analyses. (*p < 0.05, **p < 0.01, ***p < 0.001 versus control)
IRE1α inhibitors in combination with ixazomib arrest the cell cycle at G1 and induce expression of apoptotic effectors
The overexpression of the cell cycle regulator cyclin D1 is an important feature of MM cells [4, 26]. We showed that STF-083010 in combination with ixazomib causes accumulation of cells in the G0/1 phase of the cell cycle (Fig. 2A). Next, we examined cell cycle regulation of MM cells on the protein level. Combination treatment arrests cell cycle progression at G1 and significantly increases expression levels of G1 phase regulators, p21CIP1 and p27KIP1(Fig. 2B, S2A), indicating a reduction of proliferation after addition of IRE1α inhibitors to treatment with ixazomib. We then measured expression levels of the caspase substrate poly (ADP-ribose) polymerase (PARP) and its cleaved form as an indicator of caspase activation. Our combination therapy increased proteolytic cleavage of PARP in RPMI-8226 cells in comparison to single treatment (Fig. 3B, S2B). In addition, pro-apoptotic markers PUMA and NOXA as ER stress responsive genes [27, 28] were assessed by quantitative RT-PCR. Combination treatment resulted in significantly increased expression of PUMA and NOXA, suggesting that both are substantially activated in response to our combination regimen to induce cell death in MM cells (Fig. 3A).

Ixazomib in combination with STF-083010 arrests cell cycle at G1 phase. Both KMS11 and RPMI-8226 cells were incubated for 16 h in absence of FBS, followed by the treatment strategy described in Fig. 1 for 16 h), (n = 3). A Cells were harvested and DNA content was assessed by PI staining. P values were calculated by one-way analysis of variance (ANOVA). A P value of less than 0.05 was considered statistically significant for all analyses (*p < 0.05, **p < 0.01, ***p < 0.001). B Using western blot, protein levels of cell cycle negative regulators p21CIP1, p27.KIP1 and β-actin as loading control were measured in KMS11 and RPMI-8226 after 16 h of treatment

STF-083010 in combination with ixazomib initiates apoptosis in multiple myeloma cell lines. KMS11 and RPMI-8226 cells were treated as described above for 16 h, (n = 3). A Using RT-qPCR, PUMA and NOXA mRNA expression levels were measured. P values were calculated by one-way analysis of variance (ANOVA). A P value of less than 0.05 was considered statistically significant for all of the analysis (*p < 0.05, **p < 0.01, ***p < 0.001). B Protein levels of cleaved-PARP, PARP and β-actin as loading control were detected in KMS11 and RPMI-8226 using western blot
Ixazomib in combination with autophagy inhibitors synergizes cytotoxicity in multiple myeloma cells
Induction of autophagy in response to chemotherapeutic agents is known to protect cancer cells against cell death [29,30,31]. To determine the antiproliferative effect of ixazomib in combination with the autophagy inhibitors bafilomycin A1 and chloroquine in MM cell lines, we measured cell viability of KMS11 cells upon combination treatment. Autophagy inhibitors combined with ixazomib synergistically reduced viability of KMS11 cells compared to single treatment (Fig. 4A). Using the Chou-Talalay and Bliss formula, interaction efficacy of the two drugs as percentage of cell death mediated by combination treatment was verified (Fig. 4B). We also analyzed the in vitro colony formation of MNCs from bone marrow of patients with multiple myeloma (n = 7) after combination treatment. As shown in Fig. 4C, clonogenic growth of MNCs from MM patients was significantly decreased after combination treatment in comparison with monotherapy. However, the number of colonies in two patients after dual therapy showed no significant reduction when compared to single treatment.

Inhibition of autophagy in combination with ixazomib induces cell toxicity in multiple myeloma cells. KMS11 were treated with 10 nM ixazomib and in combination with autophagy inhibitors bafilomycin A1 (BAF, 5 nM) (n = 3) or chloroquine (CQ, 40 μM), (n = 2). A Cell viability was measured using PI staining after 72 h with BAF and CQ combined treatments respectively. B The table represents bliss formula calculation for drug combination analysis. The percentage of inhibitory effect of combined drugs was calculated by bliss formula in comparison with observed inhibitory effect. Based on this formula, we showed that ixazomib in combination with autophagy inhibitors (BAF or CQ) act synergistic. In addition, combination of Index was calculated based on Chou-Talalay. C Primary BM MNCs were isolated from multiple myeloma patients at diagnosis (n = 7, one representative is shown) and treated with ixazomib 10 nM, chloroquine 40 μM and ixazomib plus chloroquine. After 48 h, 5 × 10.4 of all pretreated cells were seeded in CFU culture medium. Colony numbers were counted after 14 days of culture using inverted light microscopy. P values were calculated using one-way ANOVA. A P value of less than 0.05 was considered statistically significant for all of the analysis (*p < 0.05, **p < 0.01, ***p < 0.001)
Inhibition of autophagy in combination with ixazomib inhibits cell growth and induces apoptosis
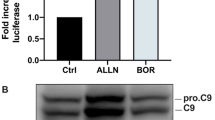
To determine if bafilomycin A1 (BAF) or chloroquine (CQ) inhibit autophagy, we measured the protein levels of LC3II as marker of autophagy flux. Expression of LC3II was notably increased upon treatment with BAF and CQ (Figure S3), indicating accumulation of LC3II at the autophagosome membrane. Next, we examined whether autophagy inhibitors in combination with ixazomib induce apoptosis in MM cells and determined the apoptotic cell fraction using Annexin-V/PI staining and observed a notable induction of apoptosis compared to single treatment (Fig. 5A). Moreover, we assessed the expression levels of caspase-3 and PARP and their cleaved forms at the protein level. Both autophagy inhibitors in combination with ixazomib increased proteolytic cleavage of caspase 3 and PARP, a signature of activated apoptosis (Fig. 5B). To provide further evidence for induction of apoptosis in MM cells upon combined treatment, we examined its effect on regulation of cell cycle progression. We noticed that expression of G1 phase negative regulators p21CIP1 and p27KIP1 increased after dual treatment, indicating an efficient blockade of cell growth and proliferation in MM (Fig. 5C).

Autophagy inhibitors in combination with ixazomib induce apoptosis and arrest cell cycle at G1 phase in multiple myeloma cells. KMS11 cells were treated with ixazomib and autophagy inhibitors as described above. A Apoptosis was measured by analysis of apoptosis fraction using annexin-V/PI staining. (BAF, 5 nM) (n = 3) or chloroquine (CQ, 40 μM), (n = 2). P values were calculated by one-way analysis of variance (ANOVA). A P value of less than 0.05 was considered statistically significant (*p < 0.05, **p < 0.01, ***p < 0.001). B Using western blot protein levels of caspase3, cleaved-caspase3, PARP, cleaved PARP, p21CIP1 and p27.KIP1 were measured. β-actin was usedas loading control, (n = 3)
JNK is activated by the combination of ixazomib and autophagy inhibition and JNK inhibition abrogates the cytotoxic effects of combined treatment
Having shown that ixazomib in combination with autophagy inhibitors induces apoptosis in MM cells, we next speculated that this effect is mediated by the cell stress response as a consequence of dual treatment. JNK kinases have been well characterized as stress activated protein kinases, which can either be induced by extracellular stimuli, protein synthesis inhibitors, cytotoxic drugs or a combination of these mechanisms. Both kinases activate caspase cascades to initiate apoptosis [32,33,34]. We examined JNK expression in our set of treatments and observed that both autophagy inhibitors BAF and CQ in combination with ixazomib increase phosphorylation levels of JNK in KMS11 cells (Fig. 6A). To analyze the contribution of JNK to apoptosis triggered by autophagy inhibitors and ixazomib, we employed the JNK inhibitor JNK-IN-8 in combination with ixazomib and BAF. Addition of JNK-IN-8 enhanced cell viability compared to ixazomib in combination with only BAF (Fig. 6B), thereby confirming that JNK is an important mediator in cell apoptosis induced by combination treatment in MM cells.

JNK inhibition rescues multiple myeloma cells from cell death mediated by combination treatment. KMS11 were treated with 10 nM ixazomib and in combination with autophagy inhibitors as described above, (n = 3). A Phosphorylation levels of JNK-T183/Y.185 and protein levels of JNK and BIM were measured using western blot with β-actin as a loading control. B KMS11 cells were treated with 10 nM ixazomib, 5 nM bafilomycin A1, 3 μM JNK inhibitor (JNK-IN-8) and combination treatment (ixa + baf and ixa + baf + JNK). Cell viability of KMS11 cells were measured using PI staining. P value were calculated by two-way analysis of variance (ANOVA). A p value of less than 0.05 was considered statistically significant (*p < 0.05, **p < 0.01, ***p < 0.001 versus control)
Discussion
Maintenance of proteostasis plays a pivotal role for the survival and proliferation of MM cells. MM cells are heavily dependent on adaptive pathways to protect them against cell death mediated by stress which is caused by their high metabolic demand for immunoglobulin secretion. Drug discovery has led to the development of various promising candidate drugs for the treatment of MM. Proteasome inhibitors (PIs) with bortezomib, carfilzomib and ixazomib as main representatives of this drug class have tremendously improved the treatment of MM. However, development of resistance and the overall goal of achieving a more profound, ideally complete remission in MM treatment are still considered major challenges. Therefore, develo** alternative therapeutic strategies is crucial for MM patients and provided the rationale for the present study.
Recently published data demonstrate the significance of the IRE1α-XBP1 axis of the UPR network in solid tumors and hematologic malignancies [10, 14, 35, 36]. Here, we tested the inhibition of the IRE1α-XBP1 pathway of the UPR in combination with ixazomib as a novel therapeutic approach toward MM. Our data show that IRE1α inhibitors significantly contributed to the cytotoxic effects of ixazomib even when MM cells are protected against the therapeutically efficient drugs by the MSC-mimicked bone marrow microenvironment. Our in vitro results reveal an induction of XBP1 expression upon ixazomib treatment in MM cells, highlighting the significance of XBP1 as a pro-survival mechanism against the induction of cell death by ixazomib. Apoptosis can be mediated by ER stress through the up-regulation of ER stress responsive genes [27, 28]. We demonstrate that mRNA expression of two pro-apoptotic markers (PUMA and NOXA) and proteolytic cleavage of PARP strongly increased in combination treatment. Cell cycle analysis revealed that inhibition of XBP1 activation in combination with ixazomib arrests the cell cycle at the G1 phase in MM cells. In addition, our finding clearly delineated that blocking XBP1 splicing in combination with ixazomib causes an additive effect on cytotoxicity in MM. Taken together, we demonstrated that ixazomib induces the expression of UPR genes to reduce ER stress. Nevertheless, this response was not limited to the UPR network and other adaptive pathways are activated to compensate for the loss of protein homeostasis upon treatment with ixazomib in MM cells. Autophagy, the other major protein degradation system, also importantly contributed to maintenance of protein homeostasis in our system and we accordingly hypothesized that autophagy might be burdened with an accumulation of proteins for degradation after ixazomib treatment as a compensatory mechanism. This provided a strong rationale for targeting autophagy in combination with ixazomib in MM. We assumed that autophagy is activated to protect MM cells from ixazomib treatment and that, in turn, blocking of autophagy along with ixazomib treatment could maximize cellular toxicity in this context. Recent studies have reported the cytotoxic effects of blockade of the autophagosome-lysosome fusion in cancer cells [37,38,39]. Here, we showed that both autophagy inhibitors (BAF, CQ) inhibit the late stage of autophagy and lead to increased levels of LC3II and disruption of autophagic flux in MM cells. Our in vitro results verify that the combination of ixazomib and autophagy inhibitors synergistically induces cell death in MM cells. We validated the efficacy of combined treatment in primary case samples from MM patients. In this regard, we revealed that combination treatment induces cell death via apoptosis, as measured by proteolytic cleavage of caspase 3 and its corresponding target PARP. We further demonstrated that our combination therapy is an efficient means to target cell cycle progression. Single agent treatment non-significantly induced cell death through induction of apoptosis, but we were able to show that combination treatment significantly induces cell toxicity in MM cells. The further analyses unveil that activation of JNK is an important molecular mechanism underlying the cytotoxic effects of our combination treatment for MM. However, JNK-mediated signaling has a contentious role across cancer types [40,41,42]. Studies have implicated that JNK acts in a tumor suppressive manner and is activated in response to cell stress inducers [43, 44]. In line with these findings, our observations hint toward an induction of apoptosis through the activation of stress-responsive protein kinase JNK in MM cells when autophagy inhibitors are combined with ixazomib. Addition of a selective inhibitor of JNK activation together with our dual treatment in turn increased viability of myeloma cells, implicating that activation of JNK in our treatment regimen is a pivotal mechanism for the targeting of MM cells with the combination of proteasome inhibitors and inhibitors of autophagy. A comprehensive investigation of alternative pathways contributing to JNK mediated apoptosis in MM for better insight into the specific role of the JNK signaling pathway and its crosstalk with other crucial molecules in response to targeted treatment of multiple myeloma cells remains to be done. Our present data delineate an innovative strategy against multiple myeloma and clear the way for preclinical and potential clinical investigation of the combination of proteostasis-directed agents in multiple myeloma.
Availability of data and materials
Extended data are available at Supplementary Material.
Abbreviations
- ATF6:
-
Activating transcription factor 6
- BAF:
-
Bafilomycin A1
- COX6B:
-
Cytochrome c oxidase subunit 6B1
- CQ:
-
Chloroquine
- DSMZ:
-
Deutsche Sammlung von Mikroorganismen und Zellkulturen
- EIF2AK3:
-
Eukaryotic translation initiation factor 2-alpha kinase 3
- IRE1α:
-
Inositol-requiring protein 1 subunit alpha
- JNK:
-
C-Jun N-terminal kinase 1
- LC3:
-
Microtubule-associated protein light chain 3
- MM:
-
Multiple meyloma
- NOXA:
-
Alternative name for PMAIP1
- PARP:
-
Poly (ADP-ribose) polymerase
- PERK:
-
PRKR-Like Endoplasmic Reticulum Kinase
- PIs:
-
Proteasome inhibitors
- PUMA:
-
P53 up-regulated modulator of apoptosis
- SAPK:
-
Stress-activated protein kinase
- Ub:
-
Ubiquitin
- UPR:
-
Unfolded protein response
- UPS:
-
Ubiquitin–proteasome system
- XBP1u:
-
X-box-binding protein 1 unspliced
- XBP1s:
-
X-box-binding protein 1 spliced
References
Pratt G, Goodyear O, Moss P. Immunodeficiency and immunotherapy in multiple myeloma. Br J Haematol. 2007;138(5):563–79.
Kyle R, Rajkumar SV. Criteria for diagnosis, staging, risk stratification and response assessment of multiple myeloma. Leukemia. 2009;23(1):3.
Paszekova H, Kryukov F, Kubiczkova L, Hajek R, Sevcikova S. High-risk multiple myeloma: different definitions, different outcomes? Clin Lymphoma Myeloma Leuk. 2014;14(1):24–30.
Agnelli L, Bicciato S, Mattioli M, Fabris S, Intini D, Verdelli D, et al. Molecular classification of multiple myeloma: a distinct transcriptional profile characterizes patients expressing CCND1 and negative for 14q32 translocations. J Clin Oncol. 2005;23(29):7296–306.
Merin N, Kelly KJP. Clinical use of proteasome inhibitors in the treatment of multiple myeloma. Pharmaceuticals. 2015;8(1):1–20.
Teicher BA. Tomaszewski JEJBp. Proteasome inhibitors Biochem Pharmacol. 2015;96(1):1–9.
Hipp MS, Kasturi P, Hartl FU. The proteostasis network and its decline in ageing. Mol Cell Biol. 2019;20:421–35.
Dong Z, Cui HJC. The autophagy-lysosomal pathways and their emerging roles in modulating proteostasis in tumors. Cells. 2019;8(1):4.
Klaips CL, Jayaraj GG, Hartl FU. Pathways of cellular proteostasis in aging and disease. J Cell Biol. 2018;217(1):51–63.
Masouleh BK, Chevet E, Panse J, Jost E, O’Dwyer M, Bruemmendorf TH, et al. Drugging the unfolded protein response in acute leukemias. J Hematol Oncol. 2015;8(1):87.
Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Mol Cell Biol. 2012;13(2):89.
Grootjans J, Kaser A, Kaufman RJ, Blumberg RS. The unfolded protein response in immunity and inflammation. Nat Rev Immunol. 2016;16(8):469.
Walter P, Ron DJS. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334(6059):1081–6.
Masouleh BK, Geng H, Hurtz C, Chan LN, Logan AC, Chang MS, et al. Mechanistic rationale for targeting the unfolded protein response in pre-B acute lymphoblastic leukemia. Proc Natl Acad Sci. 2014;111(21):E2219–28.
Vieri M, Preisinger C, Schemionek M, Salimi A, Patterson JB, Samali A, et al. Targeting of BCR-ABL1 and IRE1α induces synthetic lethality in Philadelphia-positive acute lymphoblastic leukemia. Carcinogenesis. 2021;42(2):272–84.
Hsu S-K, Chiu C-C, Dahms H-U, Chou C-K, Cheng C-M, Chang W-T, et al. Unfolded protein response (UPR) in survival, dormancy, immunosuppression, metastasis, and treatments of cancer cells. Int J Mol Sci. 2019;20(10):2518.
Harnoss JM, Le Thomas A, Reichelt M, Guttman O, Wu TD, Marsters SA, et al. IRE1α disruption in triple-negative breast cancer cooperates with antiangiogenic therapy by reversing ER stress adaptation and remodeling the tumor microenvironment. Cancer Res. 2020;80(11):2368–79.
Harnoss JM, Le Thomas A, Shemorry A, Marsters SA, Lawrence DA, Lu M, et al. Disruption of IRE1α through its kinase domain attenuates multiple myeloma. Proc Natl Acad Sci. 2019;116(33):16420–9.
Vincenz L, Jäger R, O’Dwyer M, Samali A. Endoplasmic reticulum stress and the unfolded protein response: targeting the Achilles heel of multiple myeloma. Mol Cancer Ther. 2013;12(6):831–43.
Obeng EA, Carlson LM, Gutman DM, Harrington WJ, Lee KP, Boise LH. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood. 2006;107(12):4907–16.
Sui X, Chen R, Wang Z, Huang Z, Kong N, Zhang M, et al. Autophagy and chemotherapy resistance: a promising therapeutic target for cancer treatment. Cell Death Dis. 2013;4(10): e838.
Zhu K, Dunner K Jr, McConkey DJJO. Proteasome inhibitors activate autophagy as a cytoprotective response in human prostate cancer cells. Oncogene. 2010;29(3):451.
Tang B, Cai J, Sun L, Li Y, Qu J, Snider BJ, et al. Proteasome inhibitors activate autophagy involving inhibition of PI3K-Akt-mTOR pathway as an anti-oxidation defense in human RPE cells. PLoS ONE. 2014;9(7): e103364.
Castro F, Dirks WG, Fähnrich S, Hotz-Wagenblatt A, Pawlita M, Schmitt M. High-throughput SNP-based authentication of human cell lines. Int J Cancer. 2013;132(2):308–14.
Zhao W, Sachsenmeier K, Zhang L, Sult E, Hollingsworth RE, Yang H. A new bliss independence model to analyze drug combination data. J Biomol Screen. 2014;19(5):817–21.
Avet-Loiseau H, Malard F, Campion L, Magrangeas F, Sebban C, Lioure B, et al. Translocation t (14; 16) and multiple myeloma: is it really an independent prognostic factor? Blood. 2011;117(6):2009–11.
Li J, Lee B, Lee AS. Endoplasmic reticulum stress-induced apoptosis multiple pathways and activation of p53-up-regulated modulator of apoptosis (puma) and noxa by p53. J Biol Chem. 2006;281(11):7260–70.
Fribley A, Wang C-Y. Proteasome inhibitor induces apoptosis through induction of endoplasmic reticulum stress. Cancer Biol Ther. 2006;5(7):745–8.
Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10(1):51–64.
Amaravadi RK, Yu D, Lum JJ, Bui T, Christophorou MA, Evan GI, et al. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Investig. 2007;117(2):326–36.
O’Donovan TR, O’Sullivan GC, McKenna SL. Induction of autophagy by drug-resistant esophageal cancer cells promotes their survival and recovery following treatment with chemotherapeutics. Autophagy. 2011;7(5):509–24.
Yuasa T, Ohno S, Kehrl JH, Kyriakis JM. Tumor Necrosis Factor Signaling to Stress-activated Protein Kinase (SAPK)/Jun NH2-terminal Kinase (JNK) and p38 germinal center kinase couples traf2 to mitogen-activated protein kinase/erk kinase kinase 1 and sapk while receptor interacting protein associates with a mitogen-activated protein kinase kinase kinase upstream of mkk6 and p38. J Biol Chem. 1998;273(35):22681–92.
Osborn MT, Chambers TC. Role of the stress-activated/c-Jun NH2-terminal protein kinase pathway in the cellular response to adriamycin and other chemotherapeutic drugs. J Biol Chem. 1996;271(48):30950–5.
Shimizu H, Kitajima Y, Banno Y, Sumi N, Naganawa T, Nozawa Y. Activation of p38 mitogen-activated protein kinase and caspases in UVB-induced apoptosis of human keratinocyte HaCaT cells. J Invest Dermatol. 1999;112(5):769–74.
** C, ** Z, Chen N-Z, Lu M, Liu C-B, Hu W-L, et al. Activation of IRE1α-XBP1 pathway induces cell proliferation and invasion in colorectal carcinoma. Biochem Biophys Res Commun. 2016;470(1):75–81.
Hetz C, Chevet E, Harding HP. Targeting the unfolded protein response in disease. Nat Rev Drug Discovery. 2013;12(9):703.
Golden EB, Cho H-Y, Jahanian A, Hofman FM, Louie SG, Schönthal AH, et al. Chloroquine enhances temozolomide cytotoxicity in malignant gliomas by blocking autophagy. Neurosurg Focus. 2014;37(6):E12.
Kroemer G, Jäättelä M. Lysosomes and autophagy in cell death control. Nat Rev Cancer. 2005;5(11):886–97.
Wang Z, Shi X, Li Y, Fan J, Zeng X, **an Z, et al. Blocking autophagy enhanced cytotoxicity induced by recombinant human arginase in triple-negative breast cancer cells. Cell Death Dis. 2014;5(12):e1563-e.
Lin A, Dibling B. The true face of JNK activation in apoptosis. Aging Cell. 2002;1(2):112–6.
**g L, Anning L. Role of JNK activation in apoptosis: a double-edged sword. Cell Res. 2005;15(1):36–42.
Leppä S, Bohmann D. Diverse functions of JNK signaling and c-Jun in stress response and apoptosis. Oncogene. 1999;18(45):6158–62.
Matsukawa J, Matsuzawa A, Takeda K, Ichijo H. The ASK1-MAP kinase cascades in mammalian stress response. J Biochem. 2004;136(3):261–5.
Volloch V, Gabai VL, Rits S, Force T, Sherman MY. HSP72 can protect cells from heat-induced apoptosis by accelerating the inactivation of stress kinase JNK. Cell Stress Chaperones. 2000;5(2):139.
Acknowledgements
This work was funded by courtesy of Ernst Jung Foundation and by Takeda Pharmaceutical. We thank Univ.-Prof. Dr. Michael Huber, RWTH Aachen University Hospital, for general, infrastructural and administrative support and supervision. In addition, we thank the RWTH centralizedrealizederial Bank (cBMB), RWTH Aachen University Hospital, Germany, for the access to primary patient samples.
Funding
Open Access funding enabled and organized by Projekt DEAL. This work was supported by funding from the Ernst Jung Foundation (BKM) and by Takeda Pharmaceutical (BKM, IA). In addition, IA gratefully received funding by the German Cancer Aid (Deutsche Krebshilfe, fund no. 70111714).
Author information
Authors and Affiliations
Contributions
Conceptualization: Iris Appelmann, Behzad Kharabi Masouleh. Data curation: Azam Salimi. Formal analysis: Azam Salimi, Iris Appelmann. Funding Acquisition: Iris Appelmann, Behzad Kharabi Masouleh. Investigation: Azam Salimi, Iris Appelmann, Kema Marlen Schroeder, Saskia Maletzke. Methodology: Azam Salimi, Iris Appelmann, Margherita Vieri. Project Administration: Iris Appelmann. Supervision: Iris Appelmann. Validation: Azam Salimi, Iris Appelmann. Visualization: Azam Salimi, Iris Appelmann. Writing – original draft preparation: Azam Salimi, Iris Appelmann. Writing – review & editing: Iris Appelmann, Azam Salimi, Mirle Schemionek, Deniz Gezer, Behzad Kharabi Masouleh. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Primary bone marrow cells from patients with multiple myeloma were kindly provided by RWTH Biobank, RWTH Aachen University Hospital, Germany, in accordance with the Ethics committee of Faculty Medicine RWTH Aachen after written informed consent. All methods were performed in accordance with the relevant guidelines and regulations.
Competing interests
IA and BKM received funding from Takeda Pharmaceutical®. Other authors have no competing interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Figure S1. Effect of IRE1α inhibitors in combination with ixazomib on cytotoxicity of multiple myeloma cells. Figure S2. Ixazomib in combination with A106 arrests cell cycle at G1 phase. Figure S3. Autophagy inhibitors lead to an accumulation of LC3II. Table 1. The list of human primers used for quantitative RT-PCR in this study.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Salimi, A., Schroeder, K.M., Schemionek-Reinders, M. et al. Targeting autophagy increases the efficacy of proteasome inhibitor treatment in multiple myeloma by induction of apoptosis and activation of JNK. BMC Cancer 22, 735 (2022). https://doi.org/10.1186/s12885-022-09775-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-022-09775-y