
Abstract
Background
Ocular infection with Toxoplasma gondii is a major preventable cause of blindness, especially in young people. The aim of the present study was to assess detection rate of T. gondii DNA in blood samples of clinically diagnosed of ocular toxoplasmosis using uracil DNA glycosylase-supplemented loop-mediated isothermal amplification (UDG-LAMP) and real-time quantitative PCR (qPCR) based on REP-529 and B1.
Methods
One hundred and seventeen patients with clinically diagnosed ocular toxoplasmosis (OT) were participated in the study as well as 200 control patients. Peripheral blood samples were assessed using UDG-LAMP and qPCR techniques targeting REP-529 and B1.
Results
Detection limits of qPCR using REP-529 and B1 were estimated as 0.1 and 1 fg of T. gondii genomic DNA, respectively. The limits of detection for UDG-LAMP using REP-529 and B1 were 1 and 100 fg, respectively. In this study, 18 and 16 patients were positive in qPCR using REP-529 and B1, respectively. Based on the results of UDG-LAMP, 15 and 14 patients were positive using REP-529 and B1, respectively. Results of the study on patients with active ocular lesion showed that sensitivity of REP-529 and BI targets included 64 and 63%, respectively using qPCR. Sensitivity of 62 and 61%, were concluded from UDG-LAMP using REP-529 and B1 in the blood cases of active ocular lesion. qPCR was more sensitive than UDG-LAMP for the detection of Toxoplasma gondii DNA in peripheral blood samples of patients with clinically diagnosed toxoplasmic chorioretinitis. Furthermore, the REP-529 included a better detection rate for the diagnosis of ocular toxoplasmosis in blood samples, compared to that the B1 gene did. Moreover, the qPCR and UDG-LAMP specificity assessments have demonstrated no amplifications of DNAs extracted from other microorganisms based on REP-529 and B1.
Conclusions
Data from the current study suggest that qPCR and UDG-LAMP based on the REP-529 are promising diagnostic methods for the diagnosis of ocular toxoplasmosis in blood samples of patients with active chorioretinal lesions.
Similar content being viewed by others

Background
Ocular toxoplasmosis (OT), one of the major causes of posterior uveitis globally, can lead to vision-threatening complications such as retinal detachment, choroidal neovascularization and glaucoma. The disease is associated with congenital or postnatally acquired infection caused by the ubiquitous apicomplexan parasite Toxoplasma gondii (T. gondii) and classically presents as a necrotizing retinochoroiditis that can be single [1], multiple [2] or satellite [3] to atrophic-pigmented scars. Congenitally infected people, who are asymptomatic at birth [4], may later develop toxoplasmosis symptoms [5]. Diagnosis of OT is routinely carried out through ophthalmic examinations and various clinical findings that confirm T. gondii infection of the retinochoroiditis. However, the clinical presentation can at times prove to be misleading [6, 7], requiring further biological tests [8] to be either confirmed or refuted [9]. When definitive clinical diagnosis cannot be carried out, a direct detection of T. gondii DNA using conventional polymerase chain reaction (PCR) [10] and antibody detection [11] with titer interpretation from the blood and/or ocular samples are successfully used to verify the primary diagnosis [12,13,14]. These methods cannot only confirm the OT diagnosis but can also rule out other similar infectious diseases [7, 15].
From various molecular techniques, real-time quantitative PCR (qPCR) and LAMP assays are particularly popular because of their high sensitivity, specificity and speed. The LAMP technique is carried out under isothermal reaction conditions and does not need advanced equipment [16,17,18,19]. The major problem of using molecular methods for the ocular fluids is linked to the invasive nature of these methods [14]. Significantly, peripheral blood sampling is less invasive than ocular fluid sampling. In the current study, qPCR and UDG-LAMP assays were assessed for the detection of T. gondii DNA in blood samples of clinically diagnosed OT patients using repetitive REP-529 sequence and B1 gene.
Methods
Patients and clinical sample collections
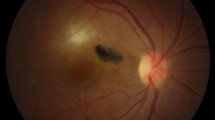
This cross-sectional study was designed using 117 immunocompetent patients with clinically diagnosed OT in Farabi Eye Hospital, Tehran, Iran, from October 2015 to August 2017. The OT was clinically diagnosed by a retina specialist (MZ). Chorioretinal involvements of T. gondii were classified into three major groups based on fundoscopic examination, including (1) Group A (active lesion): an area of white edematous retina with ill-defined borders and overlying vitritis, usually accompanied by nearby vasculitis (Fig. 1A). This appearance represents the active form of disease and when seen with no scars is considered as a consequence of the primary acquired infection [20]; (2) Group B (old scar): a chorioretinal scar with sharp borders and variable degrees of dark pigmentation (Fig. 1B). This appearance represents the inactive form of disease and is considered as a reminiscence of the active prior chorioretinitis [21, 22] and (3) Group C (reactivated disease): a combination of the two forms, including an area of active chorioretinitis in eyes with toxoplasmic chorioretinal scars, usually close to an active chorioretinitis focus (Fig. 1C). This form may be resulted from the failure of immune system to continually limit the infected foci and subsequent release of the tissue cyst content into the close tissues [23]. Moreover, 200 patients with other eye diseases other than toxoplasmosis, including viral keratitis (39 samples), amebic keratitis (38 samples), fungal keratitis (42 samples), bacterial endophthalmitis (64 samples) and cataract (17 samples) were participated as controls. Blood samples (up to 3.5 mL) were collected from the participants using EDTA-containing tubes. These blood samples have been verified using various biological techniques by the authors in previous studies [24, 25].

Clinical manifestation of ocular toxoplasmosis; A Toxoplasma retinitis (active disease), B Retinochoroiditis scar (old scar), C Toxoplasma retinitis and adjacent retinochoroiditis scar (reactivated disease)
DNA extraction
DNA was extracted from the whole blood using QIAamp Genomic DNA Blood Mini Extraction Kit (Qiagen, Hilden, Germany) based on the manufacturer instructions and kept frozen at − 20 °C for further use in qPCR and UDG-LAMP reactions [25]. The reference DNA was extracted from a virulent RH strain of T. gondii (Type I), which was previously collected from peritoneal cavity of the infected mice [26].
qPCR
A TaqMan-probe qPCR targeting REP-529 and B1 was developed using StepOne Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) with 45 cycles of amplification. REP-529 is a highly repetitive sequence with 200–300 copies in T. gondii genome [27]. The B1 gene has 35 copies in the genome and is conserved indifferent parasite strains [28]. The PCR reaction included RealQ Plus Master Mix for probes labeled with reporter fluorophor 6-carboxyfluorescein (Ampliqon, Odense, Denmark) (10 µL), TagMan probe (FAM-CCCTCGCCCTCTTCTCCACTCTTCAA-3-BHQ1) (0.2 µM), extracted DNA (5 µL), forward (5ʹ-CTTCGTCCAAGCCTCCGA-3ʹ) and reverse (5ʹ-GACGCTTTCCTCGTG GTGAT-3ʹ) primers (0.4 µL) and distilled water (4 µL). The PCR thermal cycling was carried out at 95 °C for 5 min and continued for 40 cycles of 95 °C for 1 min, annealing at 55 °C for 1 min and extension at 72 °C for 1 min. The gapdh gene was used as housekee** gene with primers common to all mammalian species to check the quality of DNA [18]. An internal control (TaqMan exogenous positive control) was used, and each run was considered valid if the positive, negative, and internal controls were acceptable. The result was considered negative if repeated atypical amplification curves with proper amplification of the internal control was present. A sample was considered inhibited if amplification of the internal control failed and there was no amplification or atypical amplification curve for the target of interest.
Uracil DNA glycosylase-supplemented loop-mediated isothermal amplification assay
UDG-LAMP assay was successfully performed in our laboratory as described previously [29]. Specific oligonucleotide primers for T. gondii used for the LAMP assay were designed based on REP-529 and B1 regions of the parasite genome (Table 1). The total reaction master mix volume was 25 μL consisting of 1 μL of template DNA, 40 picomol of each of FIP and BIP primers, 20 pmol of each LF and LB primers (used only in REP-LAMP), 5 pmol of each of F3 and B3 primers, 8 U of Bst2 DNA polymerase (New England Biolabs, USA), 1.4 mmol/L of deoxyuridine triphosphates (dUTP) instead of dTTP and 2X reaction buffer, containing 1.6 mol/L betaine (Sigma Aldrich, USA), 40 mmol/L Tris–HCl (pH 8.8), 20 mmol/L of KCL, 20 mmol/L of (NH4)2SO4, 16 mmol/L of MgSO4 and 0.2% tween 20). The LAMP assay were carried out at 60–67 °C for 30, 45, 60 and 75 min to find the optimum time and temperature conditions and finally; 63 °C and 60 min were the best temperature and time for both target genes, respectively. Addition of 3 µL of the fluorescent detection reagent, diluted SYBR Green I (Invitrogen, Carlsbad, California, USA), to the reaction tube after LAMP reaction enables visible analysis of the results under daylight and/or UV light. The positive amplification was read through observation of change in color of the reaction mixture following addition of dye to the tube and the color changes from orange (negative reaction) to green (positive reaction) (Fig. 2). The results were further verified by electrophoresis of LAMP products in 1.5% agarose gel electrophoresis stained with DNA Safe Stain (SinaClon, Iran), and visualized under UV light. To prevent self-amplification of the primers, reactions were carried out in the absence of DNA templates. The T. gondii RH-strain DNA and double distilled water were used as positive and no template controls, respectively. To assess reproducibility of the assay, all experiments were carried out in duplicate. The protocol is fully described in a previous study by the authors [29, 30].

Tubes with UDG-LAMP reaction stained with SYBR Green I. C+ , positive control; C-, negative control; 1–4, different patients with positive reactions. The color changes from orange (negative reaction) to green (positive reaction)
Analytical sensitivity and specificity of qPCR and UDG-LAMP assays
To assess analytical sensitivity of the qPCR and UDG-LAMP techniques for the detection of T. gondii (RH strain) DNA, tenfold serial dilutions of T. gondii DNA with 1 pg to 0.01 fg concentrations were prepared and used in qPCR and LAMP assays targeting REP-529 and B1. The construction is performed with various concentrations of DNA. Next, the cycle threshold (CT) values were plotted as mean (triplicate) against the standard curve values to determine the detection limit of both primer sets. Parasite concentrations are determined after the calculation of the linear regression equation (y = ax + b), where y = CT; a = curve slope (slope); x = parasite number; and b = where the curve intersects y-axis (y intercept) [31, 32].
To assess the analytical specificity of the qPCR and UDG-LAMP techniques, extracted DNAs from other microorganisms, including Leishmania tropica, L. major, Giardia lamblia, Cryptosporidium parvum, Entamoeba dispar, Trichostrongylus colubriformis, Corynosoma capsicum, Candida albicans and Acanthamoeba spp. as well as human chromosomal DNA, were used as templates in qPCR and LAMP assays using specific primers of REP-529 and B1.
Statistical analysis
Data were analyzed using SPSS Software v.21 (SPSS Inc. Chicago, IL, USA). The χ2 test was used to calculate diagnostic sensitivity and specificity of the qPCR and UDG-LAMP techniques. Agreements between the clinical and molecular assays were demonstrated using Cohen’s kappa coefficient [33]. Linear regressions were constructed from the standard curves for REP-529 and B1 and CT mean comparison of both primer pairs. Both curves were statistically analyzed using an unequal-variance t-test based on a critical value of p ≤ 0.05.
Results
Distribution of participants
Of the total samples, 55.55% (65/117) belonged to males and 44.44% (52/117) to females. Thirty six (30.76%) patients were up to 10 years old, 50 (42.73%) patients were 11–20 years old, 23 (19.65%) patients were 21–30 years old and eight (6.83%) patients were 31–40 years old. Of 117 patients, 37 (31.62%) patients included active lesions (Group A), 13 (11.11%) patients included pigmented scars (Group B) and 67 (57.26%) patients included pigmented scars with active lesions (Group C).
qPCR and UDG-LAMP of the clinical samples
Out of 37 patients in Group A (patients with active lesions), 18 and 16 patients were positive by qPCR using REP-529 and B1 and 15 and 14 patients were positive by UDG-LAMP using REP-529 and B1, respectively. All positive results using UDG-LAMP had 100% overlaps with positive results of qPCR using REP-529 and B1. None of the patients in other clinical groups (Groups B and C) were positive using molecular assays (Table 2). All patients in control group were negative for REP-529 and B1 using qPCR and UDG-LAMP. Amplification plot and derivative melt curve analysis of the T. gondii based on REP-529 and B1 targets are shown in Fig. 3. The results of chi-square test indicated that qPCR method based on REP-529 was the most sensitive than UDG-LAMP using REP-529 and B1 (P = 0.003).

qPCR amplification plot based on REP-529 and B1 targets of T. gondii from blood samples of patients suspected to ocular toxoplasmosis
Analytical sensitivity of qPCR and UDG-LAMP assays
The first step was to assess the reportable range of the reaction that was determined utilizing T. gondii DNA isolate from tachyzoites (RH strain) using the REP-529 (Fig. 4A) and B1 (Fig. 4B). The current results showed that the detection limits for UDG-LAMP using REP-529 and B1 were 1 and 100 fg, respectively. Detection limits of qPCR using REP-529 and B1 were estimated as 0.1 and 1 fg of T. gondii genomic DNA, respectively (Figs. 4, 5).

Standard curve of T. gondii, tachyzoites using REP-529 (A) and B1 (B) primer sets, respectively. Results are shown as mean cycle threshold (CT) obtained from triplicate of each DNA concentration. Standard curve analysis was performed in tenfold serial dilutions of T. gondii DNA extracted from tachyzoites, at initial concentration of 1000 fg/μL

Comparison of the sensitivity of the REP-529 bp (A) and B1 (B) genes using UDG-LAMP assay in diagnosis of T. gondii (RH strain) DNA. N: negative control, P: positive control, M: 100 bp DNA Marker, lanes 1–6: 1 pg, 100 fg, 10 fg, 1 fg, 0.1 fg and 0.01 fg of T. gondii (RH strain) DNA
Specificity of qPCR and UDG-LAMP assays
Using qPCR and UDG-LAMP, no amplifications were detected in reactions with DNA of G. lamblia, C. parvum, E. dispar, L. major, Acanthamoeba spp., T. colubriformis, C. capsicum and C. albicans as well as human genomic DNA (Fig. 6).

Analytical specificity of UDG-LAMP assay. P: positive control (T. gondii, RH strain), lanes 1–9: DNA of Giardia lamblia, Cryptosporidium parvum, Entamoeba dispar, Leishmania major, Acanthamoeba, Trichostrongylus colubriformis, Corynosoma capsicum, Candida albicans and Human Genomic DNA
Discussion
Although clinical examination is the standard method for the diagnosis of OT in the second and third forms of the disease (old scars and reactivated disease), the first form of disease (active lesions) cannot always be differentiated from other chorioretinal inflammations based on funduscopic appearance alone [34]. Based on the previous studies, ocular fluid samples are the most sensitive sources for the molecular diagnosis of OT [15, 34, 35]. Molecular examination of the aqueous humor puncture allows identification of coinfections or various etiological agents for the infectious uveitis. It is noteworthy that coinfections need various treatments in addition to anti-Toxoplasma therapy [15]. However, the most important limitation of this method is linked to its invasive nature that may hurt the eyes [20, 36].
PCR of blood samples from patients with OT produced similar results to those from PCR of aqueous humor samples, avoiding problems associated with ocular puncture [37]. More recently, Khanaliha et al. have reported that peripheral blood mononuclear cells (PBMCs) are appropriate for the assessment of toxoplasmic chorioretinitis. Furthermore they have reported that PCR with bradyzoite genes is useful for the diagnosis of toxoplasmic chorioretinitis in these cells [38]. In a previous study by the current authors, agreements between the two approaches of nested PCR and serological assays were assessed and results highlighted that nested-PCR of peripheral blood samples was a useful minimally invasive technique for providing direct evidence of the presence of T. gondii in patients with OT, especially in recently acquired infections. Nested-PCR with REP-529 target included 57 and 100% sensitivity and specificity in diagnosis of recently acquired OT, respectively [25]. In contrast, studies have revealed that molecular testing on peripheral blood samples is not sufficiently sensitive for the detection of T. gondii in patients with OT [35]. In a study, Bourdin et al. reported 35.9% sensitivity in diagnosis of OT using PCR of peripheral blood samples. These studies showed that in OT it is possible to detect T. gondii DNA in peripheral blood samples without a direct relation of Toxoplasma activity within the eye. They concluded that PCR technique was not sensitive enough for the diagnosis of OT from blood samples [39]. Several factors affect sensitivity of molecular techniques, including volumes and types of the samples, DNA markers, gene targets and types of the molecular methods [34].
In the current study, we detected T. gondii DNA in the blood of patients with active ocular lesions (Group A) using qPCR and UDG-LAMP based on REP-529 and B1, but not in the blood of patients with old scars (Group B) and reactivated disease (Group C), which suggests that parasitaemia is associated with ongoing disease. Tachyzoites of T. gondii have been isolated from the blood of immunocompetent patients with the ocular disease but not from the blood of patients with recurrent retinochoroiditis [40]. However, Silveira et al. have reported that subclinical parasitaemia is present in patients with either acute or chronic toxoplasmosis, regardless of the presence of ocular disease [41]. Although assays of this study did not include high sensitivities in all OT patients, these sensitivities were acceptably high in patients from the Group A. The presence of an old pigmented scar is usually the most useful and characteristic clinical finding in the diagnosis of OT. This clinical finding is absent in patients with active lesions resulted from recently acquired toxoplasmosis in contrast to patients with active lesions resulted from reactivation of congenitally acquired toxoplasmosis (Group C) [2, 22]. It is noteworthy that the Group B patients included old inactive scars alone with no active chorioretinitis lesions and hence did not need treatments and Group C patients included pathognomonic clinical findings of simultaneous presence of old scars and active chorioretinitis lesions and thus did not need confirmatory paraclinical diagnostic assessments [21, 23].
In the current study, detection limits of qPCR using REP-529 and B1 targets were respectively estimated as 0.1 and 1 fg of the T. gondii genomic DNA. Detection limits of UDG-LAMP using the highlighted targets were 1 and 100 fg of the T. gondii genomic DNA, respectively. Kong et al. reported detection limits of RE-LAMP, B1-LAMP and RE-nested PCR assays as 0.6, 60 and 600 fg of the DNA templates, respectively [42]. A similar study by Zhang et al. revealed that the detection limits of LAMP and conventional PCR assays using REP-529 target were 1 and 10 pg, respectively [43]. Various target genes such as SAG1, SAG2, SAG3, SAG4, ITS, B1, REP-529, P30, GRA4, GRA6 and GRA7 have been used to detect toxoplasmosis [44,45,46,47,48]. The results have shown that both REP-529 and B1 are highly sensitive; REP-529 is more sensitive than B1 gene. [32, 49, 50]. Therefore, the present technique was based on the REP-529 and B1. Based on the results from a study by da Silva et al., it has been shown that the REP-529 includes 200–300 copies in the genome of Toxoplasma parasite, the highest copy number within all studied genes that are quite specific to T. gondii [51]. Furthermore, several studies have reported that the B1 gene includes 35 copies in genome of T. gondii, which is completely specific [17, 52,53,54]. The SAG1, SAG2, SAG3, SAG4, GRA4, GRA6 and GRA7 genes include only one copy in genome of T. gondii, which are specific as well [46]. Studies have shown that the number of copies of each gene fragment includes direct relationships with the diagnosis sensitivity [48, 52]. Another factor that is important in selecting appropriate gene fragments is specificity [55]. The current results have shown no cross-reactivity with DNA corresponding to parasitic infections other than toxoplasmosis using qPCR and UDG-LAMP based on REP-529 and B1, thereby insuring high specificity for target amplification [27, 56].
Conclusion
Results of the current study have shown that samples from peripheral blood samples can potentially be used for the molecular diagnosis of OT in patients with active chorioretinal lesions. In the present study, qPCR was more sensitive than UDG-LAMP. However, UDG-LAMP can be used in fields, potentially excluding needs of expensive PCR machines and gel electrophoresis devices as well as laborious DNA extraction processes. Regarding the gene target, RE was more sensitive than B1 gene. Moreover, the qPCR and UDG-LAMP specificity assessments have demonstrated no amplifications of DNAs extracted from other microorganisms based on REP-529 and B1. Therefore, data from the current study suggest qPCR and UDG-LAMP of REP-529 as promising diagnostic techniques for the diagnosis of OT in blood samples of patients with active chorioretinal lesions.
Availability of data and materials
All data generated or analyzed during this study are included in this published article. The raw data are available from the corresponding author on reasonable request.
Abbreviations
- OT:
-
Ocular toxoplasmosis
- UDG-LAMP:
-
Uracil DNA glycosylase-supplemented loop-mediated isothermal amplification
- PCR:
-
Polymerase chain reaction
- EDTA:
-
Ethylenediaminetetraacetic acid
- SPSS:
-
Statistical package for the social sciences
References
Silveira C, Belfort R Jr, Burnier M Jr, Nussenblatt R. Acquired toxoplasmic infection as the cause of toxoplasmic retinochoroiditis in families. Am J Ophthalmol. 1988;106(3):362–4.
Nussenblatt RB, Belfort R Jr. Ocular toxoplasmosis. An old disease revisited. JAMA. 1994;271(4):304–7.
Silveira C, Ferreira R, Muccioli C, Nussenblatt R, Belfort R Jr. Toxoplasmosis transmitted to a newborn from the mother infected 20 years earlier. Am J Ophthalmol. 2003;136(2):370–1.
Shojaee S, Teimouri A, Keshavarz H, Azami SJ, Nouri S. The relation of secondary sex ratio and miscarriage history with Toxoplasma gondii infection. BMC Infect Dis. 2018;18(1):307. https://doi.org/10.1186/s12879-018-3228-0. PMID: 29976155; PMCID: PMC6034284.
Teimouri A, Mohtasebi S, Kazemirad E, Keshavarz H. Role of Toxoplasma gondii IgG avidity testing in discriminating between acute and chronic toxoplasmosis in pregnancy. J Clin Microbiol. 2020;58(9):e00505–20. https://doi.org/10.1128/JCM.00505-20. PMID: 32321784; PMCID: PMC7448626.
Johnson MW, Greven CM, Jaffe GJ, Sudhalkar H, Vine AK. Atypical, severe toxoplasmic retinochoroiditis in elderly patients. Ophthalmology. 1997;104:48–57.
Fardeau C, Romand S, Rao NA, Cassoux N, Bettembourg O, Thulliez P, et al. Diagnosis of toxoplasmic retinochoroiditis with atypical clinical features. Am J Ophthalmol. 2002;134:196–203.
Stanford MR, Gras L, Wade A, Gilbert RE. Reliability of expert interpretation of retinal photographs for the diagnosis of Toxoplasma retinochoroiditis. Br J Ophthalmol. 2002;86:636–9.
Ali-Heydari S, Keshavarz H, Shojaee S, Mohebali M. Diagnosis of antigenic markers of acute toxoplasmosis by IgG avidity immunoblotting. Parasite. 2013;20:18. https://doi.org/10.1051/parasite/2013017. PMID: 23688778; PMCID: PMC3718541.
Montoya JG, Parmley S, Liesenfeld O, Jaffe GJ, Remington JS. Use of the polymerase chain reaction for diagnosis of ocular toxoplasmosis. Ophthalmology. 1994;106:1554–63.
Montoya JG. Laboratory diagnosis of Toxoplasma gondii infection and toxoplasmosis. J Infect Dis. 2002;185(Suppl 1):e73e82.
Garweg JG, Jacquier P, Boehnke M. Early aqueous humor analysis in patients with human ocular toxoplasmosis. J Clin Microbiol. 2000;38:996–1001.
Park YH, Nam HW. Clinical features and treatment of ocular toxoplasmosis. Korean J Parasitol. 2013;51(4):393–9.
Greigert V, Foggia ED, Filisetti D, Villard O, Pfaff AW, Sauer A, Candolfi E. When biology supports clinical diagnosis: review of techniques to diagnose ocular toxoplasmosis. Br J Ophthalmol. 2019;103:1008–12.
de-la-Torre A, Valdés-Camacho J, de Mesa CL, Uauy-Nazal A, Zuluaga JD, et al. Coinfections and differential diagnosis in immunocompetent patients with uveitis of infectious origin. BMC Infect Dis. 2019;19:91.
Contini C, Seraceni S, Cultrera R, Incorvaia C, Sebastiani A, Picot S. Evaluation of a real-time PCR-based assay using the lightcycler system for detection of Toxoplasma gondii bradyzoite genes in blood specimens from patients with toxoplasmic retinochoroiditis. Int J Parasitol. 2005;35:275–83.
Kompalic-Cristo A, Frotta C, Suárez-Mutis M, Fernandes O, Britto C. Evaluation of a real-time PCR assay based on the repetitive B1 gene for the detection of Toxoplasma gondii in human peripheral blood. Parasitol Res. 2007;101(3):619–25.
Mousavi P, Mirhendi H, Mohebali M, Shojaee S, Keshavarz Valian H, Fallahi S, Mamishi S. Detection of Toxoplasma gondii in acute and chronic phases of infection in immunocompromised patients and pregnant women with real-time PCR assay using TaqMan fluorescent probe. Iran J Parasitol. 2018;13(3):373–81. PMID: 30483328; PMCID: PMC6243173.
Obande GA, Singh KKB. Current and future perspectives on isothermal nucleic acid amplification technologies for diagnosing infections. Infect Drug Resist. 2020;13:455–83.
Braakenburg A, Rothova A. Advances in ocular toxoplasmosis. In: Uveitis: an update. Cham: Springer; 2016. p. 1–7.
Grigg ME, Dubey JP, Nussenblatt RB. Ocular toxoplasmosis: lessons from Brazil. Am J Ophthalmol. 2015;159:999–1001.
Vasconcelos-Santos DV. Ocular toxoplasmosis. In: Rao N, Schallhorn J, Rodger D, editors. Posterior uveitis. Essentials in ophthalmology. Cham: Springer; 2019. https://doi.org/10.1007/978-3-030-03140-4_5.
Jones JL, Bonetti V, Holland GN, Press C, Sanislo SR, Khurana RN, Montoya JG. Ocular toxoplasmosis in the United States: recent and remote infections. Clin Infect Dis. 2015;60:271–3.
Rahimi-Esboei B, Zarei M, Mohebali M, Valian HK, Shojaee S, Mahmoudzadeh R, Salabati M. Serologic tests of IgG and IgM antibodies and IgG avidity for diagnosis of ocular toxoplasmosis. Korean J Parasitol. 2018;56(2):147–52. https://doi.org/10.3347/kjp.2018.56.2.147. PMID: 29742869; PMCID: PMC5976017.
Rahimi Esboei B, Kazemi B, Zarei M, Mohebali M, Keshavarz Valian H, Shojaee S, Zahedipour F, Fallahi S, Mousavi P, Mahmoudzadeh R, Salabati M. Evaluation of RE and B1 genes as targets for detection of Toxoplasma gondii by nested PCR in blood samples of patients with ocular toxoplasmosis. Acta Parasitol. 2019;64(2):384–9. https://doi.org/10.2478/s11686-019-00056-6. PMID: 31020496.
Azami SJ, Teimouri A, Keshavarz H, Amani A, Esmaeili F, Hasanpour H, Elikaee S, Salehiniya H, Shojaee S. Curcumin nanoemulsion as a novel chemical for the treatment of acute and chronic toxoplasmosis in mice. Int J Nanomed. 2018;13:7363–74. https://doi.org/10.2147/IJN.S181896. PMID: 30519020; PMCID: PMC6233476.
Homan WL, Vercammenb M, De Braekeleer J, Verschueren H. Identification of a 200- to 300-fold repetitive 529 bp DNA fragment in Toxoplasma gondii, and its use for diagnostic and quantitative PCR. Inter J Parasitol. 2000;30:69-75.30.
Burg JL, Grover CM, Pouletty P, Boothroyd JC. Direct and sensitive detection of a pathogenic protozoan, Toxoplasma gondii, by polymerase chain reaction. J Clin Microbiol. 1989;27:1787–92.
Keshavarz Valian H, Mirhendi H, Mohebali M, Shojaee S, Fallahi S, et al. Comparison of the RE-529 sequence and B1 gene for Toxoplasma gondii detection in blood samples of the at-risk seropositive cases using uracil DNA glycosylase supplemented loop-mediated isothermal amplification (UDG-LAMP) assay. Microb Pathog. 2020;140:103938. https://doi.org/10.1016/j.micpath.2019.103938. PMID: 31862390.
Fallahi S, Moosavi SF, Karimi A, Chegeni AS, Saki M, et al. An advanced uracil DNA glycosylase-supplemented loop-mediated isothermal amplification (UDG-LAMP) technique used in the sensitive and specific detection of Cryptosporidium parvum, Cryptosporidium hominis, and Cryptosporidium meleagridis in AIDS patients. Diagn Microbiol Infect Dis. 2018;91:6–12.
Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem. 2009;55:611–22.
Camilo LM, Pereira-Chioccola VL, Gava R, Meira-Strejevitch CDS, Vidal JE, Brandão de Mattos CC, Frederico FB, De Mattos LC, Spegiorin LCJF, FAMERP Toxoplasma Research Group. Molecular diagnosis of symptomatic toxoplasmosis: a 9-yearretrospective and prospective study in a referral laboratory in São Paulo, Brazil. Braz J Infect Dis. 2017;21(6):638–47.
Swets JA. Measuring the accuracy of diagnostic systems. Science. 1988;240:1285–93. https://doi.org/10.1126/science.3287615.
Ozgonul C, Besirli CG. Recent developments in the diagnosis and treatment of ocular toxoplasmosis. Ophthalmic Res. 2017;57:1–12.
Gómez Marín JE, Zuluaga JD, Pechené Campo EJ, Triviño J, De-la-Torre A. Polymerase chain reaction (PCR) in ocular and ganglionar toxoplasmosis and the effect of therapeutics for prevention of ocular involvement in South American setting. Acta Trop. 2018;184:83–7.
Garcia JL, Burrells A, Bartley PM, Bartley K, Innes EA, Katzer F. The use of ELISA, nPCR and qPCR for diagnosis of ocular toxoplasmosis in experimentally infected pigs. Vet Sci Res J. 2017;115:490–5.
Bou G, Figueroa MS, Martí-Belda P, Navas E, Guerrero A. Value of PCR for detection of Toxoplasma gondii in aqueous humor and blood samples from immunocompetent patients with ocular toxoplasmosis. J Clin Microbiol. 1999;37:3465–8.
Khanaliha K, Hedayatfar A, Minaeian S, Bokharaei-Salim F, Alemzadeh SA, Garshasbi S, FagheeiAghmiyuni Z, Salemi B. Detection of Toxoplasma gondii bradyzoite genes in the peripheral blood mononuclear cells among patients with toxoplasmic chorioretinitis. Trans R Soc Trop Med Hyg. 2021;Apr13:trab062.
Bourdin C, Busse A, Kouamou E, Touafek F, Bodaghi B, et al. PCR-based detection of Toxoplasma gondii DNA in blood and ocular samples for diagnosis of ocular toxoplasmosis. J Clin Microbiol. 2014;52:3987–91.
Amendoeira MR, Coutinho SG. Isolation of Toxoplasma gondii from the saliva and tonsils of a three-year-old child. J Infect Dis. 1982;145:587.
Silveira C, Vallochi AL, da Silva UR, Muccioli C, Holland GN, et al. Toxoplasma gondii in the peripheral blood of patients with acute and chronic toxoplasmosis. Br J Ophthalmol. 2011;95:396–400.
Kong QM, Lu SH, Tong QB, Lou D, Chen R, Zheng B, Kumagai T, Wen LY, Ohta N, Zhou XN. Loop-mediated isothermal amplification (LAMP): early detection of Toxoplasma gondii infection in mice. Parasit Vectors. 2012;5:2–7.
Zhang H, Thekisoe OM, Aboge GO, Kyan H, Yamagishi J, Inoue N, Nishikawa Y, Zakimi S, Xuan X. Toxoplasma gondii: sensitive and rapid detection of infection by loop-mediated isothermal amplification (LAMP) method. Exp Parasitol. 2009;122:47–50.
Selseleh MM, Keshavarz H, Mohebali M, Shojaee S, Modarressi M, Eshragian M, Selseleh MM. Production and evaluation of Toxoplasma gondii recombinant surface antigen 1 (SAG1) for serodiagnosis of acute and chronic toxoplasma infection in human sera. Iran J Parasitol. 2012;7(3):1–9. PMID: 23109955; PMCID: PMC3469165.
Selseleh M, Keshavarz H, Mohebali M, Shojaee S, Selseleh M, Eshragian MR, Mansouri F, Modarressi MH. Production and evaluation of Toxoplasma gondii recombinant GRA7 for serodiagnosis of human infections. Korean J Parasitol. 2012;50(3):233–8. https://doi.org/10.3347/kjp.2012.50.3.233. PMID: 22949752; PMCID: PMC3428570.
Holec-Gasior L. Toxoplasma gondii recombinant antigens as tools for serodiagnosis of human toxoplasmosis—the current status of studies. Clin Vaccine Immunol. 2013;20(9):1343–51.
Teimouri A, Modarressi MH, Shojaee S, Mohebali M, Rezaian M, Keshavarz H. Development, optimization, and validation of an in-house Dot-ELISA rapid test based on SAG1 and GRA7 proteins for serological detection of Toxoplasma gondii infections. Infect Drug Resist. 2019;12:2657–2669. https://doi.org/10.2147/IDR.S219281. PMID: 31695442; PMCID: PMC6717716.
Teimouri A, Abbaszadeh Afshar MJ, Mohtasebi S, Jafarpour Azami S, Alimi R, Keshavarz H. Assessment of an in-house enzyme-linked immunosorbent assay and IgG avidity test based on SAG1 and GRA7 proteins for discriminating between acute and chronic toxoplasmosis in humans. J Clin Microbiol. 2021;59(8):e0041621. https://doi.org/10.1128/JCM.00416-21. PMID: 34077255; PMCID: PMC8288279.
Cassaing S, Bessières M, Berry A, Berrebi A, Fabre R, Magnaval J. Comparison between two amplification sets for molecular diagnosis of toxoplasmosis by real-time PCR. J Clin Microbiol. 2006;44:720–4.
Robert-Gangneux F, Darde ML. Epidemiology of and diagnostic strategies for toxoplasmosis. Clin Microbiol Rev. 2012;25:264–96.
da Silva RC, Langoni H, Su C, da Silva AV. Genotypic characterization of Toxoplasma gondii in sheep from Brazilian slaughterhouses: new atypical genotypes and the clonal type II strain identified. Vet Parasitol. 2011;175:173–7.
Okay TS, Yamamoto L, Oliveira LC, Manuli ER, Andrade Junior HFD, Del Negro GMB. Significant performance variation among PCR systems in diagnosing congenital toxoplasmosis in São Paulo, Brazil: analysis of 467 amniotic fluid samples. Clinics. 2009;64:171–6.
Correia CC, Melo HR, Costa VM. Influence of neurotoxoplasmosis characteristics on real-time PCR sensitivity among AIDS patients in Brazil. Trans R Soc Trop Med Hyg. 2010;104:24–8.
Buchbinder S, Blatz R, Rodloff AC. Comparison of real-time PCR detection methods for B1 and P30 genes of Toxoplasma gondii. Diagn Microbiol Infect Dis. 2003;45:269–71.
Ossorio PN, Sibley LD, Boothroyd JC. Mitochondrial-like DNA sequences flanked by direct and inverted repeats in the nuclear genome of Toxoplasma gondii. J Mol Biol. 1991;222:525–36.
Notomi T, Hiroto O, Masubuchi H, Yonekawa T, Watanabe K, Amino N, Hase T. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 2000. https://doi.org/10.1093/nar/28.12.e63.
Acknowledgements
We would like to acknowledge all staff from the Toxoplasmosis Laboratory (Department of Medical Parasitology and Mycology, Tehran University of Medical Sciences, Tehran, Iran) and Farabi Eye Hospital, Tehran, Iran, for their useful collaboration.
Funding
This research was financially supported by Tehran University of Medical Sciences with Grant No: 94-03-160-29998.
Author information
Authors and Affiliations
Contributions
HKV and BRE conceived, designed and performed the experiments, analyzed and interpreted the data, and wrote the original draft paper. MZ, RM and MS isolated the samples. SF and BK contributed in qPCR assay. PM contributed in LAMP PCR method. MM, SF, BK, SS, RM and MS, contributed reagents, materials, analysis tools or data. BRE, MZ, MM and AT prepared the manuscript. HKV and AT review and editing of the final version of manuscript. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was carried out based on the ethical standards by institutional and/or national research committees and Helsinki Declaration, 1964. All experimental protocols were approved by the Ethical Committee of Tehran University of Medical Sciences, Tehran, Iran (Ethical no. 29998). Furthermore, informed consents were collected from all the participants before starting the study. Parental consent was obtained from the parent or guardian for participants less than 18 years old included in this study.
Consent for publication
Not applicable.
Competing interests
The authors declare that we do not have any competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Rahimi Esboei, B., Fallahi, S., Zarei, M. et al. Utility of blood as the clinical specimen for the diagnosis of ocular toxoplasmosis using uracil DNA glycosylase-supplemented loop-mediated isothermal amplification and real-time polymerase chain reaction assays based on REP-529 sequence and B1 gene. BMC Infect Dis 22, 89 (2022). https://doi.org/10.1186/s12879-022-07073-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12879-022-07073-3